
Abstract
Background
Deposition of α-synuclein and neuroinflammation are key pathological features of Parkinson’s disease (PD). There is no cure for the disease; however, targeting the pathological features might be available to modulate the disease onset and progression. Hypoestoxide (HE) has been demonstrated as a NF-κB modulator, thereby acting as a potential anti-inflammatory and anti-cancer drug.
Methods
In order to assess the effect of HE in a mouse model of PD, mThy1-α-syn transgenic mice received intraperitoneal (IP) injections of either vehicle or HE (5 mg/kg) daily for 4 weeks.
Results
Treatment of HE decreased microgliosis, astrogliosis, and pro-inflammatory cytokine gene expression in α-syn transgenic mice. HE administration also prevented the loss of dopaminergic neurons and ameliorated motor behavioral deficits in the α-syn transgenic mice, and α-synuclein pathology was significantly reduced by treatment of HE. In addition, increased levels of nuclear phosphorylated NF-κB in the frontal cortex of α-syn transgenic mice were significantly reduced by HE administration.
Conclusions
These results support the therapeutic potential of HE for PD and other α-synuclein-related diseases.
Similar content being viewed by others

Background
Intraneuronal Lewy bodies (LBs) and Lewy neurites (LNs) are key pathological features of Parkinson’s disease (PD) [1]. α-synuclein is a small neuronal protein (140 amino acids), and the pathological amyloid fibrils are a major component of LBs and LNs [2]. While the physiological functions of α-synuclein are still unclear, studies have suggested its roles in neuroplasticity and synaptic vesicle recycling [2]. Furthermore, accumulating evidence has demonstrated that abnormal deposition of α-synuclein is not only a pathological feature but also plays critical roles in the onset and progression of diseases [3].
In addition to α-synuclein deposits, neuroinflammation is another pathological feature of PD [4]. For example, accumulations of reactive microglia have been found in the brains of PD patients, and elevated levels of inflammatory cytokines, such as TNFα and IL6 have been detected in the CSF and plasma of PD patients [5]. In addition, studies have shown that neuroinflammation is not only a pathological feature but also plays critical roles in neurodegeneration. Administration of anti-inflammatory drugs prevented dopamine neuronal degeneration in substantia nigra (SN) of toxicant-induced animal models of PD [6]. Epidemiological studies have suggested that administration of non-steroidal anti-inflammatory drugs reduce the risk for PD [7–9] suggesting that reducing or preventing inflammation may reduce the risk of PD.
Microglia, a brain resident immune cell, plays a central role in the process of neuroinflammation. Microglia could be activated by various types of stimuli resulting in neuroinflammation, including systemic inflammation, brain injury, and ischemia [10]. Recent studies have also showed that extracellular α-synuclein can also induce activation of microglia. Exposure to various forms of recombinant α-synuclein can induce activation of microglia [3]. In our previous study, we demonstrated that neuron-released oligomeric forms of α-synuclein-induced microglia activation via interaction with TLR2 and β1-integrin on the surface of microglia [8, 11].
Although it is well established that the outcome of chronic neuroinflammation is neurodegeneration [6], recent studies have also suggested the roles of neuroinflammation in the deposition and accumulation of amyloid protein aggregates in the brain. For example, neuroinflammation is induced by systemic administration of lipopolysaccharide (LPS)-induced accumulation of amyloid-β in hippocampus of non-transgenic (non-tg) mice [12, 13]. Similarly, intraperitoneal injection of LPS increased accumulation of α-synuclein aggregates in substantia nigra of both non-tg and α-synuclein transgenic (α-syn-tg) mice [14]. Furthermore, inhibition of neuroinflammatory enzymes, such as NADPH oxidase and iNOS decreased nigral α-synuclein deposition in α-syn-tg mice [15] once again suggesting that blocking or preventing inflammation may prevent PD pathology.
Hypoestoxide (HE) is a natural diterpene isolated from the shrub Hypoestes rosea (Acanthaceae) [16]. Studies have demonstrated that HE may modulate the activity of NF-κB through IκB kinase inhibition [16]. Thereby, HE has been suggested as a potential anti-inflammatory and anti-cancer drug [16, 17]. The polar surface area for HE is 68.4 Å2 which is considered very good for blood brain barrier penetration. Therefore, we examined the potency of HE as an anti-neuroinflammatory drug for PD using a mouse model. In conclusion, administration of HE ameliorates neuroinflammation, neurodegeneration, and behavioral defects in a PD mouse model via modulation of NF-κB activity, thus supporting a role for HE as an anti-inflammatory drug for the treatment of PD.
