
Abstract
Juvenile idiopathic arthritis (JIA) is the most common paediatric rheumatological disorder and is classified by subtype according to International League of Associations for Rheumatology criteria. Depending on the number of joints affected, presence of extra-articular manifestations, systemic symptoms, serology and genetic factors, JIA is divided into oligoarticular, polyarticular, systemic, psoriatic, enthesitis-related and undifferentiated arthritis. This review provides an overview of advances in understanding of JIA pathogenesis focusing on aetiology, histopathology, immunological changes associated with disease activity, and best treatment options. Greater understanding of JIA as a collective of complex inflammatory diseases is discussed within the context of therapeutic interventions, including traditional non-biologic and up-to-date biologic disease-modifying anti-rheumatic drugs. Whilst the advent of advanced therapeutics has improved clinical outcomes, a considerable number of patients remain unresponsive to treatment, emphasising the need for further understanding of disease progression and remission to support stratification of patients to treatment pathways.
Similar content being viewed by others


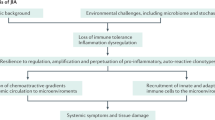
Introduction and classification
Juvenile idiopathic arthritis (JIA) unifies all forms of chronic childhood arthritis, affecting not only joints, but extra-articular structures, including eyes, skin, and internal organs, leading to disability and even associated fatality. It is defined as the presence of arthritis of unknown aetiology that begins before the age of 16 and persists for at least 6 weeks [1].
The International League of Associations for Rheumatology (ILAR) stratifies subtype of autoimmune inflammatory disorders, determined by the number of joints affected, the presence of systemic symptoms and detection of rheumatoid factor (RF). JIA is divided into the sub-forms: oligoarticular (persistent or extended), polyarticular (RF-negative or RF-positive), systemic (sJIA), psoriatic arthritis and enthesitis-related arthritis, with each differing in genetic susceptibility and severity of arthritis [1]. Any arthritis that does not fit into these categories or corresponds to > 1 subtype is considered as undifferentiated [2]. The main characteristics of arthritis, internal organ involvement, genetic predisposition, laboratory markers of each subtype and adult equivalent are shown in Table 1.
Oligoarticular JIA is characterised by inflammation of up to four joints that archetypically proceeds as asymmetrical arthritis predominantly affecting the joints of the lower extremities, such as knee and ankle, with high frequency of positivity to anti-nuclear antibody (ANA) and high risk of chronic uveitis [3]. Polyarticular JIA (pJIA) affects five or more large/ small joints and is hallmarked by injury to the metacarpophalangeal joints and wrists [4]. Both RF-positive and negative variants have characteristic clinical features. RF-negative pJIA, inflammation can be asymmetrical, but for RF-positive pJIA symmetric involvement of the large and small joints of hands and feet is the most prevalent. Enthesitis-related arthritis (ERA) resembles oligoarthritis, affecting the joints of the lower limb in association with enthesitis. Due to the association with lower limb and sacroiliac joints, enthesitis, uveitis and the association with HLA-B27, Ravelli et al. (2007) have suggested ERA to be a disease belonging to the group of spondyloarthropathies [1]. Psoriatic arthritis often proceeds as oligoarthritis or RF-negative polyarthritis and involves more commonly the small joints accompanied by dactylitis, psoriatic rash and/ or nail pitting [1, 3]. Psoriatic JIA itself is described as a heterogeneous disease where children < 6 years are more likely to be female, ANA-positive and predisposed to chronic uveitis, with arthritis of wrists and small joints of the hands and feet. In older children disease is associated with HLA-B27 positivity, enthesitis and axial disease with male predisposition [5].
Standing apart from other subtypes, sJIA manifests not only with widespread joint arthritis, but also with a significant range of systemic inflammation symptoms (Table 1) [6]. Approximately 10% of sJIA patients present systemic symptoms with associated macrophage activation syndrome (MAS), a potentially life-threatening condition with histopathological features that include the accumulation of terminally-differentiated macrophages with high hemophagocytic activity [7, 8]. Currently the limitations of the ILAR classification scheme are pertinent and include the absence of link to pathogenesis, molecular pathways and response to the therapy [9]. In addition, there is a substantial unclassified cohort of patients with JIA onset before 6 years of age and female predominance having specific features that include symmetric arthritis, iridocyclitis, ANA and HLA-DR8-positivity. In 2019 the Pediatric Rheumatology International Trials Organization (PRINTO) Consensus revised the ILAR classification criteria and proposed to identify this complex of features as early-onset ANA-positive JIA [10]. Other JIA disorders identified according to these preliminary criteria include sJIA, RF-positive JIA and enthesitis/spondylitis-related JIA (Table 2). Arthritis of more than 6 weeks duration that does not fit the criteria is grouped as ‘Other JIA’, and that fitting more than one criterion, ‘unclassified JIA’ [10]. PRINTO Consensus highlights JIA as not a single disease but a group of different disorders, of which diagnosis does not require joint count or the presence of arthritis. The onset of the disease has been changed to before 18 years of age [10].
Histopathology of JIA
The main hallmark of JIA is joint inflammation with tissue destruction [11]. Within the synovial joint, the synovial membrane thickens in response to uncontrolled proliferation of synoviocytes and immunocompetent cells, including T-cells, B-cells, natural killers, neutrophils, macrophages, dendritic cells and plasma cells that infiltrate the sub-lining layer of the synovium (Fig. 1) [11].

Schematic diagram showing the differences between the normal and JIA joint. The pathological process within the JIA synovial joint is characterised by uncontrolled proliferation of synoviocytes resulting in increased number of layers and thickening of the synovial membrane; rapid pathological angiogenesis; formation of pathological synovium, “pannus”, with uncontrolled growth and invasive properties; accumulation of granulocytes, macrophages, plasma cells, lymphocytes and the production of inflammatory mediators, provoking synovitis. Created with BioRender.com
Hyperplasia and hypertrophy of the synovium causes intraarticular hypoxia, increasing the production of pro-angiogenic mediators and initiating pathological angiogenesis [12]. Increased concentrations of vascular endothelial growth factor (VEGF), a potent endothelial cell (EC) mitogen, its soluble receptors-1 and -2 (sVEGF-R1, sVEGF-R2), and osteopontin (OPN), a chemotactic factor that activates mononuclear cells, have been correlated to synovial angiogenesis assessed by Doppler ultrasonography of JIA patients [12]. Angiopoietin-1 (Ang-1), another pro-angiogenic EC mitogen with a role in stabilisation of newly formed vessels was also shown to be upregulated in JIA. New blood vessel formation within the synovium increases blood supply and the migration of pro-inflammatory cells into the joint, forming a pathological synovium, known as ‘pannus’ (Fig. 1).
Granulocytes, macrophages, plasma cells and lymphocytes accumulate in the subintima of the joint and produce pro-inflammatory mediators including tumour necrosis factor-α (TNFα) and interleukin (IL)1β, upregulating pannus-synoviocyte production of catabolic proteases including matrix metalloproteinases (MMPs, particularly MMP1 and MMP3), aggrecanases and cathepsins, that breakdown the extracellular matrix of the articular cartilage tissue causing loss of function, biomechanical strength and the ability to smoothly articulate the joint [2, 13]. Pro-inflammatory cytokine-mediated activation of receptor activator of nuclear factor-kappaB (RANK)-expressing osteoclasts results in bone erosion [14]. Damage to cartilage and bone in the advanced stages of JIA causes ankyloses and the loss of movement in the affected joints. Considering that JIA is a disease of the developing body, patients with JIA are likely to suffer from disruption of skeletal growth [15].
