
Abstract
Myeloid-derived suppressor cells (MDSCs), which are immature heterogeneous bone marrow cells, have been described as potent immune regulators in human and murine cancer models. The distribution of MDSCs varies across organs and is divided into three subpopulations: granulocytic MDSCs or polymorphonuclear MDSCs (G-MDSCs or PMN-MDSCs), monocytic MDSCs (M-MDSCs), as well as a recently identified early precursor MDSC (eMDSCs) in humans. Activated MDSCs induce the inactivation of NK cells, CD4+, and CD8+ T cells through a variety of mechanisms, thus promoting the formation of tumor immunosuppressive microenvironment. ER stress plays an important protecting role in the survival of MDSC, which aggravates the immunosuppression in tumors. In addition, ferroptosis can promote an anti-tumor immune response by reversing the immunosuppressive microenvironment. This review summarizes immune suppression by MDSCs with a focus on the role of endoplasmic reticulum stress-mediated immune suppression in cancer and infectious disease, in particular leprosy and tuberculosis.
Similar content being viewed by others

Introduction
Myeloid-derived suppressor cells (MDSCs) are immature myeloid suppressor cell populations that are derived from the bone marrow. MDSCs accumulate and exert immune suppressive effects during pathologic conditions such as cancer, inflammation, infection, autoimmune disease, and obesity [1]. The MDSCs suppress T cell activation by downregulation of L-selectin and sequestration of cysteine, which the T cells cannot synthesize spontaneously and that they require to become activated. The development, expansion, and activation of MDSCs were triggered by the tumor microenvironment, particularly the immune microenvironment, and regulated by differential intracellular signaling molecules [2]. The microenvironment during these pathologic conditions is characterized by a low pH, hypoxia, nutrient deprivation, and free radicals. This microenvironment disrupts protein folding, which triggers cellular “endoplasmic reticulum (ER) stress” [3]. ER stress impacts inflammatory and tumor microenvironment-induced immune suppression [4,5,6]. Furthermore, tumor cells can transmit ER stress to immune cells recruited to inflammatory tissues [7, 8]. Most noteworthy, there is compelling evidence that ER stress can transform immune cell populations into immunosuppressive phenotypes [6, 9], with MDSCs from cancer patients and tumor-bearing mice producing a robust ER stress response. Many factors can induce ER stress in MDSCs. Reactive oxygen species (ROS), one of the main inducers of the ER stress response, are a significant product of MDSCs [10]. Lipids can also induce ER stress [11], with lipid accumulation associated with MDSCs [12].
Currently, MDSCs are becoming the main immuno-therapeutic targets. How ER stress regulates the biological properties of MDSCs in the tumor microenvironment is critical for MDSCs-targeted immunotherapy. This review summarises the ER stress effect on the immunosuppressive function of MDSCs in different kinds of tumors and infectious diseases, focusing on Mycobacterium leprae and Mycobacterium tuberculosis infections. We also summarized investigated molecules as the immunotherapy targets aiming to provide a more comprehensive theoretical basis for targeted MDSCs immunotherapy in the clinic.
MDSCs, their expansion, roles, and mechanisms in immunosuppressive function in pathological conditions
The terminology of MDSCs was first defined in 2007 and referred to the origin and the suppressive function of these cells. On physiological conditions, hematopoietic stem cells (HSCs) first develop into common myeloid progenitors (CMPs) and then into immature myeloid cells (IMCs). IMCs further differentiate into mature functional granulocytes, macrophages, and dendritic cells (DCs). However, during pathological conditions, IMCs differentiate into MDSCs within the bone marrow and then migrate to peripheral tissues [13] .
MDSCs are a heterogeneous population composed of monocytes, polymorphonuclear leukocytes and immature myeloid cells. MDSCs are broadly divided into three subgroups: granulocytic MDSCs or polymorphonuclear MDSCs (G-MDSCs or PMN-MDSCs), monocytic MDSCs (M-MDSCs) [14], as well as a recently identified early precursor MDSC (eMDSCs) in humans [15]. In mice, Gr-1 and CD11b are used to identify MDSCs. Ly6G and Ly6C are used to distinguish M-MDSCs (CD11b+Ly6G−Ly6Chigh) from G-MDSCs (CD11b+Ly6G+Ly6Clow) [16]. In humans, the common MDSC phenotype is CD11b+HLA-DR−/low. CD33 is the common myeloid marker for humans, while CD14 and CD15 are used to distinguish M-MDSCs (CD11b+HLA-DR−/lowCD33+CD15−CD14+), G-MDSCs (CD11b+HLA-DR−/lowCD33+CD15+CD14−), and eMDSCs (CD11b+HLA-DR−/lowCD33+CD15−CD14−). It is difficult to identify G-MDSCs from neutrophils in mice or humans, as they have a similar phenotype. However, the two cell populations can be distinguished by density gradient centrifugation, which has limitations [15, 17]. Recently, it is found that Lectin-type oxidized LDL receptor-1 (LOX-1) is a unique surface marker of human G-MDSCs, which can be used as a distinguish marker of G-MDSCs. Meanwhile, S100A9 has been used to refine identification of M-MDSCs in human [18]. Work from several groups has demonstrated that the key immunosuppressive feature does not distinguish MDSCs from conventional myeloid cells during inflammation [19]. A combination of molecular markers is considered being the most accurate means by which to identify different subtypes of MDSCs, with the caveat that different methods for collection and analysis of MDSCs can influence outcomes.
MDSCs accumulate during pathological conditions such as infection, inflammation, traumatic stress, and cancer [20]. The expansion of MDSCs to the pathological sites is induced by factors primarily produced by tumors or bone marrow stromal cells. Macrophage CSF (M-CSF), granulocyte/macrophage colony-stimulating factor (GM-CSF), granulocyte CSF (G-CSF), IL-6, IL-1, β-fibroblast growth factor (β-FGF) and vascular endothelial growth factor (VEGF) affect the mobilization and the expansion of MDSCs [21,22,23]. The primary transcription factor that regulates expansion and activation of MDSCs is STAT3, which is a downstream target of phosphorylated Janus kinases (JAKs). Essential genes for MDSC survival and proliferation are Bcl-XL, MYC, survivin, and cyclin D1, which are upregulated by STAT3. S100A8/S100A9 expression is activated by STAT3, which then modulates interferon regulatory factor-8 (IRF-8), a negative regulator of MDSCs. Further, miR-155 and miR-21 induce MDSCs by reduction of the negative regulators (SHIP-1 and PTEN), increasing STAT3 activation [24,25,26].