Methods
Antibodies and chemicals
The protease and phosphatase inhibitor cocktails were purchased from Sigma-Aldrich (St Louis, MO). Hypoestoxide was obtained from Immune Modulation, Inc. (Bloomington, CA). The following antibodies were used: α-synuclein (Syn-1; BD Bioscience, San Diego, CA); TNFα, glial fibrillary acidic protein (GFAP) (GA5), TH, and NeuN (Millipore, County Cork, Ireland); β-actin (Sigma-Aldrich, St Louis, MO); NF-κB p65 and phospho-NF-κB p65 (Cell signaling, Beverly, MA); IL-1β and IL6 (Abcam, Cambridge, MA); α-synuclein (CT, Syn105) [18]; α-synuclein (syn211) (Life Technologies, Grand Island, NY); and Iba-1 (Wako, Richimond, VA).
Animal treatment and behavioral analysis
Mice overexpressing human α-synuclein under mThy1 promoter (α-syn-tg) were used for this study because mice develop behavioral motor deficits, axonal pathology, and accumulation of α-synuclein in cortical and subcortical regions, thus mimicking PD [19–21]. The procedure for intraperitoneal injection has been described elsewhere [22]. Briefly, 5-month-old non-tg and α-syn-tg female mice were injected intraperitoneally (IP) with either vehicle (40 % captisol) or hypoestoxide (5 mg/kg) daily for 4 weeks. The right hemibrains were post-fixed in phosphate-buffered 4 % PFA at 4 °C for neuropathological analysis, while the left hemibrains were snap-frozen and stored at −70 °C for subsequent protein and messenger NA (mRNA) analysis. All animal procedures were approved by the UCSD Institutional Animal Care and Use Committee.
Following treatment, animals were assessed for gait and coordination using the open field and the round beam tests. As previously described [23], total activity was calculated as total beam breaks in 10 min. The impairment of gait and balance was accessed by round beam analysis. Three consecutive trials of 1 min each were run in 1 day. The numbers of foot slippages and distance traveled were recorded. The total errors on the beam were calculated as foot slips/distance traveled.
Immunohistochemistry and immunofluorescence and neuropathological analysis
The procedures for immunohistochemical, immunofluorescence, and neuropathological analysis have been described elsewhere [22, 24]. Briefly, blind-coded sagittal brain sections were incubated with primary antibodies at 4 °C for overnight. The next day, sections were incubated with either biotinylated- or FITC-conjugated secondary antibodies and detected with avidin D-HRP HRP (ABC elite, Vector Laboratories, Burlingame, CA) and with Tyramide Signal Amplification Direct system (PerkinElmer, Waltham, MA), respectively.
To determine the neuroinflammation, neurodegeneration, accumulation of α-synuclein, and NF-κB activation, we stained brain sections with Iba-1, GFAP, TNFα, IL-1β, IL6, human α-synuclein, NF-κB, and phosphorylated NF-κB antibodies, respectively. Sections were imaged by Olympus BX41 microscope. All immunoreactivity levels were determined by optical density analysis using Image Quant 1.43 program (NIH) except the immunoreactivity of Iba-1. The cell numbers of Iba-1-positive cells were determined per field (230 μm × 184 μm) of each animal based on cell body recognition using Image Quant 1.43 program (NIH).
Preparation of tissue extract and Western blot analysis
The procedures for tissue extract preparation and Western blot analysis have been described elsewhere [25]. Briefly, brain homogenates were prepared in the lysis buffer to separate sodium dodecyl sulfate (SDS)-soluble and SDS-insoluble fractions. Chemiluminescence detection and analysis were performed using Versadoc XL imaging apparatus and Quantity One (Bio-rad, Hercules, CA).
Quantitative polymerase chain reaction
The procedure for quantitative polymerase chain reaction (qPCR) has been described elsewhere [23]. Briefly, total mRNA was extracted from the mice frontal cortex using RNeasy Lipid mini kit (Qiagen, Germantown, MD) and reverse transcribed using SuperScript VILO cDNA synthesis kit (Life Technologies), respectively. Quantitative real-time PCR was performed using TaqMan® Fast Advanced Master Mix (Life Technologies) according to manufacturer’s instruction with gene-specific primers obtained from Life Technologies, such as TNFα (Mm00443258_m1), IL6 (Mm00446190_m1), IL-1β (Mm00434228_m1), and β-actin (Mm00607939_s1). Amplification of DNA products was measured by the StepOnePlus real-time PCR system (Applied Biosystems, Carlsbad, CA). Relative mRNA levels were calculated according to the 2-exp (ΔΔCt) method. All ΔCT values were normalized to β-actin.