Aetiology
The heterogeneity of JIA disease subtypes adds complexity to the investigation of cause and mechanism of pathogenesis, and the initiating factors of JIA remain unresolved [16].
Environmental factors, including infectious agents, vaccinations, antibiotics, vitamin D deficiency, stress and trauma have been proposed as risk factors. Infectious viruses (Epstein-Barr virus, Parvovirus B, Rubivirus, Hepatitis B virus) and bacteria (Salmonella spp., Shigella spp., Campylobacter spp., S. pyogenes, B. henselae, M. pneumoniae, Chlamydophila pneumonia) have been reported as causal factors provoking JIA [17]. Gastrointestinal infection leading to loss of gut microbiome diversity and disrupted tryptophan metabolism increases the risk of ERA [18]. Carlens et al. (2009) reported that maternal smoking during pregnancy increased the probability of an immune imbalance during foetal development leading to the onset and progression of paediatric arthritis [19]. In contrast, some beneficial factors such as breast feeding and household siblings might decrease the risk of developing JIA [20].
Several studies have documented genetic associations to JIA [21,22,23,24,25]. Genetic linkage depends on subtype and may be divided into two groups: HLA genes and non-HLA-related genes. Meta-analysis of genetic predisposition to JIA subtypes has shown association with HLA class II molecules (A2, DRB1, DPB1) mostly for non-systemic subtypes (Table 1), while for sJIA the lack of association with HLA genes has been found [21]. Oligoarticular JIA is associated with A2, DRB1*11, DRB1*08, DPB1*02, DRB1*13, DRB1*15*01 and DRB1*01, while for RF- polyarticular the most commonly associated genes are DPB1:03 and DRB1:08, and for RF+ JIA, DRB1*04 and DRB1*01 [23]. Of interest, HLA-A, HLA-B and HLA-DR were observed in females with oligoarthritis but not males, which may point to disease heterogeneity [2]. The main gene associated with ERA is HLA-B27, with other genes predisposing the development of ERA being DRB1*01, DQA1*01, and DQB1*05 [18]. HLA-B27 is also found in late-onset psoriatic JIA [5].Genetic pre-disposition of non-HLA-related genes plays a pivotal role in the onset of inflammatory response leading to tissue damage. Genes encoding cytokines TNF, IL2, IL10, IL6, macrophage migration inhibitory factor (MIF), protein tyrosine phosphatase (PTPN22), signal transducer and activator of transcription-4 (STAT4), solute carrier family-11 (proton-coupled divalent metal ion transporters), member-1 (SLC11A1), natural resistance-associated macrophage protein-1 (NRAMP1) and WNT1-inducible signalling pathway protein-3 (WISP3) have all been associated with JIA [2, 21, 23]. Polymorphism in genes encoding endoplasmic reticulum resident aminopeptidases (ERAP1 and ERAP2) predispose to ERA, while genes encoding IL1, IL6, IL10 and MIF increase the risk of sJIA, which itself is considered as a genetically distinct subtype of JIA [16].
Pathogenesis of JIA
JIA subtypes represent a heterogeneous group of diseases with multifactorial and different pathogenesis (Table 3). It is not completely understood how the combination of the environmental triggers and genetic susceptibility disrupt the balance between regulatory and effector cells in the pathogenesis of JIA.
Immunological changes
Initiation of the JIA pathophysiological cascade includes abnormal activation of T-cells, B-cells, natural killer (NK) cells, dendritic cells (DC), macrophages and neutrophils and the production of pro-inflammatory mediators that cause joint destruction and systemic complications.
Oligoarticular and pJIA are characterised by autoreactive antigen-specific T-cells and high titres of autoantibodies, and typically shows strong associations with MHC class II alleles. Breakdown of immunologic self-tolerance involves MHC class II alleles suggesting a pivotal role for CD4+ T helper (Th) cells [21]. Inflammation is considered to be a consequence of disrupted balance between pro-inflammatory Th1/Th17 and anti-inflammatory regulatory T-cells (Treg). The decrease in Treg cells is inversely correlated with an increase in the Th17 cell population and occurs from the differentiation of naïve T-cells by influence of IL1β [26]. Differentiation of naive T-cells into Th-cells results in the production of pro-inflammatory cytokine IL17, which may induce production of IL6, MMP1 and 3, IL8 (a chemoattractant for neutrophils) by synoviocytes, resulting in subsequent joint destruction [16, 26]. Prelog et al. (2008) revealed premature immunosenescence of T-cells in oligoarticular, pJIA and sJIA patients, as indicated by the loss of compensatory proliferation of naive T-cells, increased telomeric erosion, and loss of capability of the thymus to produce T-cell receptor excision circles [27].
Association with HLA class II and the presence of ANA suggests that an adaptive immune response is predominant in the pathogenesis of oligoarticular JIA. However, activated neutrophils with altered phenotype and dysfunction, and impaired synovial monocytes and macrophages with reduced capacity to phagocytose have recently been identified in synovial fluid of patients with oligoarticular JIA [28, 29]. Together with high levels of monocyte-derived cytokines this emphasises the importance of the innate immune system in oligoarticular JIA pathogenesis [28, 29].
The pathogenesis of ERA is driven by HLA-B27-mediated presentation of arthritogenic peptide following T-cell activation and IL23 and IL17 secretion. Bowel wall inflammation usually accompanied with ERA is driven by γδT cells, innate lymphoid cells type-3 or Th17 cells and IL17 and IL23 production [18]. Enthesitis is triggered by repeated biomechanical stress stimulation resulting in microtrauma and release of fibronectin, hyaluronan, and other molecular components from damaged connective tissue, which may directly activate synovial macrophages, stromal cells and IL23 production to establish a positive feedback loop. Interestingly, the pathogenesis of late-onset psoriatic JIA resembles ERA with entheses inflammation and inflammation in the bowel wall [5]. Early-onset psoriatic JIA is characterised by adaptive immune mechanisms involvement with development of dactylitis [5].
Standing apart from non-systemic subtypes that are known as autoimmune disorders, sJIA has been suggested to be an autoinflammatory pathology with different pathogenesis [16]. In sJIA, uncontrolled activation of the innate immune system results in activation of monocytes/macrophages, neutrophils and immature (CD34 + CD33+) myelomonocytic precursors, and increased production of pro-inflammatory cytokines IL1β, IL6, IL18 and phagocyte-specific S100 proteins [16, 30, 31]. MAS is a complicated sJIA, where some triggers (bacterial or viral infections, drugs) cause uncontrolled expansion of cytotoxic CD8+ T-cells that produce pro-inflammatory cytokines and propagate the induction and activation of hemophagocytic macrophages that infiltrate bone marrow and multiple organs in particular the liver and spleen [8].
