
Abstract
Background
Rho GTPase activating protein 9 (ARHGAP9) is expressed in various types of cancers and can inactivate Rho GTPases that mainly regulate cytoskeletal dynamics. However, the exact role of ARHGAP9 in acute myeloid leukemia (AML) has yet to be clarified.
Methods
We compared the transcriptional expression, prognosis, differentially expressed genes, functional enrichment, and hub genes in AML patients on the basis of the data published in the following databases: UALCAN, GEPIA, Gene Expression Omnibus, the Human Protein Atlas, Cancer Cell Line Encyclopedia, LinkedOmics, Metascape, and String. Data from the Cancer Genome Atlas database was used to evaluate the correlations between ARHGAP9 expression and various clinicopathological parameters, as well as the significantly different genes associated with ARHGAP9 expression.
Results
We found that ARHGAP9 expression was higher in the tissues and cell lines extracted from patients with AML than corresponding control tissues and other cancer types. ARHGAP9 overexpression was associated with decreased overall survival (OS) in AML. Compared with the ARHGAP9low group, the ARHGAP9high group, which received only chemotherapy, showed significantly worse OS and event-free survival (EFS); however, no significant difference was observed after treatment with autologous or allogeneic hematopoietic stem cell transplantation (auto/allo-HSCT). The ARHGAP9high patients undergoing auto/allo-HSCT also had a significantly better prognosis with respect to OS and EFS than those receiving only chemotherapy. Most overlapping genes of the significantly different genes and co-expression genes exhibited enriched immune functions, suggesting the immune regulation potential of ARHGAP9 in AML. A total of 32 hub genes were identified from the differentially expressed genes, within which the KIF20A had a significant prognostic value for AML.
Conclusions
ARHGAP9 overexpression was associated with poor OS in AML patients and can be used as a prognostic biomarker. AML patients with ARHGAP9 overexpression can benefit from auto/allo-HSCT rather than chemotherapy.
Similar content being viewed by others


Background
Acute myeloid leukemia (AML), which is caused by the malignant transformation of myeloid primordial cells, is the second most common type of leukemia diagnosed in adults and children. Despite the significant progress made in risk stratification, supportive care, multiagent chemotherapy intensification, and autologous or allogeneic hematopoietic stem cell transplantation (auto/allo-HSCT), the outcome for AML patients remains discouraging because of recurrence and refractory [1, 2]. Approximately 10% to 40% of younger patients and a larger percentage of patients aged 60 and above have refractory after standard chemotherapy (40%–60%) [3]. Therefore, identifying robust prognostic markers is crucial to provide optimal care for AML patients.
The Rho family of GTPases is a family of small (~ 21 kDa) signaling G proteins, which act as molecular switches and are tightly controlled by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs) by generating active GTP-binding and inactive GDP-binding proteins, respectively [4, 5]. Activated Rho GTPases interact with their downstream effectors to regulate the cytoskeleton of the cell membrane or other cellular compartments [6, 7]. Considerable research has supported the importance of Rho GTPases in hematopoiesis and confirmed that Rho GTPases are related to cytoskeleton rearrangement including adhesion, cytokinesis, differentiation, migration, engraftment, aging, and self-renewal in the cellular process [8,9,10,11,12,13,14]. The reorganization of the actin and microtubuleb cytoskeleton is essential for platelet adhesion and thrombus formation to avoid excessive bleeding.
A total of 80 Rho family GAPs have thus far been identified, but fewer than half of them in cancer have been clearly investigated. Rho GTPase activating protein 9 (ARHGAP9) containing RhoGAP, SH3, WW, and PH domains, is a member of the Rho GAPs family. Research indicates that ARHGAP9 suppresses the adhesion of KG-1 (a human leukemia cell line) to fibronectin and collagen via the activation of Cdc42 and Rac1 rather than RhoA [15]. ARHGAP9 is also considered as a MAP kinase docking protein, and the WW domain of ARHGAP9 interacts with the CD domains of Erk2 and p38alpha, leading to the inactivation of MAP kinases [16]. ARHGAP9 inhibits the migration and invasion of hepatocellular carcinoma cell by increasing FOXJ2/E-cadherin expression [17]. By contrast, silencing ARHGAP9 reduces the proliferation, migration, and invasion of breast and gastric cancer cells in vitro [18, 19]. These occurrences suggest that ARHGAP9 plays distinct roles in various physiological conditions or various tissues and cells.