Statistical analysis
GraphPad Prism (GraphPad Software, San Diego, CA) was used for all statistical analysis. All data are presented as means ± s.e.m. All data were analyzed for statistical significance by using either unpaired t test or two-way ANOVA (general linear model) followed by Bonferroni’s multiple comparison post-test.
Results
Administration of HE decreases neuroinflammation in a mouse model of PD
Neuroinflammation is a key pathological feature in PD that is recapitulated in the mThy-1α-synuclein transgenic (α-syn-tg) mouse model of PD [18, 23]. In order to determine if neuroinflammation could be ameliorated, we administered HE (5 mg/kg) through intraperitoneal injection (IP) to the α-syn-tg mice daily for 4 weeks. Analysis of the numbers of brain immune cells, such as Iba-1-positive microglia and GFAP-positive astrocytes were significantly increased in the neocortex of α-syn-tg mice (Fig. 1). Interestingly, administration of HE significantly decreased the numbers of microglial cells and the levels of astrogliosis in the neocortex in α-syn-tg mice to levels similar to those in non-tg mice (Fig. 1a, b, d, and e). In addition to a decrease in overall numbers of immune cells in the α-syn-tg mice, we observed a decrease in the numbers of branches per glial cell (Fig. 1a, c, d, and f) [26]. A positive interactive effect of HE treatment on Iba-1 optical density (F interaction (1, 16) = 80.48, p < 0.0001), numbers of microglia branches (F interaction (1, 16) = 83.25, p < 0.0001), GFAP optical density (F interaction (1, 16) = 15.88, p = 0.0011), and numbers of astroglial branches (F interaction (1, 16) = 4.04, p = 0.0616) was confirmed by two-way ANOVA.

Hypoestoxide reduces neuroinflammation in a mouse model of PD. Immunohistochemical analysis of Iba-1 and GFAP in neocortex of non-tg and α-syn-tg mice injected with either vehicle or hypoestoxide. a Immunoreactivity against Iba-1 was analyzed in neocortex of the brains. b, c The numbers of Iba-1-positive cells (b) and the average numbers of branches per Iba-1-positive cell (c) (n = 5 per each group; two-way ANOVA, Bonferroni’s multiple comparison post-test; ***p < 0.001). Error bars represent ±SEM. d Immunoreactivity against GFAP was analyzed in neocortex of the brains. e, f Optical density analysis of GFAP immunoreactivity (e) and the average numbers of branches per GFAP-positive cell (f) (n = 5 per each group; two-way ANOVA, Bonferroni’s multiple comparison post-test; *p < 0.05, **p < 0.01, ***p < 0.001). Error bars represent ±SEM. Scale bars = 100 μm (low magnification) and 20 μm (high magnification)
To verify our observation, we analyzed the levels of pro-inflammatory cytokines using immunohistochemical analysis and gene expression analysis (Fig. 2). The levels of TNFα, IL-1ß, and IL6 were increased in α-syn-tg mice compared to non-tg mice (Fig. 2a–d). In contrast, treatment of HE significantly reduced the levels of these pro-inflammatory cytokines in the neocortex of α-syn-tg mice (Fig. 2a–d). A positive interactive effect of HE treatment on the levels of TNFα (F interaction (1, 16) = 12.34, p = 0.0029), IL-1β (F interaction (1, 16) = 11.58, p = 0.0036), and IL6 (Finteraction (1, 16) = 31.06, p < 0.0001) was confirmed by two-way ANOVA. In addition, quantitative gene expression analysis showed the mRNA levels of TNFα, IL-1β, and IL6 were clearly decreased by HE administration in the neocortex of α-syn-tg mice (Fig. 2e–g). A positive interactive effect of HE treatment on the mRNA levels of TNFα (F interaction (1, 16) = 15.78, p = 0.0019), IL-1β (F interaction (1, 16) = 11.65, p = 0.0051), and IL6 (F interaction (1, 16) = 6.40, p = 0.0264) was confirmed by two-way ANOVA. Together, these results suggest that administration of HE inhibits activation of microglia and astrocytes, thereby reducing the production of pro-inflammatory cytokines in a mouse model of PD.