Inflammatory cytokines
There is a significant and predominant pro-inflammatory cytokine signature in the plasma and synovial fluid of patients with JIA. JIA patients demonstrate high levels of TNFα, MIF, macrophage inflammatory protein (CCL3), macrophage-derived chemokine (CCL22), monokine induced by IFNγ (CXCL9), monocyte chemoattractant protein-1 (CCL2) and IFNγ-induced protein-10 (CXCL10) in blood and synovial fluid [32]. Comparison of patients with different subtypes showed significantly higher concentrations of plasma CCL11, CXCL10 and CCL2 in oligoarticular JIA compared to sJIA, while patients with sJIA demonstrated higher level of IL1, IL6 and IL18 in serum [32, 33]. Elevated levels of IL33 are observed in patients with RF+ polyarticular JIA in comparison with oligo- and RF+ polyarticular JIA, and are correlated with disease activity, indicative of being a potential biomarker candidate for pJIA disease activity [34]. The concentrations of MIF, IL10 and IL17 in serum or synovial fluid is predictive for oligoarticular JIA (with less than 60% accuracy). MIF, IL17 and IL23 are also increased in ERA [18]. IL18 is predictive for sJIA (with 93% accuracy) [32], and plays a pivotal role in the pathogenesis of MAS, with an increased concentration reported to be predictive of MAS complication in sJIA patients [35]. Martini et al. (2012) showed the correlation between elevated levels of pro-inflammatory cytokine IL6 in sJIA with the severity of joint inflammation and microcytic anaemia due to IL6 influence on erythropoiesis by increasing the synthesis of iron-lowering hormone hepcidin [6].
Leucine-rich α2-glycoprotein (LRG), induced by pro-inflammatory cytokines IL1, IL6 and TNFα, promotes differentiation and proliferation of Th17 cells and is present in the sJIA and MAS correlated with serum CRP and ferritin levels [36]. Adenosine deaminase-2 (ADA2), released by monocytes and macrophages following stimulation with IL18 and IFNγ is considered to be a novel biomarker of MAS, being strongly correlated with ferritin, IL18 and CXCL9 [37].
Anti-inflammatory cytokines are also involved in the progression of JIA [16]. The pleotropic effects of both transforming growth factor-beta (TGFβ) and IL10 impact on the control of innate and adaptive immunity. Thus, TGFβ1 directly targets T-cells leading to the established immune tolerance to self- and environmental antigens. IL10 mediates anti-inflammatory actions by induction of heme oxygenase-1, a stress-inducible protein with anti-inflammatory properties, and through the induction of mammalian target of rapamycin (mTOR) inhibitor [38]. Thus, the clinically inactive disease, in the absence of medication in some patients, may represent compensation of the autoimmune activity by anti-inflammatory cytokines [16]. Another anti-inflammatory cytokine, IL37 was found to be significantly elevated in plasma and IL-37 mRNA expression has been shown to correlate with disease activity and production of pro-inflammatory cytokines IL6, TNFα and IL17 [39].
The collective data available to date, suggests that cytokine patterns may be appropriate for accurate disease classification in early JIA with the potential as targets for improving diagnosis and treatment strategies for patients with paediatric autoimmune disease [33].
Autoantibody production
Serological biomarkers in JIA patient tissues may be stratified into those that are stable and persistent throughout disease course (including antibodies such as RF) and those that change over time and disease activity (including cytokines such as IL18). ANA, RF, anti-cyclic citrullinated peptide (anti-CCP) and antibodies against mutated citrullinated vimentin (anti-MCV) are reported in non-systemic JIA pathogenesis [40, 41]. RF is an antibody specific to the Fc portion of IgG; it was first described as a key serological marker in patients with adult rheumatoid arthritis (RA) and then identified in a small sub-group of pJIA patients (only 5% of total JIA patients) [4]. RF-positivity is associated with severe prognosis of JIA and rapid formation of bone erosions [40]. ANA is considered to be common for oligoarticular, polyarticular, psoriatic subtypes of JIA, and associated with increase the risk of uveitis in JIA patients [42]. Anti-CCP and anti-MCV typically characterise RF-positive pJIA and may predict more severe and erosive disease progression needing earlier and more intensive therapy [40, 43]. Anti-CCP activates complement and macrophages by crosslinking TLR4 and Fc gamma receptors and inducing TNFα production by binding to the macrophage Fcγ receptor IIa in vitro. Patients double positive for Anti-CCP and RF have higher levels of TNFα, IL1β, IL6 and IL17 [41].
Establishing diagnosis and prediction of complications
Clinical symptoms, family history, laboratory markers and instrumental examinations (ultrasound and magnetic resonance imaging) are used to determine JIA subtype. Physical examination findings are paramount and include signs of arthritis (pain, tenderness, stiffness and swelling of synovial joints) and extra-articular findings (such as rash, lymphadenopathy, dactylitis, nail changes). Laboratory tests for HLA-B27, RF or anti-CCP antibody identifies the subtype of JIA and the risk of bone erosions and joint damage. Myeloid-related protein (MRP)8, MRP14 and IL18 may be used as biomarkers for active sJIA, whereas HLA-B27 is predictive of ERA [41]. ANA and RF are useful for the diagnosis of oligo and pJIA subtypes [41]. ANA is associated with increased risk of chronic non-granulomatous uveitis, which is the most common extra-articular manifestation of JIA and is typically asymptomatic but has an elevated risk of causing visual impairment. Aljaberi et al. (2020) reported higher levels of pro-inflammatory calcium-binding S100 proteins in sJIA patients compared to other autoinflammatory syndromes. However, other studies have revealed that high baseline S100A12 concentration is associated with higher disease activity and response to methotrexate (MTX) and anti-TNF therapy in patients with JIA including pJIA, ERA, oligoarticular and psoriatic arthritis [44]. Thus, S100A8/9 and S100A12 proteins are subclinical inflammation markers that may help with diagnosis and monitoring disease activity [45].
Recent history of gastrointestinal or urinary infection, gut inflammation confirmed by elevated fecal calprotectin levels, sacroiliitis with inflammatory spinal changes and enthesitis detected by MRI support diagnosis of ERA [18]. Subclinical gut inflammation has also been identified in older-onset of psoriatic JIA [5].
The diagnosis of sJIA in accordance with ILAR criteria requires arthritis and fever within the last 2 weeks, and one of the following criteria: rash, generalised lymphadenopathy, enlargement of liver or spleen, or serositis [46]. Common laboratory abnormalities suggestive of systemic inflammation include elevated erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), white blood cell count, platelet count, ferritin, transaminases, aldolase and d-dimers help to define the activity of the disease [47]. Laboratorial analysis of patients with active sJIA may reveal granulocytosis, thrombocytosis, anaemia, upregulation of acute phase reactants (elevated erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP), which are typical findings but not so essential for diagnosis in comparison with the life-threatening complication of MAS that include pancytopaenia, increased levels of ferritin, liver enzymes (aspartate and alanine transaminases), triglycerides, d-Dimers and hypofibrinogenemia [7]. Clinical findings of MAS include high non-remitting fever, generalised lymphadenopathy, hepatosplenomegaly, central nervous system dysfunction and hemorrhagic manifestations.
