
Abstract
Background
Early-onset sarcoidosis (EOS) and Blau syndrome (BS) are systemic inflammatory granulomatous diseases without visible pulmonary involvement, and are distinguishable from their sporadic and familial forms. The diseases are characterized by a triad of skin rashes, symmetrical polyarthritis, and recurrent uveitis. The most common morbidity is ocular involvement, which is usually refractory to conventional treatment. A gain-of-function mutation in the nucleotide-binding oligomerization domain-containing protein 2 (NOD2) gene has been demonstrated in this disease; however, little is known about the relationship between the activation of NOD2 and the pathophysiology of EOS/BS. Here we describe EOS/BS with a novel mutation in the NOD2 gene, as well as detection of Propionibacterium acnes (P. acnes) in the granulomatous inflammation.
Case presentation
An 8-year-old Japanese girl presented with refractory bilateral granulomatous panuveitis. Although no joint involvement was evident, she exhibited skin lesions on her legs; a skin biopsy revealed granulomatous dermatitis, and P. acnes was detected within the sarcoid granulomas by immunohistochemistry with P. acnes-specific monoclonal (PAB) antibody. Genetic analyses revealed that the patient had a NOD2 heterozygous D512V mutation that was novel and not present in either of her parents. The mutant NOD2 showed a similar activation pattern to EOS/BS, thus confirming her diagnosis. After starting oral prednisolone treatment, she experienced an anterior vitreous opacity relapse despite gradual prednisolone tapering; oral methotrexate was subsequently administered, and the patient responded positively.
Conclusions
We presented a case of EOS/BS with a novel D512V mutation in the NOD2 gene. In refractory granulomatous panuveitis cases without any joint involvement, EOS/BS should be considered as a differential diagnosis; genetic analyses would lead to a definite diagnosis. Moreover, this is the first report of P. acnes demonstrated in granulomas of EOS/BS. Since intracellular P. acnes activates nuclear factor-kappa B in a NOD2-dependent manner, we hypothesized that the mechanism of granuloma formation in EOS/BS may be the result of NOD2 activity in the presence of the ligand muramyl dipeptide, which is a component of P. acnes. These results indicate that recognition of P. acnes through mutant NOD2 is the etiology in this patient with EOS/BS.
Similar content being viewed by others


Background
Sarcoidosis is a multiorgan inflammatory disease with unknown etiology, characterized by the histological features of noncaseating epithelioid cell granulomas [1]. Two distinct types of sarcoidosis have been reported in children: classic sarcoidosis (CS) and early-onset sarcoidosis (EOS) [1, 2]. The former, clinically characterized by a triad of lung, lymph node, and eye involvement, similar to its manifestation in adults, presents in older children and is detectable via chest radiography. The latter is quite rare and found in younger children, presenting with a triad of skin, joint, and eye disorders with no apparent pulmonary involvement.
EOS and Blau syndrome (BS) are systemic inflammatory granulomatous diseases [1, 3]. The pathophysiology is the same for both diseases; however, differentiation between the sporadic and familial (autosomal dominant inheritance) forms is possible [3]. Clinically, in both diseases, the cutaneous and articular symptoms primarily appear in children aged < 4 years [4,5,6], while ocular manifestations typically appear between the ages of 7 and 12 years [7]. On the other hand, the most common morbidity associated with EOS/BS is ocular involvement that is usually refractory to conventional treatment, including continuous local and systemic glucocorticoids [8].
In 2001, NOD2 was identified on chromosome 16q12 as the gene responsible for EOS/BS [1, 3, 7, 9]. The NOD2 gene is intracellularly expressed in phagocytic cells and recognizes muramyl dipeptide (MDP), a component of the bacterial peptidoglycan, inducing an immune response through nuclear factor-kappa B (NF-κB) activation. NOD2 mutations in patients with EOS/BS enhance the self-oligomerization of NOD2, leading to augmented NOD2 activity even in the absence of the ligand MDP; further increases in activity by the addition of ligands represent gain-of-function mutations, consistent with the dominant mode of inheritance of the granulomatous disease [10, 11]. However, genetic screening of the NOD2 gene revealed no common mutations in Japanese and Caucasian CS patients [12, 13].
