
Abstract
Recent findings have led to a renewed interest and support for an active role of inflammation in neurodegenerative dementias and related neurologic disorders. Detection of neuroinflammation in vivo throughout the course of neurodegenerative diseases is of great clinical interest. Studies have shown that microglia activation (an indicator of neuroinflammation) may present at early stages of frontotemporal dementia (FTD), but the role of neuroinflammation in the pathogenesis of FTD is largely unknown. The first-generation translocator protein (TSPO) ligand ([11C]-PK11195) has been used to detect microglia activation in FTD, and the second-generation TSPO ligands have imaged neuroinflammation in vivo with improved pharmacokinetic properties. This paper reviews related literature and technical issues on mapping neuroinflammation in FTD with positron-emission tomography (PET) imaging. Early detection of neuroinflammation in FTD may identify new tools for diagnosis, novel treatment targets, and means to monitor therapeutic efficacy. More studies are needed to image and track neuroinflammation in FTD. It is anticipated that the advances of TSPO PET imaging will overcome technical difficulties, and molecular imaging of neuroinflammation will aid in the characterization of neuroinflammation in FTD. Such knowledge has the potential to shed light on the poorly understood pathogenesis of FTD and related dementias, and provide imaging markers to guide the development and assessment of new therapies.
Similar content being viewed by others
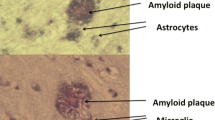


Background
Frontotemporal dementia (FTD) is a devastating neurodegenerative disorder, primarily affecting the frontal and/or temporal lobes of the brain. It is the second most frequent cause of presenile neurodegenerative dementia in those less than 65 years of age [1]. The prevalence is around 15 per 100,000 in people between 45 and 65 years of age, and the mean survival varies from 3 to 10 years from diagnosis [2]. There are mainly three types of FTD: behavioral variant FTD (bv-FTD), semantic dementia (SD), and progressive non-fluent aphasia (PNFA), and they are common in the aggregation of neuronal proteins such as the microtubule-associated protein tau (MAPT), the transactive response DNA-binding protein with molecular weight 43 kDa (TDP-43), and the fused in sarcoma protein (FUS) [3]. Of the FTD cases, 25% to 50% are inherited [4], and the mutations are in the genes for MAPT, progranulin (GRN), and in the chromosome 9 open reading frame 72 (C9orf72) [3].
The clinical diagnosis of FTD is a challenge. In general, from symptom onset, it takes 4 years for bv-FTD and SD, and 3 years for PNFA to reach a correct diagnosis [5]. FTD is often misdiagnosed as Alzheimer’s disease (AD) or psychiatric disorders. Abnormalities in structural imaging may be very subtle at early stages, while functional imaging changes may not be specific enough to differentiate FTD from AD at individual patient level. For example, failure to correctly diagnose FTD and AD often occurs in cases that present with temporoparietal hypometabolism [6]. Currently, there is no effective pharmacological treatment to slow the progression of FTD. Prognosis is therefore poor, and dependency on caregivers and eventual death typically occurs in a few years after diagnosis [2,3].
Leading models have highlighted the potential neurotoxic properties of tau aggregation in FTD [7] and related neurodegenerative dementias [8,9]. Recent findings have led to a renewed interest and support for an active role of inflammation in neurodegenerative processes [10]. In AD, chronic neuroinflammation appears to be a central process in AD pathophysiology [11,12], and there are promising targets in modulation of neuroinflammation for AD treatment [12]. Further, a link has been proposed recently between neuroinflammation and specific forms of FTD, suggesting that neuroinflammation is an important component of FTD [10,13]. Consequently, early detection of neuroinflammation in FTD may identify new tools for diagnosis, novel treatment targets, and means to monitor therapeutic efficacy.
Pathological protein aggregation and neuroinflammatory responses may begin before patients start experiencing AD or FTD symptoms [10,11]; thus, neuroinflammation could be an early marker for neurodegenerative dementias. Since the neuroinflammatory response may mediate the outcome of brain tissue in many neurologic diseases including stroke, epilepsy, and neurodegenerative disorders, it is of great clinical interest to detect neuroinflammation accurately and reliably [14]. Positron-emission tomography (PET) imaging with radioligands that label activated microglia, a key cellular component of the neuroinflammatory response, offers a potential means to characterize neuroinflammation in vivo. Increases in the translocator protein (TSPO, 18 kDa) expression detected by PET imaging with radioligands of TSPO or peripheral benzodiazepine receptor (PBR) is recognized as a biomarker of activated microglia [15], which might aid in the diagnosis of early FTD. In this paper, the existing evidence on neuroinflammation in FTD and PET imaging of neuroinflammation in FTD with TSPO ligands is reviewed and related technical issues are discussed.