Treatment
Therapeutic intervention begins at diagnosis with non-steroidal anti-inflammatory drugs (NSAIDs) followed by disease-modifying anti-rheumatic drugs (DMARDs, most often methotrexate) and/or corticosteroid intra-articular injection. In blocking prostaglandin production via inhibition of cyclooxygenase-1 and cyclooxygenase-2, NSAIDs obtain both analgesic and anti-inflammatory effects. Local corticosteroid joint injections are effective in synovitis and may be a first-line treatment for oligoarthritis alone or in addition to DMARDs. Systemic administration of high dose corticosteroids provides good short-term effect, especially in sJIA patients, but has no influence on the long-term disease outcome. Moreover, its prolonged administration is associated with severe side effects including osteoporosis, growth suppression, immunosuppression and metabolic effects (Table 4) (48).
The American College of Rheumatology (ACR) recommends early use of DMARDs, specifically MTX, leflunomide and/or sulfasalazine (Table 4) [48]. MTX is considered to be the first choice DMARD for oligo- and pJIA when NSAIDs and intraarticular steroids are insufficient [49,50,51]. MTX is also considered to be effective in children with PsJIA, though the axial manifestations limits prescription of MTX and so TNF inhibitors are typically required in these cases [5]. Leflunomide may be used as an alternative DMARD for pJIA in cases of MTX intolerance [52, 53]. Sulfasalazine is recommended for patients with moderate activity of ERA with active peripheral arthritis, but is inefficient in case of sacroiliitis [18, 54]. The Clinical Commissioning Policy Statement: Biologic Therapies for the treatment of Juvenile Idiopathic Arthritis (2015) reports that in 30–50% of patients where disease continues to progress, advanced biologics are the next therapeutic step.
The first biological drugs registered for the treatment of JIA were anti-TNFα agents, etanercept and adalimumab. Etanercept was approved for the treatment of pJIA in 1999, based on a randomised, placebo-controlled double-blind study evaluation of safety and efficacy [55]. Now TNFα inhibitors are recognized to be the most effective drugs for the treatment of JIA with influence on pain, stiffness, growth and quality of life and were first successful in the treatment of pJIA, then ERA, psoriatic and oligoarthritis subtypes [48, 56, 57]. Combination of TNFα blocking agents with MTX increases the opportunity of achieving JIA remission in patients with these subtypes and is an effective option in uveitis-associated JIA [58]. In a randomized double-blind trial anti-TNF agents namely etanercept or adalimumab have proven effective for ERA [18].
For pJIA that is nonresponsive to at least one DMARD, including TNFα inhibitors, abatacept (CTLA4-Ig) may be recommended [54, 59] following demonstration of long-term efficacy, safety and improvement of quality of life in 58 JIA patients for 7 years [59]. Another option should TNFα inhibition reach sub-optimal clinical outcomes for pJIA is the IL6 receptor inhibitor, tocilizumab. Tocilizumab might also be a treatment option for JIA-related uveitis refractory to MTX and TNF inhibitors [60].
For many years anti-TNFα therapy demonstrated improved treatment outcomes for all forms of JIA but were less effective for sJIA, where the therapeutic approach has been IL1β/IL6 signalling blockade [56, 61,62,63]. Tocilizumab (anti-IL6R) was the first approved medication for the treatment of active sJIA, demonstrating safety and efficacy in two multicentre studies of patients with sJIA and pJIA [31]. Other studies have shown the efficacy of IL1 blockade in sJIA [61]. Complete remission was obtained in seven out of nine patients with refractory sJIA treated with recombinant IL1 receptor antagonist anakinra (the other two patients demonstrating a partial treatment response) [62]. However, Quartier et al. (2011) reported a short-term effect of this drug in corticosteroid-dependent patients with sJIA in a randomised, double-blind, placebo-controlled trial and showed that anakinra is less effective on arthritis than on systemic symptoms [63]. Currently, anakinra (IL1Ra), rilonacept (IL1 inhibitor) and canakinumab (anti-IL1β) have been successfully studied in clinical research with comparable long-term efficacy where half of treated patients achieved remission [61, 64,65,66,67]. IL18 may be another target for treatment of sJIA resistant to IL1 and IL6 inhibition, as far as higher levels of IL18 have been associated with high ferritin levels reported in MAS [68, 69].
MAS as a form of hemophagocytic lymphohistiocytosis is usually treated with high-dose methylprednisolone and cyclosporine A (a calcineurin inhibitor). In relation to biological therapies, treatment of MAS has been successful using IL1 receptor antagonist anakinra (IL-1Ra) and rituximab (anti-CD20) [70], which has also demonstrated efficacy in other immunological disorders, including SLE [61]. Other promising therapeutics, tadekinig alfa (anti-IL18) and emapalumab (anti-IFNγ) are currently undergoing clinical trials with data reporting safety and potential efficacy for the treatment of sJIA and MAS [71, 72].Another new class of biological DMARDs are the Janus-associated tyrosine kinases (JAK) inhibitors. The mechanism of their action consists of blocking JAK-STAT pathways to interrupt the transduction of extracellular pro-inflammatory signals into the cell nucleus. The efficacy of first generation of JAK inhibitors (namely tofacitinib and baricitinib) were first explored in adults with RA, and then in other immune-mediated inflammatory diseases such as ankylosing spondylitis, SLE, inflammatory bowel disease and psoriasis [73]. Tofacitinib was effectively used in case of refractory sJIA [74]. Miserocchi et al. (2020) showed effectiveness of tofacitinib and baricitinib in 4 cases of JIA uveitis, defined by a reduction of intraocular inflammation according to Standardized Uveitis Nomenclature criteria, and no side effects were registered [75]. The results of a randomised phase 3, multinational, double-blind, controlled clinical trial (NCT02592434) has proven the safety and effectiveness of tofacitinib in pJIA, resulting in reduced flares and disease activity [76]. Ongoing trials are investigating baricitinib (NCT04088396, NCT03773978, NCT04088409) and tofacitinib (NCT03000439) in patients with other JIA disorders [77].Recent therapeutic advances including combination of DMARDs, corticosteroids and the biological agents reduce synovitis, tissue damage progression and systemic complications, making low disease activity an achievable goal in JIA. Early treatment with biologics may be important in controlling disease activity and avoiding steroids altogether or at least reducing the duration of use. However, long-term prescription of biologic agents due to immune suppression increases the risk of opportunistic infections and potentially even malignancy (the main adverse events are shown in the Table 4) [56]. Early aggressive treatment of JIA was shown to be beneficial with 40% of patients achieving clinical inactive disease within 6 months [78], though some serious adverse events was registered during treatment (such as pneumonia, septic joint, elevation of transaminases, peritonsillar abscess, recurrent herpes simplex). It may be suggested that some of these patients would have achieved remission even with less aggressive treatment [79].
Disease course, quality of life and functional outcome
Substantial progress in JIA treatment has been made over the last three decades. Clinical outcomes have dramatically improved, with disease control and remission possible in most patients. Nevertheless, a significant proportion of patients have ongoing disease activity. In fact, about half of patients continue to require active treatment into adult life, whereas complete remission is achieved in only 20–25% of patients [80, 81].
Administration of biological agents has decreased the mortality rate of JIA from 1 to 4% in 1970s to 0.3–1% in 2016 [82]. Improved clinical outcomes in physical disability are reflected in the Steinbrocker functional classification scale. Between 1976 and 1994, 15% of JIA patients were within Class III (limited to few or no activities of the patient’s usual occupation) and Class IV (bedridden with little or no self-care), compared to 5% in 2002 [83]. However, joint damage, occurring before treatment led to surgical intervention in 14% of patients, emphasising the importance of early aggressive treatment to achieve complete remission [84]. The prominent factor influencing treatment outcome is presence of systemic manifestation. Multi-organ failure in patients with MAS is fatal in approximately 8% of cases [8].