When MDSCs expanded to the tumor or inflammatory sites, activation signals were launched and endowed MDSCs to carry out the inhibitory function. The NF-κB signaling pathway is essential to MDSC activation. IL-1β activates MDSC recruitment and promotes IL-6 and TNF-α production through the NF-κB pathway. M-MDSCs from cancer patients produced a high level of TGF-β secretion when treated with PGE2 activating p38 MAPK/ERK signaling [27, 28]. For G-MDSCs, eIF2 and eIF4 were related to ER stress, mTOR and MAPK pathway upregulation [29]. T cell immunosuppression is due to the depletion or sequestration of amino acids. The stress of low extracellular amino acid levels promotes the activation of metabolic sensor (GCN2 kinase, FATP2, and AMPK) and the accumulation of metabolic waste products within the tumor microenvironment (TME) [2]. Furthermore, MDSC-mediated immunosuppression can be induced by the metabolic conversion of the amino acid l-arginine by arginase 1 (ARG1) or by the production of inducible nitric oxide synthase (iNOS). The iNOS degrades l-arginine to produce nitric oxide (NO) and citrulline. ARG1 uses l-arginine as a substrate to produce l-ornithine and urea. As a result of l-arginine starvation, T-cell proliferation and the synthesis of T-cell effector molecules are impaired, leading to severe T-cell dysfunction [21, 22]. Further, NO production by iNOS prevents IL-2 production by activated leukocytes in that the stability of IL-2-encoding mRNA is impaired. The loss of l-cystine and l-cysteine inhibits activated T cell synthesis of the anti-oxidant glutathione, which impairs proliferation and activation of T cells [23]. In addition, MDSCs induce tryptophan depletion via indoleamine-pyrrole 2,3-dioxygenase (IDO). IDO catalyzes extracellular l-tryptophan to kynurenine. Tryptophan depletion and kynurenine exposure hinders T cell proliferation and facilitates the expansion of regulatory T cells [2]. MDSCs also produce reactive nitrogen species (RNS) and ROS as well as other suppressive molecules that blunt TCR signaling and reduce T cell survival [30]. Furthermore, persistent ER stress promotes tumor progression by affecting malignant cells and infiltrated MDSCs. The regulation of MDSC expansion and suppressive function was shown in Table 1.
In addition, ferroptosis can promote anti-tumor immune response by reversing immunosuppressive microenvironment. A comprehensive index of ferroptosis and immune status (CIFI) was concluded from twenty-seven prognostic ferroptosis- and immune-related signatures in hepatocellualr carcinoma, which could predict a subgroup of patients with a worse prognosis. These patients have higher fractions of cancer-associated fibroblasts (CAFs) and MDSCs [31]. A manganese porphyrin-based metal-organic framework (Mn-MOF), FAP gene-engineered tumor cell-derived exosome-like nanovesicles (eNVs-FAP), NC06 or the Dihydroartemisinin (DHA) were explored to treatment hypoxic tumors or as the a candidate tumor vaccine, which was designed to reduce the number of MDSCs by targeting ferroptosis [32,33,34,35,36,37]. It is indicated that ferroptosis inducer by controlling MDSC polarization or population is a promising immuno-therapeutic strategy [38]. The ferroptosis-based immunotherapy targets affecting the MDSCs population in different kinds of cancers was shown in Table 2.
Unfolded protein response (UPR) and ER stress
ER is a closed plumbing system within the eukaryotic cytoplasm. It is divided into rough ER and smooth ER. The rough ER performs functions related to membrane synthesis and secretion of proteins and is widespread in cells with high secretory capacity. The smooth ER is responsible for the synthesis and transport of lipids. ER is a crucial cell organelle that is involved in the regulation of calcium homeostasis, protein synthesis, lipid metabolism, post-translational modification, transport, and is an essential organelle for synthesis and folding of secreted and transmembrane proteins. However, cellular stressors such as hypoxia, nutrient deficiency, and Ca2+ homeostatic induced ER function disorder which lead to unfolded or misfolded protein accumulation in the ER lumen. If proteins are not properly folded, they are ubiquitinated on the ER membrane and subsequently degraded in a process known as ER associated protein degradation (ERAD). When accumulated misfolded proteins are not eliminated by ERAD, the ER activates the unfolded protein response (UPR). UPR signaling has important roles in immunity, inflammation, and different types of cancer [39, 40].
The UPR has three important sensors: inositol requiring enzyme 1 (IRE1ɑ), protein kinase RNA-activated (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6), which are transmembrane proteins associated with the ER [41]. As well, glucose-regulated protein 78 (GRP78), also referred to Bip or HSPA5, is a key ER chaperone that binds accumulated unfolded/misfolded proteins within the ER lumen. This binding process promotes protein folding and trafficking. Thus, GRP78 is a marker of ER stress and plays an important role in combating ER stress of solid tumor cells [42, 43].
ER stress or exposure to tumor-related ER stress augments the immunosuppressive potential of MDSCs
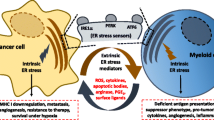
The tumor microenvironment (TME) comprises tumor cells, immune cells, the extracellular matrix and chemokines, cytokines, growth factors, and extracellular vesicles. MDSCs, DCs, and macrophages accumulate within infectious or tumor microenvironments in which hypoxia, nutrient starvation, low pH, and increased levels of free radicals trigger a state of ER stress in cancer cells and in infiltrating myeloid cells. The UPR response triggered by ER stress protects cells from damage. However, when damage is excessive, UPR signals self-destruction, which removes bacteria and prevents further damage. In response to ER stress, cancer cells and MDSCs activate the UPR to promote cell survival and adaptation during adverse environmental conditions [2]. MDSC infiltrates tumor tissues and displays immunosuppression function by suppressing NK cells, T cells, and Treg cells. MDSCs could also display ER stress to survive in the hypoxia-induced tumor microenvironments. The survival MDSCs could produce Arg1, NO, and TGF-β and play roles in immunosuppression [21, 22]. Thus ER stress plays an essential protecting role in the survival of MDSC, which aggravates the immunosuppression in tumors (Fig. 1). The role of ER stress in immune modulation has not fully characterized, but the effects of ER stress on MDSCs during infection and cancer are described below.

ER stress prolong the immunosuppressive function of MDSC cells. MDSCs induce the inactivation of NK cells, CD4+, CD8+ T, and Treg cells through the secretion of TGF-β, NO, and Arg-1. In addition, hypoxia, nutrient starvation, low pH, and increased levels of free radicals in the tumor microenvironment trigger the activation of ER stress sensors, such as IRE1ɑ, PERK, and ATF6, which results in the activation of ER stress of MDSCs. ER stress prolongs the survival of MDSCs and thus aggravates the immunosuppression in tumors
Immunosuppressive effects of ER stress and UPR on MDSCs in infectious disease
When inflammation or infection occurs, MDSCs rapidly expand and travel to the injury sites and regulate the host’s immune system. Therefore, it is crucial to have a thorough understanding of immunomodulatory mechanisms of infection and inflammatory diseases, which may also assist in exploring therapeutic targets. The current data highlight the contribution of ER stress to MDSC immuno-suppression function. Indeed, ER stress can also occur in infectious diseases. Therefore, exploring the role of ER stress in MDSC regulating inflammation helps overcome bacterial infection. By now, the research concerning the interaction of ER stress and MDSCs in infectious disease mainly focuses on Mycobacterium leprae and Mycobacterium tuberculosis infections.