Neuroinflammation in FTD
Compared with healthy subjects and AD patients, an increased prevalence of related autoimmune diseases has been reported in FTD patients with semantic variant primary progressive aphasia (svPPA) who were GRN mutation carriers [13,16]. It has also been found that a proapoptotic protein (regulated partially by vasoactive neuropeptides) in astrocytes called Bax showed immunoreactivity in FTD, which suggested autoimmunity in the pathology of FTD [17]. In tau-negative FTD, neuroinflammation may play a more important role in the pathogenesis of FTD because mutations in the progranulin (GRN) genes lead to tau-negative FTD [10]. Progranulin acts as a mediator of the inflammatory response [18], and deficiency in progranulin may lead to greater microglial activation and a dysregulated inflammatory response in microglia that could cause neuron death and disease progression in FTD [10]. In addition, head trauma that triggers neuroinflammation has been associated with behavioral variant FTD [19]. Furthermore, some biomarkers of inflammation, such as elevated cytokines (for example, tumor necrosis factor (TNF)-α) in the cerebrospinal fluid (CSF), have been observed in patients with FTD [20]. These findings support the hypothesis that neuroinflammation has a detrimental role in FTD [10]. However, the pathogenesis of FTD, in particular the role of neuroinflammation, is still poorly understood.
Microglia are the resident immune cells in the central nervous system (CNS), representing the first line of defense against pathogens: they sense subtle pathological changes and become activated before obvious functional or anatomical abnormalities occur. Normal protective microglia mediate clearance of abnormal protein (such as Aβ or tau) aggregates, remove cell debris, and promote neuroregeneration. However, activated microglia secrete inflammatory mediators (for example, interleukin (IL)-1β), coactivate astrocytes, and induce neuronal death, which further increases brain tissue damage with amplified microglial activation [14]. The neuroinflammatory reaction involves dramatic upregulation of a mitochondrial transmembrane protein, TSPO, which is a marker for microglial activation and a target for imaging neuroinflammation with PET ligands designed to bind TPSO. It has been reported that PET imaging with the widely used first-generation TSPO ligand [11C]-PK11195 detected neuroinflammation in patients with mild cognitive impairment (MCI) [21]. This and other evidence [22-25] suggests that neuroinflammation is an early and continuous process in neurodegenerative dementias [26]. It is now known that microglia detect pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) through pattern recognition receptors (PRRs), and in neurodegenerative disorders, microglia cannot discriminate between invading pathogens and aberrant molecules (or abnormal proteins) of the host, which leads to DAMP-triggered neuroinflammation through sustained and excessive release of pro-inflammatory cytokines such as IL-1β [10]. In addition, age-related microglia priming is crucial in exaggerated neuroinflammation [27], and microglial activation may be related to dementia progression [28].
Genes related to microglial activation (for example, a variant of the triggering receptor expressed on myeloid cells 2 (TREM2)) have now been associated with FTD [29,30]. Abnormal protein aggregates such as amyloid or tau deposition could cause microglial activation. New hypothesis has suggested that microglia could be functionally impaired by abnormal protein aggregates, leading to reduced microglial motility and phagocytic activity in vivo [31]. There is evidence that sustained exposure to bacterial lipopolysaccharide (LPS) or other pro-inflammatory mediators restricts microglial phagocytosis of protein aggregation and suppresses axonal transportation [10]. Several animal studies and postmortem studies have revealed that neuroinflammation stimulated neuronal degeneration [32,33]. On the other hand, microglia-driven neuroinflammation could lead to the formation of tau aggregation [34]. In FTD, the neuronal and axonal degeneration are sufficient to induce microglial activation [19].
Few studies to date have examined neuroinflammation in vivo in FTD patients. Molecular PET imaging with [11C]-PK11195 has found that compared with controls, the mean [11C]-PK11195 binding was significantly increased in patients with FTD (n = 5, including four patients with progressive non-fluent aphasia and one patient with behavioral variant FTD) in regions such as the left dorsolateral prefrontal cortex and the right hippocampus and parahippocampus [35]. The pattern of microglial activation partially overlapped with the pattern of brain atrophy, but there was also increased [11C](R)-PK11195 binding in regions contralateral to predominant lobar atrophy suggesting that microglial activation was present at early stages of FTD prior to anatomical changes [19]. Using postmortem brain tissues, Vennetic et al. found that [11C]-PK11195 binded specifically to activated microglia in FTD, and the binding was correlated with microglial activation identified by immunohistochemistry in situ [36]. Postmortem immunohistochemistry study further demonstrated that compared with controls, higher level of microglial activation detected by [11C]-PK11195 was found in the frontal and temporal cortex in patients with FTD (n = 78), and greater microglial activation was found in the temporal subcortical white matter in FTD-MART than in other FTD genetic types [37].
In neurodegenerative disorders such as FTD, there is a common pattern in the mechanisms of sensing abnormal protein aggregates, activating microglia, transducing to the release of cytokines, and amplifying the neurotoxic effects in a chronic inflammatory process [14]. A better understanding of the immune response in the brain is critical for possible modulation of microglial activity to slow down or reverse the course of neurodegeneration [14]. Further development of PET imaging with TSPO ligands represents a potential in vivo tool for tracking the progression of neuroinflammation in neurodegenerative disorders such as FTD.