Being the most frequent extraarticular manifestation JIA-associated uveitis became the main cause of vision loss in childhood, and furthermore about half of these patients suffer from active uveitis in adulthood [85]. Additionally, a high risk of osteoporosis and consequently fractures in early adulthood remain higher in JIA patients even in remission [15].
Long-term outcomes of JIA patients are dependent on subtype and disease activity, which may remain elevated for many years including into adulthood. In early adulthood about half of patients with JIA have active disease and approximately 30% suffer from some form of disability [80]. Selvaag et al. (2016) reported a 59% remission rate in patients with JIA after 30 years but noted the low quality of life in adults with JIA [81].
Comorbidities and complications highlight the status of JIA as the most important paediatric rheumatological pathology that may continue with remissions and flares throughout life, leading to impairment of connective tissue and the reduction of life quality. There is a need for more discovery science research to help the understanding of the complexity of the inflammatory process and to enable the development of treatments that may come with the promise of an actual cure.
Conclusions
JIA is a chronic rheumatic disease of childhood, characterised by progressive joint destruction and serious systemic manifestations. Complex interactions between immune cell populations, including lymphocytes, monocytes, macrophages and neutrophils, trigger the pathophysiological cascade in JIA. Our review of clinical research has demonstrated that the heterogeneity of non-systemic and sJIA pathogenesis stratifies JIA patients by subtype, with requirement for differing therapeutic approaches. A broad range of DMARDs such as T-cell inhibitors, anti-TNFα agents, IL1 and IL6 blockers, JAK inhibitors have significantly improved the clinical management of JIA. However, further research is needed to deepen our understanding of the complexity of the inflammatory process in JIA and to enable the development of effective treatments that improve upon clinical outcomes and disease remission.
Availability of data and materials
Not applicable.
Abbreviations
- JIA:
-
Juvenile idiopathic arthritis
- ILAR:
-
International League of Associations for Rheumatology
- RF:
-
Rheumatoid factor
- sJIA:
-
Systemic juvenile idiopathic arthritis
- ERA:
-
Enthesitis-related arthritis
- pJIA:
-
Polyarticular JIA
- PRINTO:
-
Pediatric Rheumatology International Trials Organization
- Early-onset ANA+ JIA:
-
Early-onset antinuclear antibody–positive juvenile idiopathic arthritis
- AOSD:
-
Adult-onset Still’s disease
- MAS:
-
Macrophage activation syndrome
- ANA:
-
Antinuclear antibodies
- RF+ JIA:
-
Rheumatoid factor–positive juvenile idiopathic arthritis
- Anti-CCP:
-
Anti-cyclic citrullinated peptide
- CRP:
-
C-reactive protein
- HLA:
-
Human leukocyte antigen system
- VEGF:
-
Vascular endothelial growth factor
- EC:
-
Endothelial cell
- sVEGF-R:
-
Soluble vascular endothelial growth factor receptor
- OPN:
-
Osteopontin
- Ang-1:
-
Angiopoietin-1
- TNF:
-
Tumour necrosis factor
- IL:
-
Interleukin
- MMPs:
-
Matrix metalloproteinases
- RANK:
-
Receptor activator of nuclear factor-kappaB
- MIF:
-
Macrophage migration inhibitory factor
- PTPN22:
-
Protein tyrosine phosphatase non-receptor type-22
- STAT4:
-
Signal transducer and activator of transcription-4
- SLC11A1:
-
Solute carrier family-11 member-1
- NRAMP1:
-
Natural resistance-associated macrophage protein-1
- WISP3:
-
WNT1-inducible signalling pathway protein-3
- ERAP:
-
endoplasmic reticulum resident aminopeptidases
- NK cells:
-
Natural killer cells
- DC:
-
Dendritic cells
- Anti-MCV:
-
Antibodies against mutated citrullinated vimentin
- Anti-CCP:
-
Anti-cyclic citrullinated peptide
- Th:
-
T helpers
- Treg:
-
Regulatory T-cells
- CCL2:
-
C-C motif chemokine ligand-2; Monocyte chemoattractant protein-1
- CCL3:
-
C-C motif chemokine ligand 3; Macrophage inflammatory protein-1α
- CCL22:
-
C-C motif chemokine ligand-22; Macrophage-derived chemokine
- CXCL9:
-
C-X-C motif chemokine ligand-9; Monokine induced by IFNγ
- CXCL10:
-
C-X-C motif chemokine ligand-10; IFNγ-induced protein 10
- LRG:
-
leucine-rich α2-glycoprotein
- ADA2:
-
adenosine deaminase-2
- TGF:
-
Transforming growth factor
- mTOR:
-
Mammalian target of rapamycin
- RA:
-
Rheumatoid arthritis
- MRP:
-
Myeloid-related protein
- ESR:
-
Erythrocyte sedimentation rate
- CRP:
-
C-reactive protein
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- DMARDs:
-
Disease modifying anti-rheumatic drugs
- MTX:
-
Methotrexate
- ACR:
-
American College of Rheumatology
- GC:
-
Glucocorticoids
References
Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369(9563):767–78. https://doi.org/10.1016/S0140-6736(07)60363-8.
Macaubas C, Nguyen K, Milojevic D, Park JL, Mellins ED. Oligoarticular and polyarticular JIA: epidemiology and pathogenesis. Nat Rev Rheumatol. 2009;5(11):616–26. https://doi.org/10.1038/nrrheum.2009.209.
Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International league of associations for rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31(2):390–2.
Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet. 2011;377(9783):2138–49. https://doi.org/10.1016/S0140-6736(11)60244-4.
Stoll ML, Punaro M. Psoriatic juvenile idiopathic arthritis: a tale of two subgroups. Curr Opin Rheumatol. 2011;23(5):437–43. https://doi.org/10.1097/BOR.0b013e328348b278.
Martini A. Systemic juvenile idiopathic arthritis. Autoimmun Rev. 2012;12(1):56–9. https://doi.org/10.1016/j.autrev.2012.07.022.
Ravelli A, Minoia F, Davi S, Horne A, Bovis F, Pistorio A, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European league against rheumatism/American College of Rheumatology/Paediatric rheumatology international trials organisation collaborative initiative. Arthritis & rheumatology (Hoboken, NJ). 2016;68(3):566-76.
Minoia F, Davi S, Horne A, Demirkaya E, Bovis F, Li C, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis & rheumatology (Hoboken, NJ). 2014;66(11):3160–9.
Eng SW, Duong TT, Rosenberg AM, Morris Q, Yeung RS. The biologic basis of clinical heterogeneity in juvenile idiopathic arthritis. Arthritis & rheumatology (Hoboken, NJ). 2014;66(12):3463–75.
Martini A, Ravelli A, Avcin T, Beresford MW, Burgos-Vargas R, Cuttica R, et al. Toward new classification criteria for juvenile idiopathic arthritis: first steps, pediatric rheumatology international trials organization international consensus. J Rheumatol. 2019;46(2):190–7. https://doi.org/10.3899/jrheum.180168.