Hypoestoxide reduces pro-inflammatory cytokines in the brain of α-syn-tg mice. a–d Immunofluorescence or immunohistochemical analysis of TNFα, IL-1β, and IL6 in the neocortex of non-tg and α-syn-tg mice treated with either vehicle or hypoestoxide. a Representative images. b–d Quantitative analysis of fluorescence intensities of TNFα (b), IL-1β (c), and IL6 (d) stainings were analyzed in the neocortex of the brains (n = 5 per each group; two-way ANOVA, Bonferroni’s multiple comparison post-test; **p < 0.01, ***p < 0.001). Error bars represent ±SEM. e–g Quantitative real-time PCR analysis of expression levels of TNFα (e), IL6 (f), and IL-1β (g) in the neocortex of mice presented as levels over β-actin (n = 4 per each group; two-way ANOVA, Bonferroni’s multiple comparison post-test; **p < 0.01, ***p < 0.001). Error bars represent ±SEM. Scale bar = 20 μm
Amelioration of neurodegeneration and behavioral defect by HE administration in a mouse model of PD
Neuroinflammation is one of the well-known causes of neurodegeneration [3]. Since the levels of pro-inflammatory cytokines were significantly decreased by HE administration in α-syn-tg mice, we hypothesized that administration of HE would prevent neurodegeneration and behavioral deficit through inhibition of neuroinflammation in α-syn-tg mice. To verify the hypothesis, we performed neurodegeneration analysis and behavioral tests using non-tg and α-syn-tg mice treated with either vehicle or HE (Fig. 3). Neuronal overexpression of human α-synuclein resulted in the loss of TH-positive striatal fibers in α-syn-tg mice while the numbers of nigral TH-positive cells were not altered by α-synuclein expression (Fig. 3a–c). However, administration of HE significantly decreased the loss of TH-positive striatal fibers in α-syn-tg mice (Fig. 3a, b). A positive interactive effect of HE treatment on the level of TH-positive striatal fibers (F interaction (1, 16) = 5.12, p = 0.038) was confirmed by two-way ANOVA.

Hypoestoxide ameliorates neurodegeneration and behavioral deficits in α-syn-tg mice. a–c Immunohistochemical analysis of TH in the striatum and substantia nigra (S.Nigra) of non-tg and α-syn-tg mice treated with either vehicle or hypoestoxide. a Representative images of striatum. b The levels of dopaminergic fiber in striatum were analyzed by optical density quantification (n = 5 per each group; two-way ANOVA, Bonferroni’s multiple comparison post-test; *p < 0.05, **p < 0.01). Error bars represent ±SEM. c Stereological analysis of TH-positive cells in S. Nigra. d, e Motor behavioral analysis of non-tg and α-syn-tg mice. Total activity from open field analysis (d) and the numbers of slippages from round beam test (e) were recorded (n = 5 per each group; two-way ANOVA, Bonferroni’s multiple comparison post-test; *p < 0.05, ***p < 0.001). Error bars represent ±SEM. Scale bars = 250 μm (a, upper panel) and 25 μm (a, lower panel)
To investigate the effect of HE on the anxiety-like behavior and motor behavior deficit in α-syn-tg mice, we performed open field and round beam tests, respectively (Fig. 3d, e). α-syn-tg mice showed a significant increase of the beam break numbers and the total round beam errors compared to non-tg control mice. Treatment of α-syn-tg mice with HE reduced these errors to levels observed in non-tg mice (Fig. 3d, e). A positive interactive effect of HE treatment on the beam break numbers (F interaction (1, 16) = 15.61, p = 0.0011) and total round beam errors (F interaction (1, 16) = 8.58, p = 0.0098) was confirmed by two-way ANOVA. Taken together, these results suggest that administration of HE prevents neurodegeneration and ameliorates behavioral defect in a mouse model of PD.