Molecular PET imaging with second-generation TSPO ligands
The value of TSPO PET imaging is in detecting microglia activation in the diseased brain, visualizing neuroinflammation and its progression, and monitoring treatment effect, which is highly needed in the diagnosis and treatment of FTD. TSPO is an interesting target for molecular PET imaging because it is involved in a number of neurodegenerative disorders (such as AD, FTD, and Parkinson’s disease (PD)) and neuroinflammatory disorders (such as ischemic stroke and multiple sclerosis). However, there are several limitations in the first-generation TSPO ligand [11C]-PK11195, mainly high non-specific binding, low brain penetration, and high plasma protein binding, which may explain the negative findings in several studies using [11C]-PK11195 [38,39]. In recent years, novel tracers such as [11C]-DAA1106, [11C]vinpocetine, [11C]-DPA-713, [11C]-PBR28, [18F]-FEDAA1106, [18F]-PBR06, [18F]-PBR111, [18F]-DPA-714, and [18F]-FEPPA have been developed as the second-generation TSPO radioligands. Comparative studies showed that novel TSPO ligands such as [18F]-DPA-714 have higher specific binding and lower non-specific binding than [11C]-PK11195 in rodent models [40,41]. Further, Venneti et al. reported that [3H]-DAA1106 showed a higher binding affinity than [11C]-PK11195 in postmortem brain tissues of patients with neurodegenerative disorders such as FTD [36]. Similarly, Vas et al. found higher binding of [11C]vinpocetine than [11C]-PK11195 in patients with multiple sclerosis [42]. These findings suggest that the second-generation TSPO ligands are better than the first-generation TSPO ligand [11C]-PK11195 in imaging activated microglia in vivo in neurodegenerative disorders due to improved pharmacokinetic properties [36].
In recent years, PET imaging with novel TSPO radioligands has been applied to visualizing neuroinflammation in neurodegenerative disorders, although there is very limited data on in vivo PET imaging with the second-generation TSPO tracers in FTD. Using PET imaging with [11C]-DAA1106, Miyoshi et al. examined patients with FTD (n = 3) who were presymptomatic MART gene carriers with parkinsonism linked to chromosome 17 (FTDP-17) and found increased microglial activation in regions such as frontal cortex in patients compared with controls, although such increase was not overt throughout the diseased brain in FTD [43]. In addition, regional increased [11C]-DAA1106 binding has been found in patients with MCI (n = 7) compared to healthy controls [22], and patients with AD (n = 19) had greater regional [11C]-PBR28 binding than controls, which was correlated with severity of the disease [44]. These findings suggest that neuroinflammation is an intrinsic process in tau pathology, which exists even at a presymptomatic stage.
However, research on imaging neuroinflammation with the second-generation TSPO ligands is still in its infancy, and the sample sizes of such research are usually small. In vivo and in vitro studies with the second-generation TSPO ligands have shown significant inter-subject variability because of differences in binding affinity in individual subjects [44-47]. Three affinity patterns of binding variations have been reported: high-affinity binders (HABs), low-affinity binders (LABs), and mixed-affinity binders (MABs) [47,48]. A single polymorphism (rs6971) located in the exon 4 of the TSPO gene determines the binding affinity of the second-generation ligands and causes large inter-subject variation [44,49,50], while for the first-generation TSPO ligand [11C]-PK11195, the inter-subject variation in binding affinity is little.
Compared with carbon-11-labeled TSPO ligands (for example, [11C]-PK11195), tracers labeled with fluorine 18 have a longer half-life ([18F] vs. [11C]: approximately 110 min vs. approximately 20 min), which is suitable for long-distance dissemination and larger clinical studies. Among the fluorine-18-labeled second-generation TSPO ligands, [18F]-FEDAA1106 has proved ineffective [51,52] and [18F]-PBR06 produces a metabolite that confounds quantification of TSPO binding [53], but [18F]-FEPPA (FEPPA) and [18F]-DPA-714 (DPA714) may be promising [54-56]. FEPPA PET imaging has been applied to animals [54,57-62] and humans [63-70] and has demonstrated that increased neuroinflammation was not associated with normal aging, but regional increased FEPPA uptake was associated with AD or PD [67-70]. Similarly, there are a number of PET imaging studies with [18F]-DPA-714 [40,56,71-79], and focal increase in DPA714 uptake has been found in patients with neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS) [78].
Taken together, although in its infancy, research on PET imaging with the second-generation TSPO ligands is in progress and has shown increased uptake in brain regions associated with neuroinflammation in early FTD and other neurodegenerative disorders such as those of tau pathology (even at a presymptomatic stage), suggesting that TSPO imaging is useful in detecting neuroinflammation in vivo from the early stage of the disease.
Discussion
In neurodegenerative and neuroinflammatory disorders, microglia activation may be an early phenomenon, which can be visualized by PET imaging with TSPO ligands, and the second-generation TSPO ligands have shown improved pharmacokinetic properties. However, to image neuroinflammation in FTD using PET with the second-generation TSPO ligands, several practical issues or technical details need to be considered.
First, since PET imaging with the second-generation TSPO ligands display inter-subject variability in binding affinity, the genotype at the polymorphism (rs6971) in the TSPO gene needs to be determined. Subjects may need to be screened for LABs because the second-generation TSPO ligands do not provide a measureable signal in PET studies involving LABs. The ratio of LABs in healthy subjects varies from 0% [66] to 13.3% [80], and the ratio of LABs in one sample of AD patients was 15.4% [67]. Therefore, genotype analysis for polymorphism (rs6971) in the TSPO gene is needed before PET imaging scan, and a small portion of subjects with LABs may need to be excluded from PET imaging with the second-generation TSPO ligand.