Twilt M, Pradsgaard D, Spannow AH, Horlyck A, Heuck C, Herlin T. Joint cartilage thickness and automated determination of bone age and bone health in juvenile idiopathic arthritis. Pediatric rheumatology online journal. 2017;15(1):63. https://doi.org/10.1186/s12969-017-0194-9.
Swidrowska-Jaros J, Smolewska E. A fresh look at angiogenesis in juvenile idiopathic arthritis. Central-European journal of immunology. 2018;43(3):325–30. https://doi.org/10.5114/ceji.2018.80052.
Wojdas M, Dąbkowska K, Winsz-Szczotka K. Alterations of Extracellular Matrix Components in the Course of Juvenile Idiopathic Arthritis. Metabolites. 2021;11(3).
Spelling P, Bonfá E, Caparbo VF, Pereira RM. Osteoprotegerin/RANKL system imbalance in active polyarticular-onset juvenile idiopathic arthritis: a bone damage biomarker? Scand J Rheumatol. 2008;37(6):439–44. https://doi.org/10.1080/03009740802116224.
Stagi S, Cavalli L, Signorini C, Bertini F, Cerinic MM, Brandi ML, et al. Bone mass and quality in patients with juvenile idiopathic arthritis: longitudinal evaluation of bone-mass determinants by using dual-energy x-ray absorptiometry, peripheral quantitative computed tomography, and quantitative ultrasonography. Arthritis Res Ther. 2014;16(2):R83. https://doi.org/10.1186/ar4525.
Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nat Rev Rheumatol. 2011;7(7):416–26. https://doi.org/10.1038/nrrheum.2011.68.
Rigante D, Bosco A, Esposito S. The Etiology of Juvenile Idiopathic Arthritis2014.
Mistry RR, Patro P, Agarwal V, Misra DP. Enthesitis-related arthritis: current perspectives. Open access rheumatology : research and reviews. 2019;11:19–31. https://doi.org/10.2147/OARRR.S163677.
Carlens C, Jacobsson L, Brandt L, Cnattingius S, Stephansson O, Askling J. Perinatal characteristics, early life infections and later risk of rheumatoid arthritis and juvenile idiopathic arthritis. Ann Rheum Dis. 2009;68(7):1159–64. https://doi.org/10.1136/ard.2008.089342.
Horton DB, Shenoi S. Review of environmental factors and juvenile idiopathic arthritis. Open Access Rheumatol. 2019;11:253–67. https://doi.org/10.2147/OARRR.S165916.
De Silvestri A, Capittini C, Poddighe D, Marseglia GL, Mascaretti L, Bevilacqua E, et al. HLA-DRB1 alleles and juvenile idiopathic arthritis: diagnostic clues emerging from a meta-analysis. Autoimmun Rev. 2017;16(12):1230–6. https://doi.org/10.1016/j.autrev.2017.10.007.
Hollenbach JA, Thompson SD, Bugawan TL, Ryan M, Sudman M, Marion M, et al. Juvenile idiopathic arthritis and HLA class I and class II interactions and age-at-onset effects. Arthritis Rheum. 2010;62(6):1781–91. https://doi.org/10.1002/art.27424.
Hersh AO, Prahalad S. Immunogenetics of juvenile idiopathic arthritis: a comprehensive review. J Autoimmun. 2015;64:113–24. https://doi.org/10.1016/j.jaut.2015.08.002.
Cobb JE, Hinks A, Thomson W. The genetics of juvenile idiopathic arthritis: current understanding and future prospects. Rheumatology (Oxford, England). 2014;53(4):592–9.
Prahalad S, Glass DN. A comprehensive review of the genetics of juvenile idiopathic arthritis. Pediatric rheumatology online journal. 2008;6(1):11. https://doi.org/10.1186/1546-0096-6-11.
Nistala K, Moncrieffe H, Newton KR, Varsani H, Hunter P, Wedderburn LR. Interleukin-17-producing T cells are enriched in the joints of children with arthritis, but have a reciprocal relationship to regulatory T cell numbers. Arthritis Rheum. 2008;58(3):875–87. https://doi.org/10.1002/art.23291.
Prelog M, Schwarzenbrunner N, Sailer-Höck M, Kern H, Klein-Franke A, Ausserlechner MJ, et al. Premature aging of the immune system in children with juvenile idiopathic arthritis. Arthritis Rheum. 2008;58(7):2153–62. https://doi.org/10.1002/art.23599.
Arve-Butler S, Schmidt T, Mossberg A, Berthold E, Gullstrand B, Bengtsson AA, et al. Synovial fluid neutrophils in oligoarticular juvenile idiopathic arthritis have an altered phenotype and impaired effector functions. Arthritis Res Ther. 2021;23(1):109. https://doi.org/10.1186/s13075-021-02483-1.
Schmidt T, Berthold E, Arve-Butler S, Gullstrand B, Mossberg A, Kahn F, et al. Children with oligoarticular juvenile idiopathic arthritis have skewed synovial monocyte polarization pattern with functional impairment-a distinct inflammatory pattern for oligoarticular juvenile arthritis. Arthritis Res Ther. 2020;22(1):186. https://doi.org/10.1186/s13075-020-02279-9.
Correll CK, Binstadt BA. Advances in the pathogenesis and treatment of systemic juvenile idiopathic arthritis. Pediatr Res. 2014;75(1–2):176–83. https://doi.org/10.1038/pr.2013.187.
Machado SH, Xavier RM. Safety of tocilizumab in the treatment of juvenile idiopathic arthritis. Expert Opin Drug Saf. 2017;16(4):493–500. https://doi.org/10.1080/14740338.2017.1303479.
de Jager W, Hoppenreijs EP, Wulffraat NM, Wedderburn LR, Kuis W, Prakken BJ. Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: a cross-sectional study. Ann Rheum Dis. 2007;66(5):589–98. https://doi.org/10.1136/ard.2006.061853.
Vastert SJ, Kuis W, Grom AA. Systemic JIA: new developments in the understanding of the pathophysiology and therapy. Best Pract Res Clin Rheumatol. 2009;23(5):655–64. https://doi.org/10.1016/j.berh.2009.08.003.
Ishikawa S, Shimizu M, Inoue N, Mizuta M, Nakagishi Y, Yachie A. Interleukin-33 as a marker of disease activity in rheumatoid factor positive polyarticular juvenile idiopathic arthritis. Mod Rheumatol. 2017;27(4):609–13. https://doi.org/10.1080/14397595.2016.1246118.
Yasin S, Fall N, Brown RA, Henderlight M, Canna SW, Girard-Guyonvarc’h C, et al. IL-18 as a biomarker linking systemic juvenile idiopathic arthritis and macrophage activation syndrome. Rheumatology. 2019.
Shimizu M, Inoue N, Mizuta M, Nakagishi Y, Yachie A. Serum leucine-rich α2-glycoprotein as a biomarker for monitoring disease activity in patients with systemic juvenile idiopathic arthritis. J Immunol Res. 2019;2019:3140204.
Lee PY, Schulert GS, Canna SW, Huang Y, Sundel J, Li Y, et al. Adenosine deaminase 2 as a biomarker of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Ann Rheum Dis. 2020;79(2):225–31. https://doi.org/10.1136/annrheumdis-2019-216030.
Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356(6337):513–9. https://doi.org/10.1126/science.aal3535.