ER stress activates MDSCs and mediates immunosuppression during Mycobacterium leprae and Mycobacterium tuberculosis infections
Leprosy and tuberculosis are caused by intracellular M. leprae and M. tuberculosis, respectively. Various immune system components such as M1 and M2 macrophages, natural killer (NK) cells, DCs, and diverse subtypes of lymphocytes are involved in these infections. Infection can also trigger the accumulation of MDSCs at inflammatory sites [44,45,46]. However, M. tuberculosis and M. leprae can escape and evade the host’s innate immune system [47, 48]. Tuberculoid leprosy (T-LEP) is self-limiting with few bacilli. The host response is a Th1 type. Lepromatous leprosy (L-lep) is a progressive form of disease that is characterized by a high bacillary load within macrophages. The host response to L-lep is a Th2 type [49], with the number of MDSC greater in L-lep patients than in T-lep patients. MDSCs from L-lep patients suppress T-cell proliferation of M. leprae-specific T cells and reduce the production of IFN-γ, which allows bacterial growth and disease progression. Therefore, immunosuppression by MDSCs may worsen M. leprae infection and contribute to the progression of leprosy [44, 45, 50].
GM-CSF and M-CSF drive the expansion of myeloid immune cells within the bone marrow and spleen. MDSCs can be recruited and activated by many factors, such as the proinflammatory cytokines IL-1β, IL-6, and IFN-γ. ER stress can also activate MDSCs and trigger these cells to produce iNOS, ROS, and Arg-1, that are immune suppressive [51,52,53]. Kelly-Scumpia et al. [54] found an increase in immature myeloid cells displaying a granulocytic MDSC cell-surface phenotype (HLA-DR-CD33+CD15+) and T-cell suppressive activity in the blood of patients with disseminated/progressive leprosy when compared to self-limited T-Lep. In terms of mechanism, ER stress significantly regulates the T cell inhibitory activity of MDSCs. Further, ER stress promotes IL-10 secretion, which contributes to MDSC activity and highlights the role of ER stress and IL-10 in MDSC-mediated effects during human M. leprae infection [55]. Further, MDSC ER stress can be caused by circulating IL-1α, IL-6, and IFN-γ [53] in L-lep and tuberculosis infections. These cytokines also cause ER stress in macrophages, DCs, and T cells in L-lep patients, suggesting ER stress may be another factor contributing to the exacerbation of leprosy and tuberculosis [6, 54]. Uncontrolled bacterial growth worsens the ER stress in MDSCs, resulting in increased production of IL-10 and enhanced immunosuppressive activity [5, 56]. Taken together, MDSC mediated immune-suppression is a leading cause of M. leprae and tuberculosis infection, with ER stress activating MDSC immunosuppressive activity. Crispr-cas9, ZFNs, and TALENS are new genetic tools [55, 57] that can block IRE1α and XBP1 signaling and stabilize the ER of MDSCs. Previous studies have shown that reducing the expression of CHOP in MDSCS can promote immune activity and stimulate T cells [8]. Therefore, targeting the UPR could regain or reduce ER stress in tuberculosis and leprosy, thereby reducing the immunosuppressive activity of MDSCs [58, 59]. Breaking ER homeostasis in MDSC may be a potential strategy to combat and eradicate leprosy and tuberculosis.
Immunosuppressive effects of ER stress and UPR on MDSCs in cancers
MDSC was initially described as immunosuppressive myeloid cells that evade cancer. The MDSCs, which accumulate in tumor-bearing mice and cancer patients, are site-specific inflammatory and immunosuppressive agents that contribute to cancer progression in different cancers. MDSCs accumulated in the TME under chronic inflammation conditions and cancer contributed to the growth of tumors. Furthermore, the population of immunosuppressive MDSCs decreased after radiotherapy. Thus, preventing MDSC development and/or interfering with their immunosuppressive functions in cancer could reduce immunosuppression, thereby increasing antitumor immunity. In this part, we will discuss ER stress-activated MDSCs and enhanced immunosuppression, which may serve as targets in immunotherapy for different kinds of tumors.
ER stress and MDSCs as therapeutic targets for ulcerative colitis and colorectal cancer
Colorectal cancer (CRC) is one of the primary causes of cancer-related deaths globally, with more than 2.2 million new cases projected by 2030 [60]. Ulcerative colitis is a chronic colon inflammation, a complex, recurrent, and remitting form of intestinal inflammation [61]. For ulcerative colitis and CRC, MDSCs are a main component of the inflammatory microenvironment with infiltration of the intraepithelial and lamina propria layers. When activated, MDSCs reduce T cell immune function and recruit tumor-associated macrophages (TAM) that down-regulate immune activity in the colonic epithelial barrier [62]. Furthermore, MDSCs secrete M-CSF and GM-CSF that recruit tumor-associated neutrophils (TANs) and TAMs in inflamed colon intraepithelial, lamina propria, and cancerous tissues [63]. In addition, studies have shown that UPR activation and ER stress are involved in colitis and tumorigenesis. During colitis, the stable status of the ER protein-folding environment is disrupted by physiologic, pathologic, or environmental injury, which results in the accumulation of misfolded proteins. When the accumulation of misfolded proteins exceeds the tolerance threshold, the ER-resident sensors trigger the UPR, resulting in transcriptionally enhanced ER protein folding capacity [64]. Colonic mucosal cells undergo apoptosis if these corrections are insufficient. However, if cells limit pro-apoptotic UPR successfully, ER stress can promote tumorigenesis [65]. Thus, continuous activation of robust ER stress sensors can confer tumorigenesis. Studies have shown that controlling robust ER stress is an effective therapeutic strategy for the prevention of colitis and tumorigenesis [5]. Feng Wang et al. demonstrated a new derivative of myricetin, (M10), to inhibit ulcerative colitis and colorectal neoplasms by weakening gross ER stress. Inhibition of ER stress-induced the UPR pathway by direct regulation of mTOR expression. Therefore, M10 may be a promising drug for chemo-prophylaxis of colitis and tumorigenesis [66]. In addition, MDSCs may be an effective therapeutic target in that emerging evidence suggests critical roles for GM-CSF and M-CSF in chronic, relapsing, and complex inflammatory states in colonic tissues [67]. MDSCs can also produce IL-6 and TNF-ɑ, which are involved in the IL-6/STAT3 pathway signaling, playing an immunosuppressive role in the tumor microenvironment [68, 69]. It has been reported that naringin inhibits MDSCs, proinflammatory mediators (GM-CSF/M-CSF, IL-6, TNF-ɑ), and the NF-κB/IL-6/STAT3 cascade in colorectal tissue, reducing the severity of colitis and colorectal adenoma. Naringin inhibits ER transmembrane proteins (GRP78, ATF6, and IRE1), as well as activated PERK, phosphorylated eIF-2α in colorectal mucosal cells. Further, naringin prevents the secretion of the ATG3, ATG5, ATG7, ATG12, ATG16, and ATG16L1 complex, thus preventing the occurrence of colitis and colorectal cancer [70].