Administration of hypoestoxide results in reduced neuronal α-synuclein accumulation in a mouse model of PD
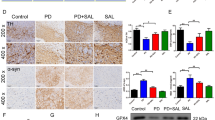
To determine if the behavioral improvements observed in the α-syn-tg mice were related to alterations of α-synuclein pathology, we performed immunohistochemical analysis for α-synuclein with brain sections from non-tg and α-syn-tg mice treated with either vehicle or HE. Immunohistochemical analysis showed overexpression of α-synuclein in neurons and the neuropil of α-syn-tg mice (Fig. 4a–e). Surprisingly, administration of HE significantly decreased the levels of α-synuclein in neurons and neuropil in α-syn-tg mice (Fig. 4a–e). A positive interactive effect of HE treatment on the optical density of α-synuclein (frontal cortex, F interaction (1, 16) = 30.74, p < 0.0001; hippocampus, F interaction (1, 16) = 13.66, p = 0.0020; striatum, F interaction (1, 16) = 7.19, p = 0.0164) was confirmed by two-way ANOVA. To confirm our observations, we performed immunofluorescence analysis with human α-synuclein-specific antibodies (Additional file 1). Immunoreactivity against human α-synuclein was not detected in the frontal cortex of non-tg mice, but it was highly detected in the frontal cortex of α-syn-tg mice (Additional file 1a, b). Similar to results from the immunohistochemical analysis, the level of human α-synuclein immunoreactivity was significantly decreased by HE administration in the frontal cortex of α-syn-tg mice (Additional file 1a, b). Recent evidence suggests the C-terminal fragments of α-synuclein are particularly neurotoxic [18]. To determine if the administration of HE affected the accumulation of these C-terminal fragments, we used an antibody that specifically recognizes the C-terminus of human α-synuclein. Immunoreactivity against C-terminus human α-synuclein was also significantly decreased by HE administration in the frontal cortex of α-syn-tg mice (Additional file 1c, d).

Hypoestoxide reduces deposition of α-synuclein in a mouse model of PD. a Representative immunohistochemical staining of α-synuclein in the frontal cortex, hippocampus, and striatum. b Optical density analysis for α-synuclein-positive neuropil in the frontal cortex (n = 5 per each group; two-way ANOVA, Bonferroni’s multiple comparison post-test; ***p < 0.001). Error bars represent ±SEM. c The numbers of α-synuclein-positive cells in the frontal cortex. d, e Optical density analysis of immunoreactivity of α-synuclein in the hippocampus (d) and in the striatum (e) (n = 5 per each group; two-way ANOVA, Bonferroni’s multiple comparison post-test; **p < 0.01, ***p < 0.001). Error bars represent ±SEM. f, g Biochemical analysis of SDS-soluble (f) and SDS-insoluble (g) fractions from the frontal cortex for α-synuclein (n = 5 per each group; two-way ANOVA, Bonferroni’s multiple comparison post-test; **p < 0.01, ***p < 0.001). Error bars represent ±SEM. Scale bars = 250 μm (low magnification) and 25 μm (high magnification)
To verify our findings, we performed biochemical analysis (Fig. 4f, g). Brain homogenates were separated into SDS-soluble and SDS-insoluble factions, and analyzed by immunoblot analysis. Interestingly, the levels of α-synuclein in SDS-soluble fractions from α-syn-tg mice were not affected by HE administration (Fig. 4f, g). However, the levels of SDS-insoluble α-synuclein were significantly decreased in the brain homogenates from HE-administrated α-syn-tg mice (Fig. 4f, g). A positive interactive effect of HE treatment on the level of SDS-insoluble α-synuclein (F interaction (1, 16) = 8.68, p = 0.0095) was confirmed by two-way ANOVA. Taken together, these results suggest that administration of HE reduces the accumulation of α-synuclein in a mouse model of PD.
HE modulates neuroinflammation via inhibition of NF-κB activity in a mouse model of PD
Previous work suggests that HE modulated the activity of NF-κB, a key immune response signaling mediator, through inhibition of IκB kinase in immune cells [16]. Therefore, we investigated the alteration of NF-κB activity in the neocortex of non-tg and α-syn-tg mice that received either vehicle or HE. Immunofluorescence analysis showed that total levels of NF-κB were not changed by HE administration in the neocortex of non-tg or α-syn-tg mice (Fig. 5a, b). However, the level of immunoreactivity against phosphorylated NF-κB, the activated form of NF-κB, was highly elevated (fourfold) in the neocortex of α-syn-tg mice (Fig. 5a, c). In addition, the elevated level of phosphorylated NF-κB was significantly decreased by administration of HE in the neocortex of α-syn-tg mice to levels observed in non-tg mice (Fig. 5a, c). A positive interactive effect of HE treatment on the immunoreactivity of phosphorylated NF-κB (F interaction (1, 16) = 27.70, p < 0.0001) was confirmed by two-way ANOVA. To verify our findings, we performed biochemical analysis using brain homogenates from the cortex of non-tg and α-syn-tg mice (Fig. 5d–f). Brain homogenates were separated into cytosolic and nuclear fractions by centrifugation, and each fraction was analyzed by Western blot analysis. Total levels of NF-κB were not altered by HE administration in non-tg and α-syn-tg mice (Fig. 5d, e). However, the level of phosphorylated NF-κB was significantly increased only in nuclear fraction from α-syn-tg mice brain homogenates (Fig. 5d, f). Similar to results observed by immunofluorescence, the level of phosphorylated NF-κB was significantly reduced by HE administration in α-syn-tg mice (Fig. 5d, f). A positive interactive effect of HE treatment on the level of phosphorylated NF-κB (F interaction (1, 16) = 11.55, p = 0.0037) was confirmed by two-way ANOVA. Taken together, these results suggest that administration of HE decreases neuroinflammation through modulation of NF-κB activity in a mouse model of PD.