Despite a number of relevant studies, little is known about the relationship between the activation of NOD2 and the pathophysiology of EOS/BS [14]. The etiologic aspect of CS as an allergic endogenous infection has recently been elucidated; CS is most likely the result of a complex interaction between infection, immunity, and allergic reaction. Sarcoid granulomas form in patients with a hypersensitive immune response to Propionibacterium acnes (P. acnes). They are formed primarily as a host defense mechanism against intracellular P. acnes activated at the sites of latent infection to prevent the spread of the infectious agent [15].
Conversely, no reports have described the association between EOS/BS and P. acnes. Contrary to the asymptomatic and sometimes naturally resolving form of the disease found in CS, EOS/BS is progressive and often leads to poor prognoses, such as blindness or joint destruction [16]. Clarifying the pathogenesis of EOS/BS is essential for preventing a poor prognosis. Considering all these factors, we describe here a case of EOS/BS with a novel mutation in the NOD2 gene as well as the presence of P. acnes in the granulomatous inflammation of the biopsied skin tissue.
Case presentation
During early infancy, a Japanese girl developed papules on her legs and the dorsum of her hands. While the condition remained undiagnosed, the papules on her hand disappeared spontaneously at the age of 4 years. At age 5, she developed both conjunctival hyperemia and photophobia in her left eye; she was diagnosed with anterior uveitis, which was effectively treated using localized therapy. At age 7.5, she developed central scotoma and sudden visual loss (left > right) that was diagnosed as granulomatous panuveitis, which was effectively treated using local glucocorticoid therapy.
By age 8, the granulomatous panuveitis had flared up again; anterior chamber inflammation was seen in both eyes, and mydriasis had developed in the right eye due to posterior synechiae (Fig. 1a). The fundus was characterized by bilateral multifocal choroiditis presenting as creamy yellow lesions, and a scar was seen in the macular region of her left eye (Fig. 2). Systemic glucocorticoid therapy was therefore needed to preserve sight in the left eye. Laboratory tests showed hemoglobin level (11.9 g/dL), white blood cell count (6.22 × 109 /L), platelet count (292 × 109 /L), erythrocyte sedimentation rate (5 mm/h; reference range [RR], 5–15 mm/h), C-reactive protein level (0.01 mg/dL; RR, < 0.27 mg/dL), and matrix metalloproteinase-3 (16.4 ng/mL; RR, < 20.0 ng/mL); the tests for antinuclear antibodies and rheumatoid factor were negative. Her immunological tests were normal, and there were no episodes of recurrent or severe bacterial or fungal infection. Therefore, no additional tests, such as a flow cytometric dihydrorhodamine neutrophil respiratory burst assay, were performed to rule out chronic granulomatous disease. Magnetic resonance imaging of the extremities was carried out to evaluate the presence of arthritis; no abnormalities were detected.

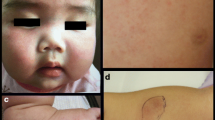
Clinical findings. a Anterior chamber inflammation in both eyes and mydriasis due to posterior synechiae in the right eye. b and c Asymptomatic diffuse papules on the extensor surfaces of the legs (arrows)

Fundus photograph. Right (a) and left (b) eyes showing bilateral multifocal choroiditis (arrowheads) presenting as creamy yellow lesions and a scar (arrow) in the macular region of the left eye
A skin biopsy from papules on her leg indicated granulomatous dermatitis (Figs. 1b and c and 3). The biopsy samples were examined by immunohistochemistry (IHC) with a P. acnes-specific monoclonal antibody (PAB antibody, D371-3, MEDICAL & BIOLOGICAL LABORATORIES CO., LTD., Aichi, Japan) and a mycobacteria-specific monoclonal antibody (LAM antibody, D372-3, MEDICAL & BIOLOGICAL LABORATORIES CO., LTD.), according to the methods described in the original study [17]. The PAB antibody detected several signals of round bodies within the sarcoid granulomas and in lesions infiltrated by inflammatory cells, while no positive signal was observed using the LAM antibody as control (Fig. 4). The sarcoid granulomas were comprised of CD4+ cells and CD68+ cells; CD8+ cells were rarely observed in the dermis and subcutaneous tissue (Fig. 5).