No studies have thus far been conducted to access the expression profile and functions of ARHGAP9 in AML, although ARHGAP9 expression in peripheral blood leukocytes has been demonstrated [15]. In the present study, we investigated ARHGAP9 expression in human AML samples and cell lines, as well as explored its associations with clinicopathological factors. We then we evaluated the prognostic significance of ARHGAP9. We also investigated the differentially expressed genes associated with ARHGAP9 expression and discussed their potential functions in AML.
Materials and methods
Analysis of The Human Protein Atlas (HPA) database
The HPA (https://www.proteinatlas.org/) is user-friendly online server, which contains the human transcriptomic and proteomic data in cells, tissues, and organs from human normal or pathological tissues via RNA sequencing (RNA-Seq) analysis and immunohistochemistry (IHC) [20]. Thus, ARHGAP9 expression was analyzed in multiple cell lines, such as various leukemia cell lines, lung cell lines, breast cell lines, and brain cell lines in HPA by using the search term “ARHGAP9”.
The Cancer Cell Line Encyclopedia (CCLE) database analysis
The CCLE project (https://portals.broadinstitute.org/ccle) is an effort to conduct comprehensive genetic characterization of a large panel of human cancer cell lines from individuals of various lineages and ethnicities [21]. It provides public access to analysis and visualization of mRNA expression, mutation data, DNA methylation, DNA copy number, and histone H3 modification for > 1100 cancer cell lines, such as breast cell lines, gastric cancer cell lines, and AML cell lines. RNA-Seq of RNA expression was used to verify ARHGAP9 expression in various cell lines.
UALCAN database analysis
The UALCAN database (http://ualcan.path.uab.edu/) contains level-3 RNA-seq and clinical data from 31 cancer types selected by the Cancer Genome Atlas (TCGA) [22]. It is an interactive web resource for the in-depth analysis of RNA-Seq expression. The function module of TCGA analysis in the UALCAN database was used in the pan-cancer analysis of ARHGAP9 expression.
GEPIA database analysis
The GEPIA database (http://gepia.cancer-pku.cn) is a comprehensive resource for the systematic analysis of gene expression [23]. This database includes 9736 tumor and 8587 normal tissue samples from the TCGA and the Genotype-Tissue Expression (GTEx) projects. In the function module, single-gene analysis was used to create plots for ARHGAP9 expression in various cancers and normal tissues. The threshold was selected as the default value.
The Gene Expression Omnibus (GEO) database analysis
ARHGAP9 expression profiles in various chromosome abnormalities were acquired from the GEO (https://www.ncbi.nlm.nih.gov/geo) database. Data on ARHGAP9 expression of GSE14468 and GSE13159 were downloaded from the profile graph of the GEO2R online program in the GEO according to the gene ID. A total of 200 and 512 AML patients were from GSE14468 and GSE13159, respectively. The data were the raw counts provided by the submitter.
The Cancer Genome Atlas (TCGA) database analysis
We studied 151 AML patients with RNA-seq and clinical data from the TCGA (https://portal.gdc.cancer.gov) to analyze the significantly different genes, prognosis, and association between ARHGAP9 expression and different clinicopathologic features [24]. The patient IDs used in the present study are listed in the Additional file 1: Table S1. Specifically, 79 patients received only chemotherapy, whereas 67 patients underwent chemotherapy and auto/allo-HSCT. The main clinical and genetic characteristics of the AML patients are presented in Table 1 and Additional file 1: Table S2. RNA-seq and clinical data are available on the TCGA website. These patients were divided into two groups (ARHGAP9low and ARHGAP9high) based on the median values of the ARHGAP9 transcript by using the RNAseq data from TCGA. The Limma package in R 3.3.3 was used to screen significantly different genes between the ARHGAP9high and ARHGAP9low groups in AML. Adjusted P < 0.05 and |log1.2 FC|≥ 1 were used as cut-off values for identifying significantly different genes.
LinkedOmics database analysis
The co-expression genes in correlated with ARHGAP9 expression were analyzed using the LinkedOmics database (http://www.linkedomics.orglogin.php) [25]. The LinkedOmics database, including mRNA sequencing data from 173 AML patients from the TCGA database, was used to determine ARHGAP9 co-expression in AML. In the LinkFinder module of LinkedOmics, Pearson’s correlation coefficient was calculated to analyze the data. Volcano plots displaying the results were generated.