ER stress, MDSCs, and breast cancer
Triple-negative breast cancer (TNBC) accounts for 15.0−25.0% of all breast cancers. TNBC cells do not express the estrogen receptor (ER), the progesterone receptor (PR), or the human epidermal growth factor receptor-2 (HER-2). TNBC is an early onset and highly aggressive malignant tumor with a poor prognosis and a high distant metastasis rate [71, 72]. Activated PERK, one of the ER-membrane-resident sensors, can phosphorylate eIF2 and induce a comprehensive stress response that results in global translation inhibition and selective translation of repair proteins [73, 74]. Overexpression of P-EIF2A has been associated with tumor progression [75, 76] and a protective clinical effect [77, 78]. Thus the effect of the tumor PERK/P/EIF2A signaling pathway is controversial. In breast cancer (BC), P-EIF2A has been reported to predict disease-free survival in patients with TNBC [79]. Zou et al. reported EIF2A mRNA levels to be negatively associated with TNBC relapse-free survival and negatively related to metastasis. P-EIF2A promotes the activity of tumor-infiltrating T cells and inhibits the activity of MDSCs by inhibiting PDL1 and CXCL5, thereby regulating TNBC metastasis. The PERK/EIF2A pathway also regulates carboplatin resistance in highly metastatic TNBC.
IRE1α, one of the ER-membrane-resident sensors, remodels the TME in TNBC by increasing pericyte levels and vascular normalization while decreasing CAFs and MDSCs [80]. Matrix cellular proteins, a group of extracellular matrix (ECM) proteins, are transducers and modulators of the interaction between cells and the extracellular microenvironment. These proteins include osteopontin (OPN), thrombospondins (TSPs), osteonectin, tenascins, periostin (POSTN), and CCNs [81]. POSTN is highly expressed in many tissues but is significantly associated with the degree of tumor malignancy, metastasis, hyperplasia, and fibrosis of inflammatory tissue. POSTN is expected to become a detection index for diagnosing and treating of many tumors and inflammatory diseases. It has recently been reported that lung fibroblast-derived POSTN is an important limiting factor of metastatic breast cancer cells within the lung by promoting of the self-renewal of breast cancer stem cells [82].
Furthermore, POSTN is reported to be associated with a poor prognosis for basal-like breast cancer, with POSTN-integrin ɑvβ3 signaling required to establish a micro-environmental niche for breast cancer stem cells [83]. It is interesting to note that POSTN can also be produced by bone MDSC cells and their derived cells, which indicates that POSTN promotes MDSC-mediated pulmonary pre-metastatic niche formation. Breast cancer metastasis could occur through the accumulation of MDSCs within the lungs. These results provide new and promising avenues to develop practical therapeutic approaches for breast cancer treatment, especially TNBC.
ER stress, a key regulator of LOX-1+ PMN-MDSCs derived from nasopharyngeal carcinoma survivors with chronic hepatitis B virus
Lectin-type oxidized LDL receptor-1 (LOX-1) is a specific marker for human PMN-MDSCs [7] that can separate and identify PMN-MDSC cells. CD15 is also a marker for neutrophils as such LOX-1+ and CD15+ cells in human blood are PMN-MDSC. In contrast, CD15+ but LOX-1− cells are normal neutrophils (PMNs) [29, 57, 84]. Levels of PMN-MDSC (LOX-1+) cells increased in the peripheral blood of nasopharyngeal carcinoma (NPC) survivors with chronic hepatitis B virus (CHB) infection. These cells may be immunosuppressive by inhibition of T cell proliferation and activation. ER stress-related gene: sXBP1, SEC61A, ATF4, ATF6, ATF3, and CHOP are significantly up-regulated in PMN-MDSCs (LOX-1+) compared with PMNs (LOX-1−) from the same NPC survivors with CHB. These observations suggest that ER stress may affect the survival of LOX-1+ PMN-MDSCs and disease progression. LOX-1+ PMN-MDSCs from NPC survivors with CHB had higher NOX2 mRNA levels, a critical ROS-related gene, suggesting that ROS mediates the immune suppressive effect of LOX-1+ PMN-MDSCs. These results suggest that PMN-MDSCs play an immunosuppressive role in the host immune response to CHB through ER stress/ROS effects [85].
ER stress may be the key regulator of PMN-MDSCs in hepatocellular carcinoma patients
PMN-MDSCs (LOX-1+CD15+) is significantly up-regulated in the peripheral blood of hepatocellular carcinoma (HCC) patients compared to healthy controls. T cell activation is significantly suppressed by LOX-1+CD15+ PMN-MDSCs, inhibiting CD4+ and CD8+ T cell proliferation as well as IFN-γ production. This immune suppression is mediated by the cellular production of ROS and by the activation of arginase I. Moreover, LOX-1 expression and suppressive function are mediated by ER stress that increases the expression of XBP1, ATF3, and CHOP [86]. These results suggest ER stress may be an essential regulator of PMN-MDSC in HCC. In addition, PMN-MDSCs of cancer patients exhibit signs of an ER stress response [29, 87], with some myeloid cells in peripheral blood exhibiting ER stress. These peripheral blood cells were distant from the tumor site, which suggests tumor-induced ER stress in myeloid cells in a remote manner. However, neither serum nor the TCM from HCC patients induced healthy donor CD15 + cells to differentiate into PMN-MDSC, nor was ER stress-induced. The underlying mechanism for this phenomenon warrants further investigation [57].
ER stress may mediate prostate cancer tumorigenesis by regulation of MDSC immune suppression
Prostate cancer is the most common urological malignancy in men, with three-quarters of cases in patients over 65. Compared with the United States, prostate cancer incidence and mortality are relatively low in China, although incidence and mortality have increased in recent years [88, 89]. The use of anti-CTLA-4 as an immune checkpoint blockade for prostate cancer treatment has not been clinically successful [90,91,92], which may be due to TME immunosuppression [93]. Myeloid-derived cells are essential components of the TME and may contribute to treatment failure in prostate cancer patients. Clinical studies have demonstrated increased numbers of infiltrating macrophages in primary prostate tumors, which may be associated with failure of androgen ablation [11]. The proportion of M-MDSCs in the peripheral blood of prostate cancer patients is significantly increased compared to age-matched controls [94, 95]. Mechanistically, T cell-suppressed proliferation, and high IL-10 levels have been confirmed in vitro [96]. Therefore, targeting MDSCs or regulating their recruitment has the potential for immunotherapeutic treatment of prostate cancer patients [97].