Histopathologic findings. a Several noncaseating epithelioid cell granulomas scattered throughout the dermis and subcutaneous tissue (arrows). b The epithelioid cell granulomas poorly infiltrated by lymphocytes (arrows). Hematoxylin and eosin stain. Scale bar; 100 µm

P. acnes detected by PAB antibody within the sarcoid granulomas of biopsied skin lesion. Hematoxylin and eosin stain and IHC with PAB antibody and LAM antibody as control are shown pairwise. a Noncaseating epithelioid cell granulomas in the dermis. b Higher magnification of the granuloma indicated by an arrow in (a). c No positive signal detected by LAM antibody in the identical granuloma. d Many round bodies detected by PAB antibody in the granuloma (arrows). Scale bar; 50 µm.
IHC, immunohistochemistry; PAB, Propionibacterium acnes-specific monoclonal; LAM, mycobacteria-specific monoclonal

Immunohistochemical findings of CD4+, CD8+, and CD68+ cells. Large granulomas in the dermis and subcutaneous tissue were comprised of CD4+ cells (a and d), and CD68+ cells (b and e). CD8+ cells were rarely detected (c and f). Scale bar; 200 µm
Genetic analyses were performed of both the patient’s and her parents’ NOD2 genes. Genomic DNA was extracted from the peripheral blood of the patient and her parents, and all 12 exons of the NOD2 gene, including exon-intron boundaries, were amplified by polymerase chain reaction and sequenced by Sanger method. Results revealed that the patient had a heterozygous c.1535A > T mutation on exon 4 in the nucleotide-binding domain of NOD2, while her parents did not. Analysis of the protein variant revealed that the mutation was D512V (p.Asp512Val) (Fig. 6). An NF-κB luciferase assay was performed. HEK293T cells (1 × 105) were cotransfected with 30 ng of the expression construct of the NOD2 variant together with an NF-κB reporter plasmid. NF-κB activity was measured after 12 h of incubation either with or without 5 µg/mL MDP. A mock vector and the wild-type NOD2 were used as controls [1]; R334W and H496L were used as positive controls [1]. An in vitro assessment of the mutation indicated an increased basal NF-κB activity that increased further after MDP stimulation [18], demonstrating a strong association with EOS/BS.

NOD2 heterozygous mutation. The electropherogram shows the sequence of heterozygous c.1535A > T transition in exon 4 of the nucleotide-binding oligomerization domain in the patient (red arrow). No such mutation was found in her parents (black arrows).
NOD2, nucleotide-binding oligomerization domain-containing protein 2
Based on her clinical, histological, and genetic features, the patient was diagnosed with EOS/BS despite the absence of arthritis during the course of the disease. She was treated with oral prednisolone (PSL) (1.5 mg/kg/day). Six months after initiating treatment, she experienced a relapse of anterior vitreous opacity while on a 0.3 mg/kg/day dose of oral PSL, which was then gradually tapered. Oral methotrexate (10 mg/m2/week) was prescribed as an additional treatment, to which there was a positive response. She has remained in clinical remission for over three years on a 0.1 mg/kg/day dose of oral PSL with methotrexate.
Discussion
This report describes a case of EOS/BS with a novel D512V mutation in the NOD2 gene and a lack of arthritis. P. acnes was also detected in the granulomatous inflammation, which leads to the elucidation of the etiology of EOS/BS.
Our patient did not show any symptoms of arthritis. However, the most common manifestation of EOS/BS is symmetric and usually painless polyarthritis [19]. Only three mutations, E383K, R587C, and R334W, were identified previously in EOS/BS patients without arthritis [18, 20]. The R334W mutation in the NOD2 gene is one of the most frequently occurring mutations in patients with EOS/BS [18, 20]. The patient with R587C mutation was a familial case with an age of onset of 216 months, and demonstrated skin rash without arthritis and uveitis [20]. Her daughter, who had the same mutation, was the proband of the disease and had rash, uveitis, and arthritis with an age of 21 months at onset [20]. It may be difficult to identify phenotypic variations, such as the timing and site of onset and the clinical severity of EOS/BS, according to the NOD2 genotype. Nongenetic factors such as environmental conditions and/or infectious agents might be involved in phenotypic variation and clinical severity. Therefore, in individuals with NOD2 mutations, P. acnes might be the trigger for onset of the disease and may determine the onset site by causing intracellular proliferation from a latently infected state. In our patient, translocation of P. acnes after local proliferation at the early onset site of the skin may have caused latent infection only in the eye (but not in the joint), where later reactivation caused uveitis (but not arthritis).