Overlapping genes between significantly different genes and co-expression genes were determined using the website Draw Venn Diagram (http://bioinformatics.psb.ugent.be/webtools/Venn/). The overlapping genes were used for the subsequent analysis, including ARHGAP9 functional enrichment and protein–protein interaction (PPI) analysis.
Functional enrichment and PPI analysis
Metascape (http://metascape.org/gp/index.html#/main/step1) is a free gene-list analysis tool for gene functional enrichment analysis [26]. The identified overlapping genes were inputted into the Metascape database for Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, tissues, and disease enrichment analysis.
We employed the String database (https://string-db.org/) to analyze the PPI network [27], which was visualized using the software Cytoscape_v3.6.1 [28]. Hub genes among the PPI were screened using the Cytoscope plugin MCODE with the following parameters: degree cutoff = 2, node score cutoff = 0.2, k-core = 2, and maximum depth = 100.
Statistical analysis
Data were Statistically analyzed using IBM SPSS 19.0.0. Pearson’s chi-square and Fisher’s exact tests were selected to compare the categorical variables, such as sex, French–American–British (FAB) classification, and cytogenetics classification. The number of samples was considerably less than 5000 in the two groups; thus, the Shapiro–Wilk test was used to explore whether the values in each group were normally distributed for the comparison of continuous variables. Two-sample Student’s t-test was used if the values in each group were normally distributed; otherwise, the Mann–Whitney U test was used. The continuous variables mainly included age, bone marrow (BM), peripheral blood (PB), white blood cell (WBC) in this study. Except GEPIA and LinkedOmics databases were used to evaluate overall survival (OS) of AML patients. The prognostic effect of ARHGAP9 expression on event-free survival (EFS) and OS were analyzed with the Log–rank and Gehan–Breslow–Wilcoxon test in GraphPad Prism 7.0.
Results
ARHGAP9 overexpression in AML cell lines
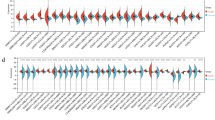
To elucidate the significance of ARHGAP9 expression in AML cells, we first analyzed ARHGAP9 expression based on the RNA-Seq data obtained from cell lines recorded in the CCLE and HPA databases. In the HPA database, the ARHGAP9 mRNA expression levels were higher in AML cell lines, such as HEL, HL60, NB4, and U937, than that in lymphoid cell lines; meanwhile, ARHGAP9 mRNA was almost not expressed in other cell lines representing the brain, breast, lung and so on (Fig. 1a). Moreover, ARHGAP9 showed the highest expression in AML cell lines in the CELL database (Fig. 1b).

ARHGAP9 expression in cell lines. a ARHGAP9 expression in leukemia cell lines, analyzed by HPA. b The expression of ARHGAP9 in leukemia Cell Lines, analyzed by CCLE
ARHGAP9 overexpression in AML
Using the UALCAN and GEPIA databases, we subsequently tested the mRNA expression of ARHGAP9 in different human tumor samples. The mRNA expression levels of ARHGAP9 was the highest among all types of human cancers (Fig. 2a, b). The transcriptional levels of ARHGAP9 in cancers were then compared with those in normal samples by using the GEPIA database. The results indicated that the expression level of ARHGAP9 was significantly upregulated in patients with AML (Fig. 2c). To further study whether ARHGAP9 expression was influenced by different chromosomal abnormalities, we retrieved two microarray data (GSE14468 and GSE13159) from the GEO database and evaluated ARHGAP9 expression among AML patients with major recurrent chromosomal translocations, including inv(16), t(8;21), t(15;17), 11q23, and complex, as well as the normal karyotype. Analysis results of both data sets showed that t(15;17) AML patients exhibited the lowest ARHGAP9 expression among the patients with cytogenetic abnormalities (Fig. 2d, e). These results for the AML samples corresponded with those in the cell lines.