Hypoestoxide inhibits NF-κB signaling in a mouse model of PD. a Representative immunohistochemical staining of NF-κB and phosphorylated NF-κB in the neocortex of non-tg and α-syn-tg mice treated with either vehicle or hypoestoxide. b, c Fluorescence intensities against NF-κB (b) and phosphorylated NF-κB (c) were analyzed in the neocortex of the brains (n = 5 per each group; two-way ANOVA, Bonferroni’s multiple comparison post-test; ***p < 0.001). Error bars represent ±SEM. d Western blot analysis of cytosolic and nuclear fractions from the neocortex for NF-κB and phosphorylated NF-κB. e The relative cytosolic phosphorylated NF-κB levels normalized against cytosolic total NF-κB. f The relative nuclear phosphorylated NF-κB levels normalized against total nuclear NF-κB (n = 5 per each group; two-way ANOVA, Bonferroni’s multiple comparison post-test; **p < 0.01, ***p < 0.001). Error bars represent ±SEM. Scale bar = 20 μm
Discussion
Previous studies have suggested an anti-inflammatory effect of HE, so we examined the potential for HE as an anti-neuroinflammatory drug in the pathogenesis of PD [16]. The levels of cytokine expression and the number of reactive glial cells were significantly reduced by HE administration in a model of PD. In addition, administration of HE prevented neurodegeneration in a mouse model of PD. The loss of TH-positive neurons was significantly decreased by administration of HE in α-syn-tg mice. Behavioral defect also has been ameliorated by HE administration in α-syn-tg mice. Furthermore, we demonstrated that HE inhibits the activity of NF-κB which results in the decrease of neuroinflammation in α-syn-tg mice.
Neuroinflammation is a typical pathological feature of PD playing a critical role in disease onset and progression [3, 4]. Recent studies have also suggested that extracellular α-synuclein is a strong inducer of neuroinflammation [8, 27]. Neurons endogenously release α-synuclein via unconventional exocytosis, and this secretion could be modulated by multiple factors including genetic defects, oxidative modification of α-synuclein, mitochondrial dysfunction, and autophagy inhibition [28–32]. Previous studies have shown that exposure to various forms of recombinant α-synuclein induces microglia activation [25, 33–35]. In addition, we have demonstrated that oligomeric forms of neuron-released α-synuclein interact with TLR2 and β1-integrin on the surface of microglia, thereby inducing pro-inflammatory responses [8, 11]. Thus, in PD and other synucleinopathies, reducing neuroinflammation may be a target for therapeutic intervention.
Intraneuronal accumulations of α-synuclein aggregates are typical pathological features of PD, and studies have demonstrated that these deposits are not only pathological but also play a critical role in the onset and development of PD. Recent studies have shown that neuronal accumulation of α-synuclein can be affected by multiple intra- and extra-neuronal factors, including genetic defects, dysfunction of protein quality control systems, secondary structural alterations, and exposure to environmental toxicants [2, 24]. In addition, neuroinflammation has been suggested as a promotable factor for α-synuclein aggregates in neurons [3]. In this study, we observed that administration of HE reduced the neuronal accumulation of α-synuclein in a model of PD. Since the levels of α-synuclein mRNA were not affected by administration of HE (data not shown), we speculate that activation of the intraneuronal autophagy process can be regulated by neuroinflammation.
We still do not know the mechanism by which microglia-mediated neuroinflammation affects neuronal accumulation of α-synuclein aggregates. However, recent studies have shown that some pro-inflammatory cytokines inhibit autophagy, an efficient intracellular process for α-synuclein elimination [2, 36]. For example, IL-10 inhibits starvation-induced, rapamycin-induced, and lipopolysaccharide-induced autophagy in murine macrophages [37–39]. In addition, IL-4 and IL-13 inhibit both starvation-induced and IFN-γ-induced autophagosome formations in human and murine macrophages [40]. These observations suggest that cytokines from activated microglia may inhibit the autophagy process of neighboring neurons in the brain, thereby resulting in a neuronal α-synuclein accumulation. Considering the previous results together with our current study, we speculate that administration of HE decreases neuronal accumulation of α-synuclein via reduction of neuroinflammation in a model of PD leading to increased autophagic degradation of α-synuclein.