Recently, ER stress has been shown to be transmitted from tumor cells to myeloid cells. When cultured in the conditioned medium of ER-stressed tumor cells, macrophages also demonstrate an ER stress response with Hspa5 and XBP1 up-regulated. The proliferation of prostate cancer cell lines can be regulated by XBP1s [98], but how XBP1s regulate MDSCs is unknown and requires future investigation. ER stress-sensitive factor, XBP1, can induce the expression of Arg1 and Nos2, which are essential regulators of the immunosuppressive function of MDSCs [99]. ER stress may play an important role in prostate cancer, mediating tumorigenesis and tumor development by regulating the immunosuppressive phenotype of prostate cancer MDSCs [100, 101].
ER stress and MDSCs as therapeutic targets in cancer and inflammatory disease
MDSCs play an essential role in tumor immunosuppression. More and more studies have shown that MDSCs are closely related to the effect of tumor immunotherapy. Therefore, it is of great significance to change tumor immunosuppression by inhibiting the function of MDSCs. Tumor-derived ER stress in MDSCs mediates the immunosuppressive activity. Therefore, researchers predicted ER stress-related proteins in MDSCs could be potential therapeutic targets in infectious diseases and cancers. ERK, AKT, and STAT3 decreased in Periostin (POSTN) -deficient MDSCs. The the pro-metastatic role of POSTN is limited to ER-negative breast cancer patients, which indicates that POSTN is a potential target for the prevention and treatment of breast tumor metastasis [91]. M10, a novel derivative of Myricetin, prevents ER stress-induced autophagy in inflamed colonic mucosal cells by targeting the NF-κB/IL-6/STAT3 pathway, which develops M10 as a promising regimen in the chemoprevention of colitis and colorectal cancer [66]. Insights from studies might substantiate PMN-MDSCs as a potential therapeutic target for lung carcinoma [97], hepatocellular carcinoma 6, and Chronic hepatitis B (CHB) with nasopharyngeal carcinoma (NPC) [85]. Further research were warranted to confirm ER stress-related proteins, including PERK, CHOP, IRE1α, and XBP1s, as potential therapeutic targets in cancers [102,103,104]. ER stress sensors or signals triggering MDSC activation could be investigated as therapeutic targets in cancers and infectious or inflammatory diseases as shown in Table 3; Fig. 2.

ER stress sensors or signals triggering MDSC activation and immunosuppression may serve as targets in immunotherapy in cancers. In lung carcinoma or metastasis cancer, CHOP, PERK, and TRAIL-Rs could be targeted by inhibitors that reduce MDSCs by inhibiting ER stress. sXBP1/ATF3/CHOP/ROS/Arg I signaling activated LOX-1+CD15+ PMN-MDSCs in Hepatocellular carcinoma. sXBP1 is one of the most important sensors in ER stress. Targeting sXBP1 signaling could reduce the immunosuppression of MDSCs in HCC and Melanoma. IRE1α, one of ER stress sensors, could also be an inhibitory target in Ovarian carcinoma. 4-Phenylbutyric acid (4-PBA), a chemical chaperone widely used as an ER stress reducer, attenuated Tg-induced MDSC expansion and tumor growth in melanoma
Future perspectives
The pathologic microenvironment associated with inflammation and tumors is characterized by hypoxia, nutrient deprivation, low pH, and free radicals that can trigger ER stress and the accumulation of MDSCs, resulting in immunosuppression. Reactive oxygen species and lipids are significantly elevated in MDSCs and are the main causes of the ER stress response. Inhibition of MDSC has been shown to be a potential and promising cancer therapy based on its complex role in promoting tumor genesis, development, and metastasis in the tumor microenvironment. Over the past few years, many preclinical studies have focused on exploring drugs, such as Sunitinib [105] and 5-phosphodiesterase inhibitors [106], to inhibit its immunosuppressive activity. New strategies that remodel tumor-associated myeloid cells into mature immune cells will greatly improved the efficacy of tumor-targeted therapies.
References
Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5(1):3–8. https://doi.org/10.1158/2326-6066.CIR-16-0297.
Dysthe M, Parihar R. Myeloid-derived suppressor cells in the tumor microenvironment. Adv Exp Med Biol. 2020;1224:117–40. https://doi.org/10.1007/978-3-030-35723-8_8.
Bettigole SE, Glimcher LH. Endoplasmic reticulum stress in immunity. Annu Rev Immunol. 2015;33:107–38. https://doi.org/10.1146/annurev-immunol-032414-112116.
Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161(7):1527–38. https://doi.org/10.1016/j.cell.2015.05.025.
Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. 2017;168(4):692–706. https://doi.org/10.1016/j.cell.2016.12.004.
Cubillos-Ruiz JR, Mohamed E, Rodriguez PC. Unfolding anti-tumor immunity: ER stress responses sculpt tolerogenic myeloid cells in cancer. J Immunother Cancer. 2017;5:5. https://doi.org/10.1186/s40425-016-0203-4.
Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci USA. 2011;108(16):6561–6. https://doi.org/10.1073/pnas.1008942108.
Zhang H, Yue Y, Sun T, Wu X, Xiong S. Transmissible endoplasmic reticulum stress from myocardiocytes to macrophages is pivotal for the pathogenesis of CVB3-induced viral myocarditis. Sci Rep. 2017;7:42162. https://doi.org/10.1038/srep42162.
Thevenot PT, Sierra RA, Raber PL, et al. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity. 2014;41(3):389–401. https://doi.org/10.1016/j.immuni.2014.08.015.
Santos CX, Tanaka LY, Wosniak J, Laurindo FR. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal. 2009;11(10):2409–27. https://doi.org/10.1089/ars.2009.2625.
Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metabol. 2012;15(5):623–34. https://doi.org/10.1016/j.cmet.2012.03.007.
Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, Corzo A, Cho HI, Celis E, Lennox B, Knight SC, Padhya T, McCaffrey TV, McCaffrey JC, Antonia S, Fishman M, Ferris RL, Kagan VE, Gabrilovich DI. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010;16(8):880–6. https://doi.org/10.1038/nm.2172.
Gabrilovich DI, Bronte V, Chen SH, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67(1):425–6. https://doi.org/10.1158/0008-5472.CAN-06-3037.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68. https://doi.org/10.1038/nri3175.
Bronte V, Brandau S, Chen SH, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. https://doi.org/10.1038/ncomms12150.