ARHGAP9 expression in AML. a ARHGAP9 expression in pan-cancer, analyzed by UALCAN. b ARHGAP9 expression in various cancer tissues and normal tissues analyzed by GEPIA; The height of the bar represents the median expression of a certain tumor type or normal tissue. c ARHGAP9 expression in AML and normal tissues analyzed by GEPIA. d and e ARHGAP9 expression in AML with various chromosomal abnormalities in the microarray data of GSE14468 and GSE13159 from the GEO database, respectively
Relationships between ARHGAP9 and clinicopathological characteristics of patients with AML
In this study, the clinical and molecular characteristics of all patients from the TCGA database are summarized in Table 1. No significant differences in age, sex, BM blasts, WBC, and PB blasts (p > 0.05) were found between ARHGAP9low and ARHGAP9high groups (Table 1). Significant differences were determined in the distribution of FAB classification, cytogenetics, risk stratification, and gene mutations. High expression of ARHGAP9 was significantly correlated with FAB-M1 (p = 0.019), whereas low expression of ARHGAP9 was significantly correlated with FAB-M3 (p < 0.001), FAB-M4 (p = 0.011), and FAB-M5 (p = 0.005) in the distribution of FAB classifications. For cytogenetics, ARHGAP9 overexpression was particularly associated with cytogenetically normal AML (CN-AML) (p = 0.026), and low expression of ARHGAP9 was associated with the t(15;17) (p < 0.01) and complex (p = 0.041) subtypes. Moreover, ARHGAP9low cases tended to be associated with a good prognosis, whereas ARHGAP9high cases were obviously correlated with intermediate risk. Among the mutated genes, high expression of ARHGAP9 was only correlated with WT1 mutation (Additional file 1: Table S2).
Prognostic values of ARHGAP9 in AML
We investigated whether ARHGAP9 expression was associated with the prognosis of AML patients. The OS against ARHGAP9 expression was evaluated using the GEPIA and LinkedOmics databases. As shown on the GEPIA and LinkedOmics databases, high expression of ARHGAP9 was correlated with poor OS in AML (Fig. 3a, 3b). We further analyzed the survival data from the TCGA database and found ARHGAP9 overexpression was related to shorter event-free survival (EFS) (Fig. 3c), albeit statistical significance was not achieved.

Effect of ARHGAP9 on the survival of AML patients. a Prognostic value of ARHGAP9 mRNA level in AML, analyzed by GEPIA. b Prognostic value of ARHGAP9 mRNA level in AML, analyzed by LinkedOmics. c Survival curves of EFS in AML patients from the TCGA cohort
No significant difference in OS (Log-rank p = 0.8792) and EFS (p = 0.7039) was found between the ARHGAP9low and ARHGAP9high groups in CN-AML (Fig. 4a, b). To determine whether AML patients with high expression of ARHGAP9 could benefit from chemotherapy or auto/allo-HSCT, we divided the tested AML patients into two groups by their treatment regimens. Significantly shorter OS (Log-rank p = 0.0046) and EFS (Log-rank p = 0.0057) were found in AML patients who received with only chemotherapy (Fig. 4c, d). Among the patients who received auto/allo-HSCT, no significant differences in OS (Log-rank p = 0.6830) and EFS (Log-rank p = 0.7368) were found between the ARHGAP9low and ARHGAP9high groups (Fig. 4e, f). Patients undergoing auto/allo-HSCT had higher OS (p < 0.0001) and EFS (p = 0.0015) than those who received only chemotherapy (Fig. 4g, h). Acute promyelocytic leukemia (APL) (FAB M3) is a unique subtype of AML associated with peculiar clinical features and treatment strategies and the prognosis of APL is good. The prognostic values of ARHGAP9 expression in CN-AML, chemotherapy, and auto/allo-HSCT after the exclusion of FAB M3 from the AML cases were the same as those without the exclusion (Additional file 2: Figure S1), which indicated that the aforementioned prognosis results were not subject to the effect of FAB M3.

Prognostic values of ARHGAP9 with different factors on the survival of AML patients from the TCGA cohort. a OS and b EFS in CN-AML. c OS and d EFS of patients undergoing chemotherapy. e OS and f EFS of patients treated with auto/allo-HSCT. g OS and h EFS of patients in the ARHGAP9high group
Overall, ARHGAP9 expression is a poor factor for AML rather than CN-AML. Moreover, chemotherapy alone showed no prognostic influence on ARHGAP9high cases. And AML patients with high expression of ARHGAP9 could benefit from auto/allo-HSCT.
ARHGAP9-associated gene analysis between ARHGAP9high and ARHGAP9low in AML patients
To further explore the role of ARHGAP9 in AML, we firstly compared the transcriptomes of the ARHGAP9low and ARHGAP9high groups base on the TCGA database. A total of 2,948 genes were identified as significantly different between the ARHGAP9low and ARHGAP9high groups (p ≤ 0.05, |log1.2 FC|≥ 1, Additional file 1: Table S3); 1173 genes denoted by red circles and 1,775 genes denoted by green circles, were significantly upregulated and downregulated in the ARHGAP9 high group, respectively (Fig. 5a).