Second, most of the human studies with fluorine-18-labeled second-generation TSPO ligands used scan acquisition times of 1.5 to 2 h which might be long for patients with FTD. In attempts to reduce FEPPA PET scan time, Rusjan et al. have found increased variability and decreased identifiability (with the average coefficient of variance ratio 14% for total distribution volume V(T)) with 1.5-h scan time [64]. However, the average of V(T) were not significantly biased compared with those of 2-h scanning, and the V(T) values at 1.5 h were correlated with the values at 3 h (r 2 = 0.91) [64], suggesting that 1.5-h or shorter scan time might be possible if relative measures (for example, distribution volume ratio) are used.
Third, current PET imaging with the second-generation TSPO ligands usually requires arterial blood sampling to determine the blood radioactivity time-activity curve (TAC) and plasma radioactivity curve so that the input function for the kinetic analysis can be created to compute the ligand uptake and binding potential. Such invasive kinetic analysis is technically demanding, and uncomfortable and potentially painful to patients. To avoid the need for arterial blood sampling, it is necessary to apply non-invasive quantitative models that use radioactivity concentration in a reference region (that is, brain area with negligible binding) to approximate concentration of tracer in the non-displaceable compartment as an indirect input function [80-86]. However, microglia cells and the TPSO ligand binding sites are distributed throughout the entire brain, and a reference region can hardly be found in the brain.
In the past, the first-generation TSPO ligand [11C]-PK11195 (PK11195) faced similar difficulties. Turkheimer et al. developed a non-invasive analysis method for PK11195 PET using reference and target region modeling where predefined kinetic classes were used to extract a gray matter reference tissue [87]. They found that binding potential values obtained by a plasma input were highly correlated with those of a reference input [86]. Since then, non-invasive kinetic analysis has been increasingly adopted in PK11195 PET imaging, and arterial blood sampling is no longer needed. Therefore, it is possible to apply non-invasive kinetic analyses such as the reference approach to PET imaging with the second-generation TSPO ligands [87]. However, there are mainly two challenges: (1) Since the kinetics of HAB and MAB genetic groups may differ, a database of reference for both HAB and MAB groups may be needed; (2) The second-generation TSPO ligands show high level of vascular binding [88], which may make it difficult to separate the normal gray matter from other tissue classes.
Finally, it is unclear how accurate TSPO PET imaging is in detecting neuroinflammation in neurodegenerative dementias. There are on-going clinical trials with TSPO ligands such as FEPPA [89-91], [11C]PBR28 [92], and [18F]PDA-714 [93] in neurodegenerative disorders (one study including FTD). However, published results of these studies are not available yet. There is a need not only for tracking neuroinflammation in FTD with TSPO PET imaging, but also for assessing the accuracy of TSPO imaging in detecting neuroinflammation in FTD. This is especially needed for subtypes within FTD that may have unique molecular mechanisms related to neuroinflammation (that is, progranulin mutations).
Conclusions
Recent molecular advances in the pathophysiology of the FTD have led to new disease models highlighting the potential role of pathologic neuroinflammation in disease onset and progression. PET imaging with the second-generation TSPO ligands offers a potentially powerful means to identify neuroinflammatory patterns in vivo across the FTD subtypes. More studies are needed to image and track neuroinflammation in FTD, which may aid in the diagnosis of early FTD. It is anticipated that the advances of molecular imaging of brain TSPO will overcome technical difficulties and that molecular imaging of neuroinflammation could aid in the characterization of neuroinflammation in FTD, increase our understanding of disease pathogenesis, and inform development and testing of novel therapeutic interventions.
Abbreviations
- AD:
-
Alzheimer’s disease
- ALC:
-
acetyl-L-carnitine
- ALS:
-
amyotrophic lateral sclerosis
- bv-FTD:
-
behavioral variant FTD
- CNS:
-
central nervous system
- DPA714:
-
[18F]-DPA714
- FEPPA:
-
[18F]-FEPPA
- FTD:
-
frontotemporal dementia
- FUS:
-
fused in sarcoma protein
- GRN:
-
progranulin
- HAB:
-
high-affinity binders
- IL:
-
interleukin
- LAB:
-
low-affinity binders
- MAB:
-
mixed-affinity binders
- MAPT:
-
microtubule-associated protein tau
- MCI:
-
mild cognitive impairment
- MS:
-
multiple sclerosis
- PD:
-
Parkinson’s disease
- PNFA:
-
progressive non-fluent aphasia
- svPPA:
-
semantic variant primary progressive aphasia
- TDP:
-
transactive response DNA-binding protein
- TNF:
-
tumor necrosis factor
- TSPO:
-
translocator protein
References
Neary D, Snowden JS, Mann DMA. Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol. 2007;114:31–8.
Hodges JR, Davies R, Xuereb J, Kril J, Halliday G. Survival in frontotemporal dementia. Neurology. 2003;61(3):349–54.
Riedl L, Mackenzie IR, Förstl H, Kurz A, Diehl-Schmid J. Frontotemporal lobar degeneration: current perspectives. Neuropsychiatric Disease Treatment. 2014;10:297–310.
Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 2012;8(8):423–34.
Diehl-Schmid J, Pohl C, Perneczky R, Hartmann J, Förstl H, Kurz A. Frühsymptome, Überlebenszeit und Todesursachen. Initial symptoms, survival and causes of death in 115 patients with frontotemporal lobar degeneration. Fortschr Neurol Psychiatr. 2007;75(12):708–13.