Damuzzo V, Pinton L, Desantis G, et al. Complexity and challenges in defining myeloid-derived suppressor cells. Cytometry B Clin Cytom. 2015;88(2):77–91. https://doi.org/10.1002/cyto.b.21206.
Mandruzzato S, Brandau S, Britten CM, et al. Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: results from an interim study. Cancer Immunol Immunother. 2016;65(2):161–9. https://doi.org/10.1007/s00262-015-1782-5.
Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, Rodriguez PC, Sica A, Umansky V, Vonderheide RH, Gabrilovich DI. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. https://doi.org/10.1038/ncomms12150.
Hegde S, Leader AM, Merad M. MDSC: markers, development, states, and unaddressed complexity. Immunity. 2021;54(5):875–84. https://doi.org/10.1016/j.immuni.2021.04.004.
Yang Y, Li C, Liu T, Dai X, Bazhin AV. Myeloid-derived suppressor cells in tumors: from mechanisms to antigen specificity and microenvironmental regulation. Front Immunol. 2020;11:1371. https://doi.org/10.3389/fimmu.2020.01371.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68.
Ostrand-Rosenberg S, Sinha P. Myeloidderived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–506.
Umansky V, Blattner C, Gebhardt C, Utikal J. The role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccine. 2016;4:36.
Cheng P, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–49.
Waight JD, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest. 2013;123:4464–78.
Li L, et al. MicroRNA-155 and microRNA-21 promote the expansion of functional myeloidderived suppressor cells. J Immunol. 2014;192:1034–43.
Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol. 2015;98:913–22.
Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32:19–25.
Condamine T, Dominguez GA, Youn JI, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. 2016;1(2):aaf8943. https://doi.org/10.1126/sciimmunol.aaf8943.
Mandula JK, Rodriguez PC. Tumor-related stress regulates functional plasticity of MDSCs. Cell Immunol. 2021;363:104312. https://doi.org/10.1016/j.cellimm.2021.104312.
Liu Y, Zhang X, Zhang J, Tan J, Li J, Song Z. Development and validation of a combined ferroptosis and immune prognostic classifier for hepatocellular carcinoma. Front Cell Dev Biol. 2020;8:596679. https://doi.org/10.3389/fcell.2020.596679.
Xu Q, Zhan G, Zhang Z, Yong T, Yang X, Gan L. Manganese porphyrin-based metal-organic framework for synergistic sonodynamic therapy and ferroptosis in hypoxic tumors. Theranostics. 2021;11(4):1937–52. https://doi.org/10.7150/thno.45511.
Hu S, Ma J, Su C, Chen Y, Shu Y, Qi Z, Zhang B, Shi G, Zhang Y, Zhang Y, Huang A, Kuang Y, Cheng P. Engineered exosome-like nanovesicles suppress tumor growth by reprogramming tumor microenvironment and promoting tumor ferroptosis. Acta Biomater. 2021;135:567–81. https://doi.org/10.1016/j.actbio.2021.09.003. (Epub 2021 Sep 8).
Zhao YY, Lian JX, Lan Z, Zou KL, Wang WM, Yu GT. Ferroptosis promotes anti-tumor immune response by inducing immunogenic exposure in HNSCC. Oral Dis. 2021. https://doi.org/10.1111/odi.14077.
Zhu H, Klement JD, Lu C, Redd PS, Yang D, Smith AD, Poschel DB, Zou J, Liu D, Wang PG, Ostrov D, Coant N, Hannun YA, Colby AH, Grinstaff MW, Liu K. Asah2 represses the p53-Hmox1 axis to protect myeloid-derived suppressor cells from ferroptosis. J Immunol. 2021;206(6):1395–404. https://doi.org/10.4049/jimmunol.2000500. (Epub 2021 Feb 5).
Zhang H, Zhuo Y, Li D, Zhang L, Gao Q, Yang L, Yuan X. Dihydroartemisinin inhibits the growth of pancreatic cells by inducing ferroptosis and activating antitumor immunity. Eur J Pharmacol. 2022;926:175028. https://doi.org/10.1016/j.ejphar.2022.175028. (Epub 2022 May 13).
Li S, Li F, Xu L, Liu X, Zhu X, Gao W, Shen X. TLR2 agonist promotes myeloid-derived suppressor cell polarization via Runx1 in hepatocellular carcinoma. Int Immunopharmacol. 2022;111:109168. https://doi.org/10.1016/j.intimp.2022.109168. (Epub 2022 Aug 20).
Yan J, Ye G, Shao Y. High expression of the ferroptosis-associated MGST1 gene in relation to poor outcome and maladjusted immune cell infiltration in uterine corpus endometrial carcinoma. J Clin Lab Anal. 2022;36(4):e24317. https://doi.org/10.1002/jcla.24317. (Epub 2022 Feb 26).
Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol. 2016;16(8):469–84. https://doi.org/10.1038/nri.2016.62.
Cao S, Tang J, Huang Y, Li G, Li Z, Cai W, Yuan Y, Liu J, Huang X, Zhang H. The road of solid tumor survival: from drug-induced endoplasmic reticulum stress to drug resistance. Front Mol Biosci. 2021;8:620514. https://doi.org/10.3389/fmolb.2021.620514.
Urra H, Dufey E, Avril T, Chevet E, Hetz C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer. 2016;2(5):252–62. https://doi.org/10.1016/j.trecan.2016.03.007.
Gifford JB, Huang W, Zeleniak AE, Hindoyan A, Wu H, Donahue TR, Hill R. Expression of GRP78, master regulator of the unfolded protein response, increases chemoresistance in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2016;15(5):1043–52. https://doi.org/10.1158/1535-7163.MCT-15-0774.
Luo C, Fan W, Jiang Y, Zhou S, Cheng W. Glucose-related protein 78 expression and its effects on cisplatin-resistance in cervical cancer. Med Sci Monit Int Med J Exp Clin Res. 2018;24:2197–209. https://doi.org/10.12659/msm.906413.
Geluk A. Correlates of immune exacerbations in leprosy. Semin Immunol. 2018;39:111–8.
Sadhu S, Mitra DK. Emerging concepts of adaptive immunity in leprosy. Front Immunol. 2018;9:604.
De Sousa JR, Sotto MN, Simoes Quaresma JA. Leprosy as a complex infection: breakdown of the Th1 and Th2 immune paradigm in the immunopathogenesis of the disease. Front Immunol. 2017;8:1635.
Mi Z, Liu H, Zhang F. Advances in the immunology and genetics of leprosy. Front Immunol. 2020;11:567.
De Martino M, Lodi L, Galli L, Chiappini E. Immune response to Mycobacterium tuberculosis: a narrative review. Front Pediatr. 2019;7:350.
Rea TH, Modlin RL. Immunopathology of leprosy skin lesions. Semin Dermatol. 1991;10(3):188–93.