Genome-wide genes associated with ARHGAP9 expression. a Volcano plot of different gene-expression profiles between the ARHGAP9low and ARHGAP9high groups. b Volcano plots for the analysis of the co-expression genes associated with ARHGAP9 expression (LinkedOmics). c Overlapping genes between positively correlated genes and significantly increased genes. d Overlapping genes between negatively correlated genes and significantly reduced genes
Subsequently, we analyzed the co-expressed genes in conjunction with the ARHGAP9 genes by using the LinkedOmics database. As shown in Fig. 5b, a total of 3,310 genes represented by dark red dots were positively correlated with ARHGAP9 expression, and 4,268 genes represented by dark green dots were negatively correlated with ARHGAP9 in AML. Among the genes correlated with ARHGAP9 expression, 3,121 co-expression genes were significant correlations with ARHGAP9 in AML (False discovery rate, FDR ≤ 0.05, p ≤ 0.05, and |cor.|≥ 0.3; Additional file 1: Table S4).
Comparison of the significantly different genes and co-expressed genes led to the determination of 963 overlapping genes. These 963 genes contained 537 positively upregulated genes and 426 negatively downregulated genes (Fig. 5c, d and Additional file 1: Table S5). The overlapping genes were used for subsequent studies.
Functional analysis of the overlapping genes
We then sought to investigate the possible biological function of ARHGAP9 in patients with AML. The 963 genes were analyzed by using tools in Metascape to study KEGG and GO annotation. The top 20 clusters of enriched sets are shown in Fig. 6a. ARHGAP9 mediated the function of Rho GTPases, including the regulation of small GTPases mediated signal transduction, regulation of cell adhesion, actin cytoskeleton organization, cell cycle, microtubule cytoskeleton organization, organelle localization, phagocytosis and regulation of G2/M transition of mitotic cell cycle. Notably, seven clusters belonged to the immune system including leukocyte activation involved in the immune response, signaling by interleukins, the adaptive immune system, regulation of leukocyte-mediated immunity, regulation of myeloid leukocyte-mediated immunity, interferon signaling, and interferon signaling. ARHGAP9 expression was also associated with protein autophosphorylation, regulation of phosphatidylinositol 3-kinase signaling (PI3K), positive regulation of protein kinase activity, CXCR4 pathway, and hemostasis. In addition, the overlapping genes were enriched in blood, spleen, and bone marrow (Fig. 6b), further suggesting immunological function of ARHGAP9 in leukemogenesis. Moreover, among different diseases, the overlapping genes participated primarily in APL (Fig. 6c).

Enrichment of functions and signaling pathways of the overlapping genes in AML. a Analysis of GO and KEGG pathway associated with ARHGAP9 expression. b Enrichment of differentially expressed genes in tissues and cells. c Enrichment of overlapping genes in diseases
Validation of hub genes
The PPI network was constructed using the String website and the figures were generated by Cytoscape (MCODE plug-in). A total of 963 common genes were imported into the PPI network. We obtained 881 nodes and 5103 edges (Fig. 7a). The most significant module (MCODE score = 29.613) contained 32 genes. These genes are identified in Fig. 7b. Subsequently, OS analysis of the hub genes was performed using the GEPIA database. Of the 32 hub genes, 31 genes exerted no effect on OS in AML patients. Only KIF20A was significantly related to OS in AML (p = 0.02, Fig. 7c). KIF20A in the AML samples was significantly lower than that in normal tissues (Fig. 7d).

PPI network of the overlapping genes. a Network view of PPI for the overlapping genes. b MCODE analysis of the most significant interactions from a (MCODE score = 29.806); c Prognostic value of hub genes in AML, analyzed by GEPIA. d KIF20A expression in AML patients and healthy tissues, analyzed by GEPIA
Discussion
Rho GAPs inactivate Rho GTPases by the conservative GAP domain that promotes GTP hydrolysis and accelerates intrinsic GTPase activity, limiting the duration of the regulated reaction [29, 30]. Numerous reports suggest that the abnormal expression of Rho GAPs is observed in various tissues from patients with cancer and immunological diseases [31,32,33,34]. The expression of Rho GAPs is found to be heterogeneous among different tumors. For instance, ARHGAP30 was downregulated in lung cancer and colorectal cancer but overexpressed in pancreatic cancer [35,36,37]. Low expression of ARHGAP9 was found in hepatocellular carcinoma and bladder cancer, whereas high expression of ARHGAP9 was observed in breast cancer [17, 19, 38]. In the current study, ARHGAP9 is overexpressed in both AML samples and cell lines compared, relative to that in their normal counterparts [19]. Nonetheless, the low expression of ARHGAP9 was found in t(15; 17) AML.