Womack KB, Diaz-Arrastia R, Aizenstein HJ, Arnold SE, Barbas NR, Boeve BF, et al. Temporoparietal hypometabolism in frontotemporal lobar degeneration and associated imaging diagnostic errors. Arch Neurol. 2011;68(3):329–37.
Ho SW, Tsui YT, Wong TT, Cheung SK, Goggins WB, Yi LM, et al. Effects of 17-allylamino-17-demethoxygeldanamycin (17-AAG) in transgenic mouse models of frontotemporal lobar degeneration and Alzheimer’s disease. Transl Neurodegener. 2013;2(1):24.
Neumann K, Farias G, Slachevsky A, Perez P, Maccioni RB. Human platelets tau: a potential peripheral marker for Alzheimer’s disease. J Alzheimers Dis. 2011;25:103–9.
Borza LR. A review on the cause-effect relationship between oxidative stress and toxic proteins in the pathogenesis of neurodegenerative diseases. Rev Med Chir Soc Med Nat Iasi. 2014;118(1):19–27.
Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14(7):463–77.
Morales I, Guzmán-Martínez L, Cerda-Troncoso C, Farías GA, Maccioni RB. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front Cell Neurosci. 2014;8:112.
Birch AM, Katsouri L, Sastre M. Modulation of inflammation in transgenic models of Alzheimer’s disease. J Neuroinflammation. 2014;11:25–38.
Miller ZA, Rankin KP, Graff-Radford NR, Takada LT, Sturm VE, Cleveland CM, et al. TDP-43 frontotemporal lobar degeneration and autoimmune disease. J Neurol Neurosurg Psychiatry. 2013;84(9):956–62.
Jacobs AH, Tavitian B. INMiND consortium. Noninvasive molecular imaging of neuroinflammation. J Cereb Blood Flow Metab. 2012;32(7):1393–415.
Venneti S, Wiley CA, Kofler J. Imaging microglial activation during neuroinflammation and Alzheimer’s disease. J Neuroimmune Pharmacol. 2009;4(2):227–43.
Kleinberger G, Capell A, Haass C, Van Broeckhoven C. Mechanisms of granulin deficiency: lessons from cellular and animal models. Mol Neurobiol. 2013;47:337–60.
Nichol KE, Kim R, Cotman CW. Bcl-2 family protein behaviour in frontotemporal dementia implies vascular involvement. Neurology. 2001;56(11 Suppl 4):S35–40.
Pickford F, Marcus J, Camargo LM, Xiao Q, Graham D, Mo JR, et al. Progranulin is a chemoattractant for microglia and stimulates their endocytic activity. Am J Pathol. 2011;178:284–95.
Rosso S, Landweer E, Houterman M, Donker Kaat L, van Duijn CM, van Swieten JC. Medical and environmental risk factors for sporadic frontotemporal dementia: a retrospective case–control study. J Neurol Neurosurg Psychiatry. 2003;74:1574–6.
Sjögren M, Folkesson S, Blennow K, Tarkowski E. Increased intrathecal inflammatory activity in frontotemporal dementia: pathophysiological implications. J Neurol Neurosurg Psychiatry. 2004;75:1107–11.
Cagnin A, Kassiou M, Meikle SR, Banati RB. In vivo evidence for microglial activation in neurodegenerative dementia. Acta Neurol Scand. 2006;114 Suppl 185:107–14.
Okello A, Koivunen J, Edison P, Archer HA, Turkheimer FE, Någren K, et al. Conversion of amyloid positive and negative MCI to AD over 3 years: an 11C-PIB PET study. Neurology. 2009;73(10):754–60.
Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19–31.
Yasuno F, Kosaka J, Ota M, Higuchi M, Ito H, Fujimura Y, et al. Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment-dementia converters measured by positron emission tomography with [11C]DAA1106. Psychiatry Res. 2012;203(1):67–74.
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–51.
Zimmer ER, Leuzy A, Benedet AL, Breitner J, Gauthier S, Rosa-Net P. Tracking neuroinflammation in Alzheimer’s disease: the role of positron emission tomography imaging. J Neuroinflammation. 2014;11:120.
Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23(3):309–17.
Hommet C, Mondon K, Camus V, Ribeiro MJ, Beaufils E, Arlicot N, et al. Neuroinflammation and β amyloid deposition in Alzheimer’s disease: in vivo quantification with molecular imaging. Dement Geriatr Cogn Disord. 2014;37(1–2):1–18.
Guerreiro RJ, Lohmann E, Brás JM, Gibbs JR, Rohrer JD, Gurunlian N, et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 2013;70:78–84.
Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener. 2013;8:19.
Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15(5):300–12.
Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, et al. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–9.
McGeer PL, McGeer EG. Inflammation and the degenerative diseases of aging. Ann N Y Acad Sci. 2004;1035:104–16.
Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–53.
Cagnin A, Rossor M, Sampson EL, Mackinnon T, Banati RB. In vivo detection of microglial activation in frontotemporal dementia. Ann Neurol. 2004;56:894–7.
Venneti S, Wang G, Nguyen J, Wiley CA. The positron emission tomography ligand DAA1106 binds with high affinity to activated microglia in human neurological disorders. J Neuropathol Exp Neurol. 2008;67:1001–10.
Lant SB, Robinson AC, Thompson JC, Rollinson S, Pickering-Brown S, Snowden JS, et al. Patterns of microglial cell activation in frontotemporal lobar degeneration. Neuropathol Appl Neurobiol. 2014;40(6):686–96.