Pinheiro RO, Schmitz V, Silva BJ de. A et al. Innate immune responses in leprosy. Front Immunol. 2018;9:518.
Korb VC, Chuturgoon AA, Moodley D. Mycobacterium tuberculosis: manipulator of protective immunity. Int J Mol Sci. 2016;17(3):131.
Kotz´e LA, Young C, Leukes VN, et al. Mycobacterium tuberculosis and myeloid-derived suppressor cells: insights into caveolin rich lipid rafts. EBioMedicine. 2020;53:102670.
Hirai KE, de Sousa JR, Silva LM, et al. Endoplasmic reticulum stress markers and their possible implications in leprosy’s pathogenesis. Dis Mark. 2018. https://doi.org/10.1155/2018/7067961.
Kelly-Scumpia KM, Choi A, Shirazi R, Bersabe H, Park E, Scumpia PO, Ochoa MT, Yu J, Ma F, Pellegrini M, Modlin RL. ER stress regulates immunosuppressive function of myeloid derived suppressor cells in leprosy that can be overcome in the presence of IFN-γ. iScience. 2020;23(5):101050. https://doi.org/10.1016/j.isci.2020.101050.
Gaj T, Gersbach CA, Barbas CF III. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31(7):397–405. https://doi.org/10.1016/j.tibtech.2013.04.004.
Li A, Song N-J, Riesenberg BP, Li Z. The emerging roles of endoplasmic reticulum stress in balancing immunity and tolerance in health and diseases: mechanisms and opportunities. Front Immunol. 2020;10:3154.
Gaj T, Sirk SJ, Shui SL, Liu J. Genome-editing technologies: principles and applications. Cold Spring Harb Perspect Biol. 2016;8(12):a023754. https://doi.org/10.1101/cshperspect.a023754.
Leukes V, Walzl G, du Plessis N. Myeloid-derived suppressor cells as target of phosphodiesterase-5 inhibitors in host-directed therapeutics for tuberculosis. Front Immunol. 2020;11:451. https://doi.org/10.3389/fimmu.2020.00451.
Li A, Song NJ, Riesenberg BP, Li Z. The emerging roles of endoplasmic reticulum stress in balancing immunity and tolerance in health and diseases: mechanisms and opportunities. Front Immunol. 2020;10:3154. https://doi.org/10.3389/fimmu.2019.03154.
Arnold M, et al. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66:683–91.
Khalili H, et al. Identification of a common variant with potential pleiotropic effect on risk of inflammatory bowel disease and colorectal cancer. Carcinogenesis. 2015;36:999–1007.
Colangelo T, et al. Friend or foe? The tumour microenvironment dilemma in colorectal cancer. Biochim Biophys Acta. 2017;1867:1–18.
Ma N, et al. MDSCs are involved in the protumorigenic potentials of GM-CSF in colitis-associated cancer. Int J Immunopathol Pharmacol. 2017;30:152–62.
Cao SS, et al. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology. 2013;144:989–1000.
Walter F, et al. Imaging of single cell responses to ER stress indicates that the relative dynamics of IRE1/XBP1 and PERK/ATF4 signalling rather than a switch between signalling branches determine cell survival. Cell Death Differ. 2015;22:1502–16.
Wang F, Song ZY, Qu XJ, Li F, Zhang L, Li WB, Cui SX. M10, a novel derivative of Myricetin, prevents ulcerative colitis and colorectal tumor through attenuating robust endoplasmic reticulum stress. Carcinogenesis. 2018;39(7):889–99. https://doi.org/10.1093/carcin/bgy057.
Griseri, et al. Granulocyte macrophage colony-stimulating factor-activated eosinophils promote interleukin-23 driven chronic colitis. Immunity. 2015;43:187–99.
Nagaraj S, et al. Regulatory myeloid suppressor cells in health and disease. Cancer Res. 2009;69:7503–6.
Grivennikov SI, et al. Inflammation and oncogenesis: a vicious connection. Curr Opin Genet Dev. 2010;20:65–71.
Zhang YS, Wang F, Cui SX, Qu XJ. Natural dietary compound naringin prevents azoxymethane/dextran sodium sulfate-induced chronic colorectal inflammation and carcinogenesis in mice. Cancer Biol Ther. 2018;19(8):735–44. https://doi.org/10.1080/15384047.2018.1453971.
Brown M, Tsodikov A, Bauer KR, Parise CA, Caggiano V. The role of human epidermal growth factor receptor 2 in the survival of women with estrogen and progesterone receptor-negative, invasive breast cancer: the California Cancer Registry, 1999–2004. Cancer. 2008;112:737–47.
Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–48.
Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389.
Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519.
Hart LS, Cunningham JT, Datta T, et al. ER stress mediated autophagy promotes myc-dependent transformation and tumor growth. J Clin Invest. 2012;122:4621.
Koshikawa N, Maejima C, Miyazaki K, et al. Hypoxia selects for high-metastatic Lewis lung carcinoma cells overexpressing Mcl-1 and exhibiting reduced apoptotic potential in solid tumors. Oncogene. 2006;25:917.
Meurs EF, Galabru J, Barber GN, et al. Tumor suppressor function of the interferon-induced double-stranded RNA activated protein kinase. Proc Natl Acad Sci USA. 1993;90:232.
Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112.
Guo L, Chi Y, Xue J, et al. Phosphorylated eIF2alpha predicts disease-free survival in triple-negative breast cancer patients. Sci Rep. 2017;7:44674.
Harnoss JM, Le Thomas A, Reichelt M, Guttman O, Wu TD, Marsters SA, Shemorry A, Lawrence DA, Kan D, Segal E, Merchant M, Totpal K, Crocker LM, Mesh K, Dohse M, Solon M, Modrusan Z, Rudolph J, Koeppen H, Walter P, Ashkenazi A. IRE1α disruption in triple-negative breast cancer cooperates with antiangiogenic therapy by reversing ER stress adaptation and remodeling the tumor microenvironment. Cancer Res. 2020;80(11):2368–79. https://doi.org/10.1158/0008-5472.CAN-19-3108.
Wang Z, Xiong S, Mao Y, Chen M, Ma X, Zhou X, Ma Z, Liu F, Huang Z, Luo Q, Ouyang G. Periostin promotes immunosuppressive premetastatic niche formation to facilitate breast tumour metastasis. J Pathol. 2016;239(4):484–95. https://doi.org/10.1002/path.4747.
Malanchi I, Santamaria-Martínez A, Susanto E, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2012;481:85–9.
Lambert AW, Wong CK, Ozturk S, et al. Tumor cell-derived periostin regulates cytokines that maintain breast cancer stem cells. Mol Cancer Res. 2016;14:103–13.
Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC: their biological role and interaction with stromal cells. Semin Immunol. 2018;35:19–28.