Several studies investigating the role of Rho GAPs in cancer have found that most Rho GAPs were associated with good outcomes in various kinds of solid tumors [33, 36, 37, 39]. ARHGAP9 also showed a good prognosis in bladder cancer, hepatocellular carcinoma, and gastric cancer [17, 18, 38]. However, we found that, ARHGAP9 overexpression was associated with a poor prognosis in AML. These results suggest that ARHGAP9 can play different roles in various cancers. Meanwhile, DOCK2 belonging to the Rho GEFs family activates GTPase, which has a function contrary to that of Rho GAPs. DOCK2 was also an independent favorable prognostic factor for both EFS and OS in AML [40]. Thus, some regulators of Rho GTPase may have other functions in cancer, in addition to being associated with GTPase.
Despite the association of the high expression of ARHGAP9 with CN-AML, no relationship between ARHGAP9 expression and prognosis of CN-AML was found in the current study. In addition, several studies have shown that abnormal expression of some genes such as NCALD, IL2RA, and BCL2 are associated with prognosis in AML patients with auto/allo-HSCT and/or chemotherapy [41,42,43]. Our findings showed that ARHGAP9high groups had poor prognosis in post-chemotherapy AML patients, whereas no significant differences in OS and EFS were found between the ARHGAP9high group and ARHGAP9low group in patients who underwent after auto/allo-HSCT. These results suggest that the effects of ARHGAP9 over-expression can be eliminated by auto/allo-HSCT, instead of chemotherapy.
Rho GAPs, regulators of Rho GTPases, are expressed mainly in hematopoietic cells [8, 44,45,46]. Costa et al. showed that inadequate ARHGAP15 leads to enhanced chemotactic responses, straightened directional migration, amplified reactive oxygen species production, increased phagocytosis, as well as improved bacterial killing in neutrophils [44]. ARHGAP25 was found to negatively regulate leukocyte transendothelial migration in mice and phagocytosis in human neutrophilic granulocytes [45, 47]. Study in vitro and in vivo indicated that ARHGAP21 knockdown could impair the function of T cells, reduce erythroid commitment and differentiation, and enhance RhoC activity [12]. ARHGAP19, a hematopoietic-specific Rho GAP, affected the stiffness and shape of lymphocytes by regulating cytokinesis and chromosome segregation in T lymphocytes [8]. These combined findings suggest that Rho GAPs play an important role in hematopoietic cells and regulate cell motility, cell cycle, adhesion, phagocytosis, NADPH oxidase, auto/allo-HSCT development, inflammatory responses, and neutrophil chemotaxis. Consistent with the previously published reports, the present study showed that differentially expressed genes associated with ARHGAP9 expression were involved in microtubule cytoskeleton organization, actin cytoskeleton organization, phagocytosis, regulation of cell adhesion, cell cycle.
Although the other members of the Rho family GAPs were shown to be involved in several vital functions of neutrophils, our study showed that differentially expressed genes associated with ARHGAP19 expression were enriched in APL. Meanwhile, 95% of APL consisted of the abnormalities of t(15;17), which encodes the promyelocytic leukemia–retinoic acid receptor alpha (PML-RARA) fusion protein [48]. Moreover, ARHGAP9 expression was the lowest in t(15;17) AML among other chromosome abnormalities in AML, and all patients with t(15;17) were in the ARHGAP9low group in the current study. Therefore, ARHGAP9 expression may be suppressed by the PML-RARA fusion protein. The physiological role of ARHGAP9 in APL requires further investigation..
Upon immunization, Rho GAPs are critical for innate immunity and adaptive immunity [49,50,51]. Meanwhile, more than 11 members of Rho GAPs participate in various neutrophil functions that belong to adaptive immunity [52]. Given that the overlapping genes were mainly enriched in the immune system and immune tissues, we speculated that ARHGAP9 plays a significant role in the immune response in AML. The phosphorylation of Rho GTPase not only affects Rho GTPase activity by altering their conformation, but also regulate Rho GTPase ability to interact with the effector proteins, and thereby influencing their subcellular localization [53,54,55]. This finding would explain the enrichment of some overlapping genes in protein autophosphorylation, positive regulation of protein kinase activity, regulation of PI3K, and organelle localization, which displayed the activity of ARHGAP9 may be regulated by the phosphorylation in the AML.