Our patient had a novel D512V mutation of the NOD2 gene. Functional analysis showed elevated background activity and augmented activity after ligand stimulation, demonstrating a strong association with EOS/BS. A recent report elucidated the etiological aspect of CS as an allergic endogenous infection caused by P. acnes [15]. Histological localization of P. acnes within sarcoid granulomas of the lymph nodes has been demonstrated by in situ hybridization [21] and IHC with PAB antibodies [17], suggesting that P. acnes is related to the cause of granuloma formation in CS. P. acnes has also been found in the cutaneous sarcoid granulomas of CS patients by IHC with PAB antibodies [22,23,24,25,26]. PAB antibody is a P. acnes-specific monoclonal antibody that reacts with lipoteichoic acids that are membrane-anchored molecules in the cell envelopes of gram-positive bacteria. In our case, using the same PAB antibody used in the previous studies, IHC detected several round bodies of P. acnes both within and around the sarcoid granulomas. The granulomas were comprised of CD4+ and CD68+ cells, indicating that they formed by the accumulation of CD4+ T-lymphocytes and macrophages. Therefore, the granulomas may have developed as a Th1 immune response to P. acnes.
P. acnes has also been demonstrated in ocular CS lesions. Nagata et al. [27] reported that P. acnes was present within granulomas in 10 (83 %) of 12 retinal biopsy samples from 9 (82 %) of 11 patients with ocular CS, whereas it was not detected in any (0 %) of the control group; the bacteria were identified as round bodies that reacted with PAB antibody. The retina is an aseptic environment and the presence of P. acnes within the retina is extremely unusual; therefore, detection of P. acnes within the retina was a critical observation, confirming the role of P. acnes as an etiological agent for CS. P. acnes is commensal on the skin, including sebaceous follicles, but usually absent in the dermis; therefore, we considered that detection of P. acnes within the sarcoid granulomas in the dermis strongly suggested P. acnes as an etiological agent for EOS/BS.
Tanabe et al. [28] found that both NOD1 and NOD2 proteins recognized intracellular P. acnes. Furthermore, systematic searches for NOD1 gene polymorphisms in Japanese CS patients identified an increased frequency of G796A. Functional analysis revealed that NOD1 G796A was associated with a lower expression at the protein level, leading to reduced NF-κB activation in response to intracellular P. acnes. These results indicated that impaired recognition of intracellular P. acnes through NOD1 variants might cause susceptibility to CS within the Japanese population. We, therefore, hypothesized that the mechanism of granuloma formation in EOS/BS may be the result of NOD2 hyperactivity in the presence of the ligand MDP, a component of P. acnes.
We found that EOS/BS and CS share a common feature of P. acnes in granulomatous pathology; however, the reason for the differing clinical features (especially the different triad of symptoms) between EOS/BS and CS remains unknown. It may be related to the presence or absence of NOD2 mutations, or different sites of preceding latent infection of P. acnes in EOS/BS and CS. The lack of arthritis in our patient may indicate the lack of latent infection. Additionally, it remains unknown how the hyperactivity of NF-κB without MDP stimulation is involved in the pathophysiology of EOS/BS. The gain-of-function NOD2 mutations found in EOS/BS patients may cause an excessive immune response to P. acnes, with increased NOD2 activity in the presence of MDP; this excessive immune response to P. acnes would lead to poor prognosis.
Nagakura et al. [29] reported that tumor necrosis factor inhibitors provide long-term clinical benefits in EOS/BS. Thus, it would be one of the candidate treatments in the case of a later relapse. Furthermore, according to the identification of P. acnes in our patient, a combination of antibiotic and symptomatic therapy might be considered as a novel treatment for EOS patients. Antibiotics against P. acnes may be effective for patients with progressive CS by preventing relapses of inflammation caused by repeated reactivation of the latent bacteria [15]. The result of a nationwide questionnaire survey, performed by a Japanese research group in 2005, indicated that antibiotic therapy was effective in 43 % of patients with CS treated with several kinds of antibiotics, including minocycline, doxycycline, and clarithromycin [15]. This study therefore provides scope for future research.