Conclusions
In conclusion, our work suggests that administration of HE modulates the activity of NF-κB in a model of PD; therefore, HE may be a potent anti-PD drug that can reduce neuroinflammation, neurodegeneration, and α-synucleinopathy.
Abbreviations
- ANOVA:
-
analysis of variance
- GFAP:
-
glial fibrillary acidic protein
- Iba1:
-
ionized calcium-binding adapter molecule 1
- IL:
-
interleukin
- TH:
-
tyrosine hydroxylase
- TNFα:
-
tumor necrosis factor α
References
Forno LS. Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol. 1996;55:259–72.
Kim C, Lee SJ. Controlling the mass action of alpha-synuclein in Parkinson’s disease. J Neurochem. 2008;107:303–16.
Sanchez-Guajardo V, Tentillier N, Romero-Ramos M. The relation between alpha-synuclein and microglia in Parkinson’s disease: recent developments. Neuroscience. 2015;302:47–58.
Wake H, Moorhouse AJ, Miyamoto A, Nabekura J. Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci. 2013;36:209–17.
Hunot S, Hirsch EC. Neuroinflammatory processes in Parkinson’s disease. Ann Neurol. 2003;53 Suppl 3:S49–58. discussion S58-60.
Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, et al. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–71.
Long-Smith CM, Sullivan AM, Nolan YM. The influence of microglia on the pathogenesis of Parkinson’s disease. Prog Neurobiol. 2009;89:277–87.
Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562.
Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–97.
Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–94.
Kim C, Cho ED, Kim HK, You S, Lee HJ, Hwang D, et al. beta1-integrin-dependent migration of microglia in response to neuron-released alpha-synuclein. Exp Mol Med. 2014;46:e91.
Ifuku M, Katafuchi T, Mawatari S, Noda M, Miake K, Sugiyama M, et al. Anti-inflammatory/anti-amyloidogenic effects of plasmalogens in lipopolysaccharide-induced neuroinflammation in adult mice. J Neuroinflammation. 2012;9:197.
Kahn MS, Kranjac D, Alonzo CA, Haase JH, Cedillos RO, McLinden KA, et al. Prolonged elevation in hippocampal Abeta and cognitive deficits following repeated endotoxin exposure in the mouse. Behav Brain Res. 2012;229:176–84.
Liu Y, Qin L, Wilson B, Wu X, Qian L, Granholm AC, et al. Endotoxin induces a delayed loss of TH-IR neurons in substantia nigra and motor behavioral deficits. Neurotoxicology. 2008;29:864–70.
Gao HM, Zhang F, Zhou H, Kam W, Wilson B, Hong JS. Neuroinflammation and alpha-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environ Health Perspect. 2011;119:807–14.
Ojo-Amaize EA, Kapahi P, Kakkanaiah VN, Takahashi T, Shalom-Barak T, Cottam HB, et al. Hypoestoxide, a novel anti-inflammatory natural diterpene, inhibits the activity of IkappaB kinase. Cell Immunol. 2001;209:149–57.
Ojo-Amaize EA, Nchekwube EJ, Cottam HB, Bai R, Verdier-Pinard P, Kakkanaiah VN, et al. Hypoestoxide, a natural nonmutagenic diterpenoid with antiangiogenic and antitumor activity: possible mechanisms of action. Cancer Res. 2002;62:4007–14.
Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A, et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci. 2014;34:9441–54.
Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, et al. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–78.
Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, et al. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci. 2004;24:9434–40.
Rockenstein E, Crews L, Masliah E. Transgenic animal models of neurodegenerative diseases and their application to treatment development. Adv Drug Deliv Rev. 2007;59:1093–102.
Spencer B, Emadi S, Desplats P, Eleuteri S, Michael S, Kosberg K, et al. ESCRT-mediated uptake and degradation of brain-targeted alpha-synuclein single chain antibody attenuates neuronal degeneration in vivo. Mol Ther. 2014;22:1753–67.
Valera E, Mante M, Anderson S, Rockenstein E, Masliah E. Lenalidomide reduces microglial activation and behavioral deficits in a transgenic model of Parkinson’s disease. J Neuroinflammation. 2015;12:93.