Conclusions
High expression of ARHGAP9 was found in AML tissues and cells, and elevated ARHGAP9 was significantly correlated with poor outcome in AML. Auto/allo-HSCT, rather than chemotherapy, can overcome the adverse outcomes related to high ARHGAP9 expression. Functional analysis of differentlially expressed genes between those exhibiting high and low expression of ARHGAP9 indicated that ARHGAP9 could perform multiple functions in AML. The physiological role of ARHGAP9 in AML requires further study. Moreover, ARHGAP9 was downregulated in the t(15;17) patients, and differentially expressed genes associated with ARHGAP9 expression were enriched in APL. PML-RARA may act as negative regulators of ARHGAP9 expression in AML.
Availability of data and materials
The datasets used and/or analyzed during the current study are available. Please contact the author to get the datasets.
Abbreviations
- ARHGAP9:
-
Rho GTPase activating protein 9
- AML:
-
Acute myeloid leukemia
- APL:
-
Acute promyelocytic leukemia
- CN-AML:
-
Cytogenetic normal acute myeloid leukemia
- TCGA:
-
The Cancer Genome Atlas
- EFS:
-
Event-free survival
- OS:
-
Overall survival
- FAB:
-
French-American-British subtypes
- BM-blast:
-
Bone marrow blast
- PB-blast:
-
Peripheral blood blast
- WBC:
-
White blood cell
- allo-HSCT:
-
Allogeneic hematopoietic stem cell transplantation
- RNA-Seq:
-
RNA sequencing
- GEO:
-
Gene Expression Omnibus
- CCLE:
-
Cancer cell cine encyclopedia
- HPA:
-
Human Protein Atlas
- PPI:
-
Protein–protein interaction
- GEFs:
-
Guanine nucleotide exchange factors
- GAPs:
-
GTPase activating proteins
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- n:
-
Number of patients
- FAB:
-
French–American–British subtypes
- BM-blast:
-
Bone marrow blast
- PB-blast:
-
Peripheral blood blast
- WBC:
-
White blood cell
- WBC:
-
Acute promyelocytic leukemia
- PML-RARA:
-
Promyelocytic leukemia–retinoic acid receptor alpha
- PI3K:
-
Phosphatidylinositol 3-kinase signaling
References
Burnett A, Stone R. AML: New Drugs but New Challenges. Clin Lymphoma Myeloma Leuk. 2020;20:341–50.
Thol F, Ganser A. Treatment of Relapsed Acute Myeloid Leukemia. Curr Treat Options Oncol. 2020;21:66.
Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373:1136–52.
Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701.
Humphries BA, Wang Z, Yang C: MicroRNA Regulation of the Small Rho GTPase Regulators-Complexities and Opportunities in Targeting Cancer Metastasis. Cancers (Basel) 2020, 12.
Buchsbaum RJ. Rho activation at a glance. J Cell Sci. 2007;120:1149–52.
Schwartz M. Rho signalling at a glance. J Cell Sci. 2004;117:5457–8.
David MD, Petit D, Bertoglio J. The RhoGAP ARHGAP19 controls cytokinesis and chromosome segregation in T lymphocytes. J Cell Sci. 2014;127:400–10.
Florian MC, Dörr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, Filippi MD, Hasenberg A, Gunzer M, Scharffetter-Kochanek K, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell. 2012;10:520–30.
Mulloy JC, Cancelas JA, Filippi MD, Kalfa TA, Guo F, Zheng Y. Rho GTPases in hematopoiesis and hemopathies. Blood. 2010;115:936–47.
Wang L, Yang L, Filippi MD, Williams DA, Zheng Y. Genetic deletion of Cdc42GAP reveals a role of Cdc42 in erythropoiesis and hematopoietic stem/progenitor cell survival, adhesion, and engraftment. Blood. 2006;107:98–105.
Xavier-Ferrucio J, Ricon L, Vieira K, Longhini AL, Lazarini M, Bigarella CL, Franchi G Jr, Krause DS, Saad STO. Hematopoietic defects in response to reduced Arhgap21. Stem Cell Res. 2018;26:17–27.