Conclusions
In summary, we presented a case of EOS/BS with a novel D512V mutation in the NOD2 gene. In refractory granulomatous panuveitis cases without joint involvement, EOS/BS should be considered as a differential diagnosis; genetic analyses would lead to a definite diagnosis. Moreover, this is the first report of P. acnes demonstrated in granulomas of EOS/BS. Since intracellular P. acnes activates NF-κB in a NOD2-dependent manner, we hypothesized that the mechanism of granuloma formation in EOS/BS may be the result of NOD2 activity in the presence of the ligand MDP, which is a component of P. acnes. These results indicate that recognition of P. acnes through mutant NOD2 is the etiology in this patient with EOS/BS.
Additional information
This patient is the same as the D512V case reported in reference number 18. There was no polyarthritis or renal calcification in this patient. In fact, the statement that polyarthritis and renal calcification were present is an error made by the authors of reference number 18, and is currently being corrected by them for submission of an erratum.
Availability of data and material
Not applicable.
Abbreviations
- BS:
-
Blau syndrome
- CS:
-
Classic sarcoidosis
- EOS:
-
Early-onset sarcoidosis
- IHC:
-
Immunohistochemistry
- LAM:
-
Mycobacteria-specific monoclonal
- MDP:
-
Muramyl dipeptide
- NF-κB:
-
Nuclear factor-kappa B
- NOD2 :
-
Nucleotide-binding oligomerization domain-containing protein 2
- PAB:
-
Propionibacterium acnes-specific monoclonal
- P. acnes :
-
Propionibacterium acnes
- PSL:
-
Prednisolone
- RR:
-
Reference range
References
Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood. 2005;105:1195–7.
Shetty AK, Gedalia A. Sarcoidosis: a pediatric perspective. Clin Pediatr (Phila). 1998;37:707–17.
Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Häfner R, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29:19–20.
Becker ML, Rose CD. Blau syndrome and related genetic disorders causing childhood arthritis. Curr Rheumatol Rep. 2005;7:427–33.
Rose CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, et al. Pediatric granulomatous arthritis: an international registry. Arthritis Rheum. 2006;54:3337–44.
Aróstegui JI, Arnal C, Merino R, Modesto C, Carballo MA, Moreno P, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 2007;56:3805–13.
Sfriso P, Caso F, Tognon S, Galozzi P, Gava A, Punzi L. Blau syndrome, clinical and genetic aspects. Autoimmun Rev. 2012;12:44–51.
Sarens IL, Casteels I, Anton J, Bader-Meunier B, Brissaud P, Chédeville G, et al. Blau syndrome-associated uveitis: preliminary results from an international prospective interventional case series. Am J Ophthalmol. 2018;187:158–66.
Achille M, Ilaria P, Teresa G, Roberto C, Ilir A, Piergiorgio N, et al. Successful treatment with adalimumab for severe multifocal choroiditis and panuveitis in presumed (early-onset) ocular sarcoidosis. Int Ophthalmol. 2016;36:129–35.
Tanabe T, Chamaillard M, Ogura Y, Zhu L, Qiu S, Masumoto J, et al. Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J. 2004;23:1587–97.
Yamaguchi N, Suzuki Y, Mahbub MH, Takahashi H, Hase R, Ishimaru Y, et al. The different roles of innate immune receptors in inflammation and carcinogenesis between races. Environ Health Prev Med. 2017;22:70.
Akahoshi M, Ishihara M, Namba K, Kitaichi N, Ando Y, Takenaka S, et al. Mutation screening of the CARD15 gene in sarcoidosis. Tissue Antigens. 2008;71:564–7.
Schurmann M, Valentonyte R, Hampe J, Muller-Quernheim J, Schwinger E, Schreiber S. CARD15 gene mutations in sarcoidosis. Eur Respir J. 2003;22:748–54.
Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–72.
Eishi Y. Etiologic aspect of sarcoidosis as an allergic endogenous infection caused by Propionibacterium acnes. Biomed Res Int. 2013;2013:935289.
Fink CW, Cimaz R. Early onset sarcoidosis: not a benign disease. J Rheumatol. 1997;24:174–7.
Negi M, Takemura T, Guzman J, Uchida K, Furukawa A, Suzuki Y, et al. Localization of Propionibacterium acnes in granulomas supports a possible etiologic link between sarcoidosis and the bacterium. Mod Pathol. 2012;25:1284–97.