Wiley CA, Lopresti BJ, Venneti S, Price J, Klunk WE, DeKosky ST, et al. Carbon 11-labeled Pittsburgh compound B and carbon 11-labeled (R)-PK11195 positron emission tomographic imaging in Alzheimer disease. Arch Neurol. 2009;66:60–7.
Schuitemaker A, Kropholler MA, Boellaard R, van der Flier WM, Kloet RW, van der Doef TF, et al. Microglial activation in Alzheimer’s disease: an (R) [11C]PK11195 positron emission tomography study. Neurobiol Aging. 2013;34:128–36.
Chauveau F, Van Camp N, Dollé F, Kuhnast B, Hinnen F, Damont A, et al. Comparative evaluation of the translocator protein radioligands 11C-DPA-713, 18F-DPA-714, and 11C-PK11195 in a rat model of acute neuroinflammation. J Nucl Med. 2009;50(3):468–76.
Van Camp N, Boisgard R, Kuhnast B, Thézé B, Viel T, Grégoire MC, et al. In vivo imaging of neuroinflammation: a comparative study between [(18)F]PBR111, [(11)C]CLINME and [(11)C]PK11195 in an acute rodent model. Eur J Nucl Med Mol Imaging. 2010;37(5):962–72.
Vas A, Shchukin Y, Karrenbauer VD, Cselényi Z, Kostulas K, Hillert J, et al. Functional neuroimaging in multiple sclerosis with radiolabelled glia markers: preliminary comparative PET studies with [11C]vinpocetine and [11C]PK11195 in patients. J Neurol Sci. 2008;264(1–2):9–17.
Miyoshi M, Shinotoh H, Wszolek ZK, Strongosky AJ, Shimada H, Arakawa R, et al. In vivo detection of neuropathologic changes in presymptomatic MAPT mutation carriers: a PET and MRI study. Parkinsonism Relat Disord. 2010;16(6):404–8.
Kreisl WC, Lyoo CH, McGwier M, Snow J, Jenko KJ, Kimura N, et al. Biomarkers Consortium PET Radioligand Project Team. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain. 2013;136(Pt 7):2228–38.
Oh U, Fujita M, Ikonomidou VN, Evangelou IE, Matsuura E, Harberts E, et al. Translocator protein PET imaging for glial activation in multiple sclerosis. J Neuroimmune Pharmacol. 2011;6(3):354–61.
Fujita M, Imaizumi M, Zoghbi SS, Fujimura Y, Farris AG, Suhara T, et al. Kinetic analysis in healthy humans of a novel positron emission tomography radioligand to image the peripheral benzodiazepine receptor, a potential biomarker for inflammation. Neuroimage. 2008;40(1):43–52.
Owen DR, Gunn RN, Rabiner EA, Bennacef I, Fujita M, Kreisl WC, et al. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J Nucl Med. 2011;52(1):24–32.
Owen DR, Howell OW, Tang SP, Wells LA, Bennacef I, Bergstrom M, et al. Two binding sites for [3H]PBR28 in human brain: implications for TSPO PET imaging of neuroinflammation. J Cereb Blood Flow Metab. 2010;30(9):1608–18.
Mizrahi R, Rusjan PM, Kennedy J, Pollock B, Mulsant B, Suridjan I, et al. Translocator protein (18 kDa) polymorphism (rs6971) explains in-vivo brain binding affinity of the PET radioligand [(18)F]-FEPPA. J Cereb Blood Flow Metab. 2012;32(6):968–72.
Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012;32(1):1–5.
Varrone A, Mattsson P, Forsberg A, Takano A, Nag S, Gulyás B, et al. In vivo imaging of the 18-kDa translocator protein (TSPO) with [18 F]FEDAA1106 and PET does not show increased binding in Alzheimer’s disease patients. Eur J Nucl Med Mol Imaging. 2013;40(6):921–31.
Takano A, Piehl F, Hillert J, Varrone A, Nag S, Gulyás B, et al. In vivo TSPO imaging in patients with multiple sclerosis: a brain PET study with [18 F]FEDAA1106. EJNMMI Res. 2013;3(1):30.
Fujimura Y, Zoghbi SS, Simeon FG, Taku A, Pike VW, Innis RB, et al. Quantification of translocator protein (18 kDa) in the human brain with PET and a novel radioligand, 18F-PBR06. J Nucl Med. 2009;50:1047–53.
Wilson AA, Garcia A, Parkes J, McCormick P, Stephenson KA, Houle S, et al. Radiosynthesis and initial evaluation of [18F]-FEPPA for PET imaging of peripheral benzodiazepine receptors. Nucl Med Biol. 2008;35(3):305–14.
James ML, Fulton RR, Vercoullie J, Henderson DJ, Garreau L, Chalon S, et al. DPA-714, a new translocator protein-specific ligand: synthesis, radiofluorination, and pharmacologic characterization. J Nucl Med. 2008;49:814–22.
Arlicot N, Vercouillie J, Ribeiro MJ, Tauber C, Venel Y, Baulieu JL, et al. Initial evaluation in healthy humans of [18F]DPA-714, a potential PET biomarker for neuroinflammation. Nucl Med Biol. 2012;39(4):570–8.