Feng M, Kang M, He F, Xiao Z, Liu Z, Yao H, et al. Plasma interleukin-37 is increased and inhibits the production of inflammatory cytokines in peripheral blood mononuclear cells in systemic juvenile idiopathic arthritis patients. J Transl Med. 2018;16(1):277. https://doi.org/10.1186/s12967-018-1655-8.
Berntson L, Nordal E, Fasth A, Aalto K, Herlin T, Nielsen S, et al. Anti-type II collagen antibodies, anti-CCP, IgA RF and IgM RF are associated with joint damage, assessed eight years after onset of juvenile idiopathic arthritis (JIA). Pediatric rheumatology online journal. 2014;12(1):22. https://doi.org/10.1186/1546-0096-12-22.
Mahmud SA, Binstadt BA. Autoantibodies in the Pathogenesis, Diagnosis, and Prognosis of Juvenile Idiopathic Arthritis. Frontiers in immunology. 2019;9:3168-.
Saurenmann RK, Levin AV, Feldman BM, Laxer RM, Schneider R, Silverman ED. Risk factors for development of uveitis differ between girls and boys with juvenile idiopathic arthritis. Arthritis Rheum. 2010;62(6):1824–8. https://doi.org/10.1002/art.27416.
Brunner J, Sitzmann FC. The diagnostic value of anti-cyclic citrullinated peptide (CCP) antibodies in children with juvenile idiopathic arthritis. Clin Exp Rheumatol. 2006;24(4):449–51.
Gohar F, Anink J, Moncrieffe H, Van Suijlekom-Smit LWA, Prince FHM, van Rossum MAJ, et al. S100A12 is associated with response to therapy in juvenile idiopathic arthritis. J Rheumatol. 2018;45(4):547–54. https://doi.org/10.3899/jrheum.170438.
Aljaberi N, Tronconi E, Schulert G, Grom AA, Lovell DJ, Huggins JL, et al. The use of S100 proteins testing in juvenile idiopathic arthritis and autoinflammatory diseases in a pediatric clinical setting: a retrospective analysis. Pediatric rheumatology online journal. 2020;18(1):7. https://doi.org/10.1186/s12969-020-0398-2.
Cimaz R. Systemic-onset juvenile idiopathic arthritis. Autoimmun Rev. 2016;15(9):931–4. https://doi.org/10.1016/j.autrev.2016.07.004.
Lee JJY, Schneider R. Systemic juvenile idiopathic arthritis. Pediatr Clin N Am. 2018;65(4):691–709. https://doi.org/10.1016/j.pcl.2018.04.005.
Ringold S, Weiss PF, Beukelman T, Dewitt EM, Ilowite NT, Kimura Y, et al. 2013 update of the 2011 American College of Rheumatology recommendations for the treatment of juvenile idiopathic arthritis: recommendations for the medical therapy of children with systemic juvenile idiopathic arthritis and tuberculosis screening among children receiving biologic medications. Arthritis care & research. 2013;65(10):1551–63.
Klein A, Kaul I, Foeldvari I, Ganser G, Urban A, Horneff G. Efficacy and safety of oral and parenteral methotrexate therapy in children with juvenile idiopathic arthritis: an observational study with patients from the German methotrexate registry. Arthritis care & research. 2012;64(9):1349–56. https://doi.org/10.1002/acr.21697.
Ferrara G, Mastrangelo G, Barone P, La Torre F, Martino S, Pappagallo G, et al. Methotrexate in juvenile idiopathic arthritis: advice and recommendations from the MARAJIA expert consensus meeting. Pediatr Rheumatol. 2018;16(1):46. https://doi.org/10.1186/s12969-018-0255-8.
Papadopoulou C, Kostik M, Böhm M, Nieto-Gonzalez JC, Gonzalez-Fernandez MI, Pistorio A, et al. Methotrexate therapy may prevent the onset of uveitis in juvenile idiopathic arthritis. J Pediatr. 2013;163(3):879–84. https://doi.org/10.1016/j.jpeds.2013.03.047.
Ayaz NA, Karadag SG, Cakmak F, Cakan M, Tanatar A, Sonmez HE. Leflunomide treatment in juvenile idiopathic arthritis. Rheumatol Int. 2019;39(9):1615–9. https://doi.org/10.1007/s00296-019-04385-7.
Blazina Š, Markelj G, Avramovič MZ, Toplak N, Avčin T. Management of Juvenile Idiopathic Arthritis: a clinical guide. Paediatric drugs. 2016;18(6):397–412. https://doi.org/10.1007/s40272-016-0186-0.
Stoll ML, Cron RQ. Treatment of juvenile idiopathic arthritis: a revolution in care. Pediatric rheumatology online journal. 2014;12(1):13. https://doi.org/10.1186/1546-0096-12-13.
Lovell DJ, Giannini EH, Reiff A, Cawkwell GD, Silverman ED, Nocton JJ, et al. Etanercept in children with polyarticular juvenile rheumatoid arthritis. Pediatric rheumatology collaborative study group. N Engl J Med. 2000;342(11):763–9. https://doi.org/10.1056/NEJM200003163421103.
Vanoni F, Minoia F, Malattia C. Biologics in juvenile idiopathic arthritis: a narrative review. Eur J Pediatr. 2017;176(9):1147–53. https://doi.org/10.1007/s00431-017-2960-6.
Ruperto N, Lovell DJ, Cuttica R, Woo P, Meiorin S, Wouters C, et al. Long-term efficacy and safety of infliximab plus methotrexate for the treatment of polyarticular-course juvenile rheumatoid arthritis: findings from an open-label treatment extension. Ann Rheum Dis. 2010;69(4):718–22. https://doi.org/10.1136/ard.2009.100354.
Balevic SJ, Rabinovich CE. Profile of adalimumab and its potential in the treatment of uveitis. Drug Des Devel Ther. 2016;10:2997–3003.
Lovell DJ, Ruperto N, Mouy R, Paz E, Rubio-Perez N, Silva CA, et al. Long-term safety, efficacy, and quality of life in patients with juvenile idiopathic arthritis treated with intravenous abatacept for up to seven years. Arthritis & rheumatology (Hoboken, NJ). 2015;67(10):2759–70.
Ramanan AV, Dick AD, Guly C, McKay A, Jones AP, Hardwick B, et al. Tocilizumab in patients with anti-TNF refractory juvenile idiopathic arthritis-associated uveitis (APTITUDE): a multicentre, single-arm, phase 2 trial. The Lancet Rheumatology. 2020;2(3):e135–e41. https://doi.org/10.1016/S2665-9913(20)30008-4.
Nigrovic PA, Mannion M, Prince FH, Zeft A, Rabinovich CE, van Rossum MA, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011;63(2):545–55. https://doi.org/10.1002/art.30128.
Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201(9):1479–86. https://doi.org/10.1084/jem.20050473.
Quartier P, Allantaz F, Cimaz R, Pillet P, Messiaen C, Bardin C, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis. 2011;70(5):747–54. https://doi.org/10.1136/ard.2010.134254.
Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat NM, Horneff G, et al. Canakinumab in patients with systemic juvenile idiopathic arthritis and active systemic features: results from the 5-year long-term extension of the phase III pivotal trials. Ann Rheum Dis. 2018;77(12):1710–9. https://doi.org/10.1136/annrheumdis-2018-213150.