Kim C, Rockenstein E, Spencer B, Kim HK, Adame A, Trejo M, et al. Antagonizing neuronal toll-like receptor 2 prevents synucleinopathy by activating autophagy. Cell Rep. 2015;13:771–82.
Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 2005;19:533–42.
Kongsui R, Beynon SB, Johnson SJ, Walker FR. Quantitative assessment of microglial morphology and density reveals remarkable consistency in the distribution and morphology of cells within the healthy prefrontal cortex of the rat. J Neuroinflammation. 2014;11:182.
Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, et al. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 2010;285:9262–72.
Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–24.
Bae EJ, Ho DH, Park E, Jung JW, Cho K, Hong JH, et al. Lipid peroxidation product 4-hydroxy-2-nonenal promotes seeding-capable oligomer formation and cell-to-cell transfer of alpha-synuclein. Antioxid Redox Signal. 2013;18:770–83.
Lee HJ, Baek SM, Ho DH, Suk JE, Cho ED, Lee SJ. Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp Mol Med. 2011;43:216–22.
Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem. 2010;113:1263–74.
Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ. Autophagic failure promotes the exocytosis and intercellular transfer of alpha-synuclein. Exp Mol Med. 2013;45:e22.
Park JY, Paik SR, Jou I, Park SM. Microglial phagocytosis is enhanced by monomeric alpha-synuclein, not aggregated alpha-synuclein: implications for Parkinson’s disease. Glia. 2008;56:1215–23.
Reynolds AD, Glanzer JG, Kadiu I, Ricardo-Dukelow M, Chaudhuri A, Ciborowski P, et al. Nitrated alpha-synuclein-activated microglial profiling for Parkinson’s disease. J Neurochem. 2008;104:1504–25.
Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging. 2008;29:1690–701.
Harris J. Autophagy and cytokines. Cytokine. 2011;56:140–4.
Park HJ, Lee SJ, Kim SH, Han J, Bae J, Kim SJ, et al. IL-10 inhibits the starvation induced autophagy in macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway. Mol Immunol. 2011;48:720–7.
Van Grol J, Subauste C, Andrade RM, Fujinaga K, Nelson J, Subauste CS. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS One. 2010;5:e11733.
Ni Cheallaigh C, Keane J, Lavelle EC, Hope JC, Harris J. Autophagy in the immune response to tuberculosis: clinical perspectives. Clin Exp Immunol. 2011;164:291–300.
Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, et al. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity. 2007;27:505–17.
Acknowledgements
This work was supported by National Research Foundation grants funded by the Korean Government 2013R1A6A3A03023385 to CK and by NIH grants AG18440, AG043384, and NS057096 to EM.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Authors affiliated with Immune Modulation, Inc. (HC, EO, OO, JO, EN) have performed previous studies with hypoestoxide in addition to these studies and have promoted hypoestoxide as a new agent for the treatment and prevention of a variety of disease indications.
Author’s contributions
EO, HC, and EM conceived the study. CK, EO, HC, and EM designed the experiments. ER, MM, and EM designed and performed the mouse experiments. CK, BS, PD, WW, AA, EN, OO, JO, and MC performed all other experiments. CK, BS, and EM wrote the paper. All authors read and approved the final manuscript.
Additional file
Additional file 1:
Hypoestoxide reduces human α-synuclein accumulation in a mouse model of PD. Mice brain sections were inmmunostained against human α-synuclein (Syn211 antibody) or C-terminal of human α-synuclein (Syn105 antibody). a Immunofluorescence analysis of human α-synuclein in the frontal cortex of non-tg and α-syn-tg mice treated with either vehicle or hypoestoxide. (n = 5 per group; unpaired t test; *p < 0.05). Error bars represent ± SEM. b Fluorescence intensity against human α-synuclein was analyzed in frontal cortex of the brains. c Immunohistochemical analysis of C-terminal of human α-synuclein in the frontal cortex of non-tg and α-syn-tg mice. d Optical density analysis for C-terminal of α-synuclein in frontal cortex. (n = 5 per group; unpaired t test; **p < 0.01). Error bars represent ± SEM. Scale bars = 250 μm (low magnification) and 25 μm (high magnification). (TIF 13692 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kim, C., Ojo-Amaize, E., Spencer, B. et al. Hypoestoxide reduces neuroinflammation and α-synuclein accumulation in a mouse model of Parkinson’s disease. J Neuroinflammation 12, 236 (2015). https://doi.org/10.1186/s12974-015-0455-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-015-0455-9