Hatano K, Yamada T, Toyama H, Kudo G, Nomura M, Suzuki H, et al. Correlation between FEPPA uptake and microglia activation in 6-OHDA injured rat brain. Neuroimage. 2010;52 Suppl 1:S138–8.
Liu F, Zhang X, Patterson TA, Liu S, Ali SF, Paule MG, et al. Assessment of potential neuronal toxicity of inhaled anesthetics in the developing nonhuman primate. J Drug Alcohol Res. 2012;1:1–9.
Zhang X, Paule MG, Newport GD, Liu F, Callicott R, Liu S, et al. MicroPET/CT imaging of [18F]-FEPPA in the nonhuman primate: a potential biomarker of pathogenic processes associated with anesthetic-induced neurotoxicity. ISRN Anesthesiology. 2012;261640:11.
Zhang X, Liu S, Paule MG, Newport GD, Callicott R, Berridge MS, et al. Protective effects of acetyl L-carnitine on inhalation anesthetic-induced neuronal damage in the nonhuman primate. J Mol Pharm Org Process Res. 2013;1:1.
Bennacef I, Salinas C, Horvath G, Gunn R, Bonasera T, Wilson A, et al. Comparison of [11C]PBR28 and [18F]FEPPA as CNS peripheral benzodiazepine receptor PET ligands in the pig. J Nucl Med. 2008;49(Supplement 1):81P.
Vasdev N, Green DE, Vines DC, McLarty K, McCormick PN, Moran MD, et al. Positron-emission tomography imaging of the TSPO with [(18)F]FEPPA in a preclinical breast cancer model. Cancer Biother Radiopharm. 2013;28(3):254–9.
Jennings D, Evaluation of [18F] FEPPA and PET imaging as a marker of inflammation in subjects with neurological conditions, [Clinical Trial: http://clinicaltrials.gov/show/NCT00970333], 2009–2012.
Rusjan PM, Wilson AA, Bloomfield PM, Vitcu I, Meyer JH, Houle S, et al. Quantitation of translocator protein binding in human brain with the novel radioligand [18F]-FEPPA and positron emission tomography. J Cereb Blood Flow Metab. 2011;31(8):1807–16.
Suridjan I, Rusjan PM, Kenk M, Verhoeff NP, Voineskos AN, Rotenberg D, et al. Quantitative imaging of neuroinflammation in human white matter: a positron emission tomography study with translocator protein 18 kDa radioligand, [18F]-FEPPA. Synapse. 2014;68(11):536–47.
Suridjan I, Rusjan PM, Voineskos AN, Selvanathan T, Setiawan E, Strafella AP, et al. Neuroinflammation in healthy aging: a PET study using a novel translocator protein 18 kDa (TSPO) radioligand, [(18)F]-FEPPA. Neuroimage. 2014;1(84):868–75.
Suridjan I, Pollock BG, Voineskos AN, Verhoeff P, Chow T, Mulsant BH, Rusjan PM, Houle S, Mizrahi R. Mapping neuroinflammation in vivo in healthy aging and Alzheimer’s disease: a PET study using a novel translocator protein 18kDA (TSPO) radioligand, [18F]-FEPPA, 2012. (poster), Alzheimer’s & Dementia, 8(4), Suppl 693:P4-164
Suridjan I, Pollock BG, Voineskos AN, Verhoeff P, Chow T, Mulsant BH, Rusjan PM, Houle S, Mizrahi R. Mapping neuroinflammation in-vivo in Alzheimer’s disease: a PET study using a novel TSPO radioligand, [18F]FEPPA. 39th Annual Harvey Stancer Research Day, University of Toronto, Toronto, 2013 (poster).
Koshimori Y, Ko JH, Mizrahi R, Rusjan PM, Wilson AA, Houle S, et al. Activated microglia in Parkinson’s disease: a PET study with a novel radiotracer, 18FFEPPA [abstract]. Mov Disord. 2012;27 Suppl 1:741.
Ko JH, Koshimori Y, Mizrahi R, Rusjan P, Wilson AA, Lang AE, et al. Voxel-based imaging of translocator protein 18 kDa (TSPO) in high-resolution PET. J Cereb Blood Flow Metab. 2013;33(3):348–50.
Bernards N, Pottier G, Thézé B, Dollé F, Boisgard R. In vivo evaluation of inflammatory bowel disease with the aid of μPET and the translocator protein 18 kDa radioligand [18F]DPA-714. Mol Imaging Biol. 2015;17:67–75.
Wu C, Yue X, Lang L, Kiesewetter DO, Li F, Zhu Z, et al. Longitudinal PET imaging of muscular inflammation using 18F-DPA-714 and 18F-Alfatide II and differentiation with tumors. Theranostics. 2014;4(5):546–55.
Doorduin J, Klein HC, Dierckx RA, James M, Kassiou M, de Vries EF. [11C]-DPA-713 and [18F]-DPA-714 as new PET tracers for TSPO: a comparison with [11C]-(R)-PK11195 in a rat model of herpes encephalitis. Mol Imaging Biol. 2009;11(6):386–98.
Boutin H, Prenant C, Maroy R, Galea J, Greenhalgh AD, Smigova A, et al. [18F]DPA-714: direct comparison with [11C]PK11195 in a model of cerebral ischemia in rats. PLoS One. 2013;8(2):e56441.