Matsuda T, Kambe N, Ueki Y, Kanazawa N, Izawa K, Honda Y, et al. Clinical characteristics and treatment of 50 cases of Blau syndrome in Japan confirmed by genetic analysis of the NOD2 mutation. Ann Rheum Dis. 2020. doi:https://doi.org/10.1136/annrheumdis-2020-217320.
Milman N, Ursin K, Rødevand E, Nielsen FC, Hansen TVO. A novel mutation in the NOD2 gene associated with Blau syndrome: a Norwegian family with four affected members. Scand J Rheumatol. 2009;38:190–7.
Rosé CD, Aróstegui JI, Martin TM, Espada G, Scalzi L, Yagüe J, Rosenbaum JT, et al. NOD2-associated pediatric granulomatous arthritis, an expanding phenotype: Study of an International Registry and a National Cohort in Spain. Arthritis Rheum. 2009;60:1797–803.
Yamada T, Eishi Y, Ikeda S, Ishige I, Suzuki T, Takemura T, et al. In situ localization of Propionibacterium acnes DNA in lymph nodes from sarcoidosis patients by signal amplification with catalysed reporter deposition. J Pathol. 2002;198:541–7.
Asahina A, Miura K, Saito I, Oshikata C, Ishii N, Eishi Y. Cutaneous sarcoidosis with livedoid lesions: evidence of the involvement of Propionibacterium acnes. J Dermatol. 2013;40:501–2.
Takama H, Yanagishista T, Muto J, Ohshima Y, Takahashi E, Tsuzuki T, et al. Granulomatous pigmented purpuric dermatosis containing Propionibacterium acnes. Eur J Dermatol. 2018;28:540–2.
Sasaki S, Kato M, Nakamura K, Namba Y, Nagashima O, Takahashi K. Management of skin sarcoidosis with minocycline monotherapy. Respirol Case Rep. 2019;7:e00413.
Noda S, Maeda A, Komiya Y, Soejima M. A Patient with necrotizing vasculitis related to sarcoidosis, which was diagnosed via immunohistochemical methods using P. acnes-specific monoclonal antibodies. Intern Med. 2020;10:4918–20.
Inoue Y, Teraki Y. Association of Propionibacterium acnes with the efficacy of minocycline therapy for cutaneous sarcoidosis. Int J Dermatol. 2020;59:704–8.
Nagata K, Eishi Y, Uchida K, Yoneda K, Hatanaka H, Yasuhara T, et al. Immunohistochemical detection of Propionibacterium acnes in the retinal granulomas in patients with ocular sarcoidosis. Sci Rep. 2017;7:15226.
Tanabe T, Ishige I, Suzuki Y, Aita Y, Furukawa A, Ishige Y, et al. Sarcoidosis and NOD1 variation with impaired recognition of intracellular Propionibacterium Acnes. Biochim Biophys Acta. 2006;1762:794–801.
Nagakura T, Wakiguchi H, Kubota T, Yamatou T, Yamasaki Y, Nonaka Y, et al. Tumor necrosis factor inhibitors provide longterm clinical benefits in pediatric and young adult patients with Blau syndrome. J Rheumatol. 2017;44:536–8.
Acknowledgements
We thank the patient, her family, and their friends for their cooperation.We would like to thank Editage (www.editage.com) for English language editing.
Funding
This work was supported in part by JSPS KAKENHI Grant Number JP19K17908.
Author information
Authors and Affiliations
Contributions
OF, WH, TT, EY, and HS wrote the paper; OF, WH, KY, NT, YH, US, and YR treated the patient; AN, HY, UK, and EY performed the pathological studies; TT, IK, HY, and NR performed the genetic study; WH, OS, and HS conducted and designed the research; all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The need for formal approval was waived.
Consent for publication
Informed consent was obtained from the patient’s guardian for publication of this case report.
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Okazaki, F., Wakiguchi, H., Korenaga, Y. et al. A novel mutation in early‐onset sarcoidosis/Blau syndrome: an association with Propionibacterium acnes. Pediatr Rheumatol 19, 18 (2021). https://doi.org/10.1186/s12969-021-00505-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12969-021-00505-5