Ruperto N, Martini A. Current and future perspectives in the management of juvenile idiopathic arthritis. The Lancet Child & adolescent health. 2018;2(5):360–70. https://doi.org/10.1016/S2352-4642(18)30034-8.
Giancane G, Minoia F, Davì S, Bracciolini G, Consolaro A, Ravelli A. IL-1 inhibition in systemic juvenile idiopathic arthritis. Front Pharmacol. 2016;7:467.
Ilowite NT, Prather K, Lokhnygina Y, Schanberg LE, Elder M, Milojevic D, et al. Randomized, double-blind, placebo-controlled trial of the efficacy and safety of rilonacept in the treatment of systemic juvenile idiopathic arthritis. Arthritis & rheumatology (Hoboken, NJ). 2014;66(9):2570–9.
Martini A. Are there new targets for juvenile idiopathic arthritis? Semin Arthritis Rheum. 2019;49(3s):S11–s3. https://doi.org/10.1016/j.semarthrit.2019.09.017.
Krei JM, Møller HJ, Larsen JB. The role of interleukin-18 in the diagnosis and monitoring of hemophagocytic lymphohistiocytosis/macrophage activation syndrome - a systematic review. Clin Exp Immunol. 2021;203(2):174–82. https://doi.org/10.1111/cei.13543.
Sakamoto AP, Pinheiro MM, Barbosa CM, Fraga MM, Len CA, Terreri MT. Rituximab use in young adults diagnosed with juvenile idiopathic arthritis unresponsive to conventional treatment: report of 6 cases. Rev Bras Reumatol. 2015;55(6):536–41. https://doi.org/10.1016/j.rbr.2014.12.015.
Benedetti FD, Brogan P, Grom A, Quartier P, Schneider R, Graaf KD, et al. OP0204 EMAPALUMAB, AN INTERFERON GAMMA (IFN-Y)-BLOCKING MONOCLONAL ANTIBODY, IN PATIENTS WITH MACROPHAGE ACTIVATION SYNDROME (MAS) COMPLICATING SYSTEMIC JUVENILE IDIOPATHIC ARTHRITIS (SJIA). Annals of the Rheumatic Diseases. 2019;78(Suppl 2):178-.
Gabay C, Fautrel B, Rech J, Spertini F, Feist E, Kötter I, et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still's disease. Ann Rheum Dis. 2018;77(6):840–7. https://doi.org/10.1136/annrheumdis-2017-212608.
Nash P, Kerschbaumer A, Dörner T, Dougados M, Fleischmann RM, Geissler K, et al. Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors: a consensus statement. Ann Rheum Dis. 2021;80(1):71–87. https://doi.org/10.1136/annrheumdis-2020-218398.
Huang Z, Lee PY, Yao X, Zheng S, Li T. Tofacitinib Treatment of Refractory Systemic Juvenile Idiopathic Arthritis. Pediatrics. 2019;143(5).
Miserocchi E, Giuffrè C, Cornalba M, Pontikaki I, Cimaz R. JAK inhibitors in refractory juvenile idiopathic arthritis-associated uveitis. Clin Rheumatol. 2020;39(3):847–51. https://doi.org/10.1007/s10067-019-04875-w.
Ruperto N, Synoverska O, Ting T, Abud-Mendoza C, Spindler A, Vyzhga Y, et al. OP0291 TOFACITINIB for the treatment of POLYARTICULAR course juvenile idiopathic arthritis: results of a phase 3, randomised, double-blind, placebo-controlled withdrawal study. Ann Rheum Dis. 2020;79(Suppl 1):180–1.
Welzel T, Winskill C, Zhang N, Woerner A, Pfister M. Biologic disease modifying antirheumatic drugs and Janus kinase inhibitors in paediatric rheumatology - what we know and what we do not know from randomized controlled trials. Pediatric rheumatology online journal. 2021;19(1):46. https://doi.org/10.1186/s12969-021-00514-4.
Wallace CA, Giannini EH, Spalding SJ, Hashkes PJ, O'Neil KM, Zeft AS, et al. Trial of early aggressive therapy in polyarticular juvenile idiopathic arthritis. Arthritis Rheum. 2012;64(6):2012–21. https://doi.org/10.1002/art.34343.
Blazina Š, Markelj G, Avramovič MZ, Toplak N, Avčin T. Management of Juvenile Idiopathic Arthritis: a clinical guide. Pediatr Drugs. 2016;18(6):397–412. https://doi.org/10.1007/s40272-016-0186-0.
Minden K. Adult outcomes of patients with juvenile idiopathic arthritis. Horm Res. 2009;72(Suppl 1):20–5. https://doi.org/10.1159/000229759.
Selvaag AM, Aulie HA, Lilleby V, Flato B. Disease progression into adulthood and predictors of long-term active disease in juvenile idiopathic arthritis. Ann Rheum Dis. 2016;75(1):190–5. https://doi.org/10.1136/annrheumdis-2014-206034.
Momah T, Ray L. Juvenile idiopathic arthritis: old disease, new tactics. J Fam Pract. 2019;68(2):E8–e13.
Oen K, Malleson PN, Cabral DA, Rosenberg AM, Petty RE, Cheang M. Disease course and outcome of juvenile rheumatoid arthritis in a multicenter cohort. J Rheumatol. 2002;29(9):1989–99.
Anink J, Prince FH, Dijkstra M, Otten MH, Twilt M, ten Cate R, et al. Long-term quality of life and functional outcome of patients with juvenile idiopathic arthritis in the biologic era: a longitudinal follow-up study in the Dutch Arthritis and Biologicals in Children Register. Rheumatology (Oxford, England). 2015;54(11):1964–9.
Barisic Kutija M, Peric S, Knezevic J, Juratovac Z, Vukojevic N. Complication and prognosis of juvenile idiopathic arthritis associated uveitis in the era of modern immunomodulatory treatment. Psychiatr Danub. 2019;31(Suppl 1):44–9.
Acknowledgements
This review article was undertaken as part of research funded by a Bolashak Scholarship Award, Kazakhstan Government, to Dr Lina Zaripova to undertake a PhD at the University of Liverpool, Liverpool, UK, and also by the Wellcome Trust Institutional Strategic Support Fund awarded to the University of Liverpool (grant number 097826/z/11/z). Work was supported by the NIHR Alder Hey Clinical Research Facility. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Funding
This review article was undertaken as part of research funded by a Bolashak Scholarship Award, Kazakhstan Government, to Dr Lina Zaripova to undertake a PhD at the University of Liverpool, Liverpool, UK, and also by the Wellcome Trust Institutional Strategic Support Fund awarded to the University of Liverpool (grant number 097826/z/11/z).
Author information
Authors and Affiliations
Contributions
LNZ, AM, SEC, MWB, EMB, RAO were involved in the design of the scope of the review. LNZ performed literature searches and analysis of content for review. LNZ, AM, SEC, MWB, EMB, RAO were involved in the creation and revision of manuscript drafts. LNZ, AM, SEC, MWB, EMB, RAO authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zaripova, L.N., Midgley, A., Christmas, S.E. et al. Juvenile idiopathic arthritis: from aetiopathogenesis to therapeutic approaches. Pediatr Rheumatol 19, 135 (2021). https://doi.org/10.1186/s12969-021-00629-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12969-021-00629-8