Awde AR, Boisgard R, Thézé B, Dubois A, Zheng J, Dollé F, et al. The translocator protein radioligand 18F-DPA-714 monitors antitumor effect of erufosine in a rat 9L intracranial glioma model. J Nucl Med. 2013;54(12):2125–31.
Pottier G, Bernards N, Dollé F, Boisgard R. [18F]DPA-714 as a biomarker for positron emission tomography imaging of rheumatoid arthritis in an animal model. Arthritis Res Ther. 2014;16(2):R69.
Gent YY, Weijers K, Molthoff CF, Windhorst AD, Huisman MC, Kassiou M, et al. Promising potential of new generation translocator protein tracers providing enhanced contrast of arthritis imaging by positron emission tomography in a rat model of arthritis. Arthritis Res Ther. 2014;16(2):R70.
Corcia P, Tauber C, Vercoullie J, Arlicot N, Prunier C, Praline J, et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS One. 2012;7(12):e52941.
Ribeiro MJ, Vercouillie J, Debiais S, Cottier JP, Bonnaud I, Camus V, et al. Could (18) F-DPA-714 PET imaging be interesting to use in the early post-stroke period? EJNMMI Res. 2014;4:28.
Mizrahi R, Rusjan PM, Vitcu I, Ng A, Wilson AA, Houle S, et al. Whole body biodistribution and radiation dosimetry in humans of a new PET ligand, [(18)F]-FEPPA, to image translocator protein (18 kDa). Mol Imaging Biol. 2013;15(3):353–9.
Farde L, Hall H, Ehrin E, Sedvall G. Quantitative analysis of D2 dopamine receptor binding in the living human brain by PET. Science. 1986;231(4735):258–61.
Lammertsma AA, Bench CJ, Hume SP, Osman S, Gunn K, Brooks DJ, et al. Comparison of methods for analysis of clinical [11C]raclopride studies. J Cereb Blood Flow Metab. 1996;16(1):42–52.
Lammertsma AA, Hume SP. Simplified reference tissue model for PET receptor studies. Neuroimage. 1996;4(3 Pt 1):153–8.
Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16(5):834–40.
Ichise M, Liow JS, Lu JQ, Takano A, Model K, Toyama H, et al. Linearized reference tissue parametric imaging methods: application to [11C]DASB positron emission tomography studies of the serotonin transporter in human brain. J Cereb Blood Flow Metab. 2003;23(9):1096–112.
Csele’nyi Z, Olsson H, Halldin C, Gulya’s B, Farde L. A comparison of recent parametric neuroreceptor mapping approaches based on measurements with the high affinity PET radioligands [11C]FLB 457 and [11C]WAY 100635. Neuroimage. 2006;32(4):1690–708.
Turkheimer FE, Edison P, Pavese N, Roncaroli F, Anderson AN, Hammers A, et al. Reference and target region modeling of [11C]-(R)-PK11195 brain studies. J Nucl Med. 2007;48:158–67.
Rizzo G, Veronese M, Tonietto M, Zanotti-Fregonara P, Turkheimer FE, Bertoldo A. Kinetic modeling without accounting for the vascular component impairs the quantification of [(11)C]PBR28 brain PET data. J Cereb Blood Flow Metab. 2014;34(6):1060–9.
Mizrahi R, 2012. Imaging neuroinflammation in mild cognitive impairment using a novel translocator protein 18kDA (TSPO) PET radioligand: [18F]-FEPPA. [Research Project] http://westonbraininstitute.ca/funded-research/projects/imaging-neuroinflammation-mild-cognitive-impairment-using-novel-translocator-protein-18kda-tspo-pet-radioligand-18f-feppa/. Accessed 20 June 2014.
Mizrahi R, 2012. F-18-FEPPA: a novel in-vivo molecular biomarker of neuroinflammation in Alzheimer’s disease. [Research Project] http://www.alzheimer.ca/en/algoma/Research/Alzheimer-Society-Research-Program/Alzheimer-Society-Research-Program/ASRP-Award-recipients-2012/Biomedical-grants. Accessed 20 June 2014.
Strafella A. 2012. Can neuroinflammation be used as a biomarker for Parkinson’s disease? [Research project] http://westonbraininstitute.ca/funded-research/projects/can-neuroinflammation-used-biomarker-parkinsons-disease/. Accessed 20 June 2014.
Innis RB, PET Evaluation of brain peripheral benzodiazepine receptors using [11C]PBR28 in frontotemporal dementia. [Clinical Trial: http://clinicaltrials.gov/show/NCT00613119], 2008–2014.
Hommet C, PET imaging of the translocator proteine ligands (TSPO) with [18F] DPA-714 biomarker of neuroinflammation in cognitive decline (NIDECO). [Clinical Trial: http://clinicaltrials.gov/show/NCT02062099], 2014.
Acknowledgements
The author would like to thank Drs. Turkheimer, Rusjan, and Jennings for addressing questions of their work in this area and their related discussion. This work was supported by the University of Western Ontario, Schulich School of Medicine & Dentistry Research Initiative Grant.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares no competing interests.
Author’s contributions
JZ contributed to the drafting of the main text and producing the references. The author read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Zhang, J. Mapping neuroinflammation in frontotemporal dementia with molecular PET imaging. J Neuroinflammation 12, 108 (2015). https://doi.org/10.1186/s12974-015-0236-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-015-0236-5