Li X, Li JL, Jiang N, Chen J, Liang ZM, Zhao ZL, Xing YF. Accumulation of LOX-1+ PMN-MDSCs in nasopharyngeal carcinoma survivors with chronic hepatitis B might permit immune tolerance to Epstein-Barr virus and relate to tumor recurrence. Aging. 2020;13(1):437–49. https://doi.org/10.18632/aging.202149.
Nan J, Xing YF, Hu B, Tang JX, Dong HM, He YM, Ruan DY, Ye QJ, Cai JR, Ma XK, Chen J, Cai XR, Lin ZX, Wu XY, Li X. Endoplasmic reticulum stress induced LOX-1+CD15+ polymorphonuclear myeloid-derived suppressor cells in hepatocellular carcinoma. Immunology. 2018;154(1):144–55. https://doi.org/10.1111/imm.12876. (Epub 2017 Dec 21).
Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, El-Deiry WS, Winograd R, Vonderheide RH, English NR, Knight SC, Yagita H, McCaffrey JC, Antonia S, Hockstein N, Witt R, Masters G, Bauer T, Gabrilovich DI. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Investig. 2014;124(6):2626–39. https://doi.org/10.1172/JCI74056.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30.
Nemunaitis JJ, Valone FH, Verjee SS, Jones LA, Hershberg RM. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–94.
Slovin SF, Higano CS, Hamid O, Tejwani S, Harzstark A, Alumkal JJ, Scher HI, Chin K, Gagnier P, McHenry MB, Beer TM. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: results from an open label, multicenter phase I/II study. Ann Oncol. 2013;24:1813–21.
Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, Krainer M, Houede N, Santos R, Mahammedi H, Ng S, Maio M, Franke FA, Sundar S, Agarwal N, Bergman AM, Ciuleanu TE, Korbenfeld E, Sengeløv L, Hansen S, Logothetis C, Beer TM, McHenry MB, Gagnier P, Liu D, Gerritsen WR, CA184-043 Investigators. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:700–12.
Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, Ganju V, Polikoff J, Saad F, Humanski P, Piulats JM, Mella G, Ng P, Jaeger SS, Parnis D, Franke FX, Puente FA, Carvajal J, Sengeløv R, McHenry L, Varma MB, van den Eertwegh A, Gerritsen AJ. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castrationresistant prostate cancer. J Clin Oncol. 2017;35:40–7.
Santegoets SJ, Stam AG, Lougheed SM, Gall H, Jooss K, Sacks N, Hege K, Lowy I, Scheper RJ, Gerritsen WR, van den Eertwegh AJ, de Gruijl TD. Myeloid derived suppressor and dendritic cell subsets are related to clinical outcome in prostate cancer patients treated with prostate GVAX and ipilimumab. J Immunother Cancer. 2014;2:31.
Nuhn P, Vaghasia AM, Goyal J, Zhou XC, Carducci MA, Eisenberger MA, Antonarakis ES. Association of pretreatment neutrophil-to-lymphocyte ratio (NLR) and overall survival (OS) in patients with metastatic castration-resistant prostate cancer (mCRPC) treated with first-line docetaxel. BJU Int. 2014;114(6b):E11–7.
Brusa D, Simone M, Gontero P, Spadi R, Racca P, Micari J, Degiuli M, Carletto S, Tizzani A, Matera L. Circulating immunosuppressive cells of prostate cancer patients before and after radical prostatectomy: profile comparison. Int J Urol. 2013;20:971–8.
Vuk-Pavlovi´c S, Bulur PA, Lin Y, Qin R, Szumlanski CL, Zhao X, Dietz AB. Immunosuppressive CD14+HLA-DRlow/− monocytes in prostate cancer. Prostate. 2010;70:443–55.
Tcyganov EN, Hanabuchi S, Hashimoto A, et al. Distinct mechanisms govern populations of myeloid-derived suppressor cells in chronic viral infection and cancer. J Clin Invest. 2021;131(16):e145971. https://doi.org/10.1172/JCI145971.
Thorpe JA, Schwarze SR. IRE1a controls cyclin A1 expression and promotes cell proliferation through XBP-1. Cell Stress Chaperones. 2010;15:497–508.
Lee BR, Chang SY, Hong EH, Kwon BE, Kim HM, Kim YJ, Lee J, Cho HJ, Cheon JH, Ko HJ. Elevated endoplasmic reticulum stress reinforced immunosuppression in the tumor microenvironment via myeloid-derived suppressor cells. Oncotarget. 2014;5:12331–45.
Storm M, Sheng X, Arnoldussen YJ, Saatcioglu F. Prostate cancer and the unfolded protein response. Oncotarget. 2016;7:54051–66.
Mohamed E, Sierra RA, Trillo-Tinoco J, et al. The unfolded protein response mediator PERK governs myeloid cell-driven immunosuppression in tumors through inhibition of STING signaling. Immunity. 2020;52(4):668-682.e7. https://doi.org/10.1016/j.immuni.2020.03.004.
Kim YJ, Chang SY, Ko HJ. Myeloid-derived suppressor cells in inflammatory bowel disease. Intest Res. 2015;13(2):105–11. https://doi.org/10.5217/ir.2015.13.2.105. (Epub 2015 Apr 27).
Liu F, Li X, Lu C, Bai A, Bielawski J, Bielawska A, Marshall B, Schoenlein PV, Lebedyeva IO, Liu K. Ceramide activates lysosomal cathepsin B and cathepsin D to attenuate autophagy and induces ER stress to suppress myeloid-derived suppressor cells. Oncotarget. 2016;7(51):83907–25. https://doi.org/10.18632/oncotarget.13438.
Yang Z, Huo Y, Zhou S, Guo J, Ma X, Li T, Fan C, Wang L. Cancer cell-intrinsic XBP1 drives immunosuppressive reprogramming of intratumoral myeloid cells by promoting cholesterol production. Cell Metab. 2022. https://doi.org/10.1016/j.cmet.2022.10.010.
Chen HM, Ma G, Gildener-Leapman N, et al. Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clin Cancer Res. 2015;21(18):4073–85. https://doi.org/10.1158/1078-0432.CCR-14-2742.
Serafini P, Meckel K, Kelso M, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203(12):2691–702. https://doi.org/10.1084/jem.20061104.
Acknowledgements
The authors thank Professor Zhaoyuan Hou for helpful comments and suggestions.
Funding
This work was supported by the Shanghai Natural Science Foundation (21ZR1457900), Shanghai Municipal Commission of Health and Family Planning Project (202240095 and 201940025).
Author information
Authors and Affiliations
Contributions
XL and YH discussed and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lou, X., Gao, D., Yang, L. et al. Endoplasmic reticulum stress mediates the myeloid-derived immune suppression associated with cancer and infectious disease. J Transl Med 21, 1 (2023). https://doi.org/10.1186/s12967-022-03835-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-022-03835-4