Yang FC, Atkinson SJ, Gu Y, Borneo JB, Roberts AW, Zheng Y, Pennington J, Williams DA. Rac and Cdc42 GTPases control hematopoietic stem cell shape, adhesion, migration, and mobilization. Proc Natl Acad Sci USA. 2001;98:5614–8.
Yang L, Wang L, Geiger H, Cancelas JA, Mo J, Zheng Y. Rho GTPase Cdc42 coordinates hematopoietic stem cell quiescence and niche interaction in the bone marrow. Proc Natl Acad Sci USA. 2007;104:5091–6.
Furukawa Y, Kawasoe T, Daigo Y, Nishiwaki T, Ishiguro H, Takahashi M, Kitayama J, Nakamura Y. Isolation of a novel human gene, ARHGAP9, encoding a rho-GTPase activating protein. Biochem Biophys Res Commun. 2001;284:643–9.
Ang BK, Lim CY, Koh SS, Sivakumar N, Taib S, Lim KB, Ahmed S, Rajagopal G, Ong SH. ArhGAP9, a novel MAP kinase docking protein, inhibits Erk and p38 activation through WW domain binding. J Mol Signal. 2007;2:1.
Zhang H, Tang QF, Sun MY, Zhang CY, Zhu JY, Shen YL, Zhao B, Shao ZY, Zhang LJ, Zhang H. ARHGAP9 suppresses the migration and invasion of hepatocellular carcinoma cells through up-regulating FOXJ2/E-cadherin. Cell Death Dis. 2018;9:916.
Sun L, Zhang Y, Lou J. ARHGAP9 siRNA inhibits gastric cancer cell proliferation and EMT via inactivating Akt, p38 signaling and inhibiting MMP2 and MMP9. Int J Clin Exp Pathol. 2017;10:11979–85.
Wang T, Ha M. Silencing ARHGAP9 correlates with the risk of breast cancer and inhibits the proliferation, migration, and invasion of breast cancer. J Cell Biochem. 2018;119:7747–56.
Thul PJ, Lindskog C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018;27:233–44.
Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7.
Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, Varambally S. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19:649–58.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–102.
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, Baty JD, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018;46:D956-d963.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447-452.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Ligeti E, Welti S, Scheffzek K. Inhibition and termination of physiological responses by GTPase activating proteins. Physiol Rev. 2012;92:237–72.
Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13–22.
Chen WX, Lou M, Cheng L, Qian Q, Xu LY, Sun L, Zhu YL, Dai H. Bioinformatics analysis of potential therapeutic targets among ARHGAP genes in breast cancer. Oncol Lett. 2019;18:6017–25.
Citi S, Guerrera D, Spadaro D, Shah J. Epithelial junctions and Rho family GTPases: the zonular signalosome. Small GTPases. 2014;5:1–15.
Liu L, Xie D, Xie H, Huang W, Zhang J, Jin W, Jiang W, Xie D. ARHGAP10 Inhibits the Proliferation and Metastasis of CRC Cells via Blocking the Activity of RhoA/AKT Signaling Pathway. Onco Targets Ther. 2019;12:11507–16.
Németh T, Futosi K, Hably C, Brouns MR, Jakob SM, Kovács M, Kertész Z, Walzog B, Settleman J, Mócsai A. Neutrophil functions and autoimmune arthritis in the absence of p190RhoGAP: generation and analysis of a novel null mutation in mice. J Immunol. 2010;185:3064–75.
Mao X, Tong J. ARHGAP30 suppressed lung cancer cell proliferation, migration, and invasion through inhibition of the Wnt/β-catenin signaling pathway. Onco Targets Ther. 2018;11:7447–57.
Wang J, Qian J, Hu Y, Kong X, Chen H, Shi Q, Jiang L, Wu C, Zou W, Chen Y, et al. ArhGAP30 promotes p53 acetylation and function in colorectal cancer. Nat Commun. 2014;5:4735.
Zhou Y, Hua Z, Zhu Y, Wang L, Chen F, Shan T, Zhou Y, Dai T. Upregulation of ARHGAP30 attenuates pancreatic cancer progression by inactivating the β-catenin pathway. Cancer Cell Int. 2020;20:225.
Piao XM, Jeong P, Yan C, Kim YH, Byun YJ, Xu Y, Kang HW, Seo SP, Kim WT, Lee JY, et al. A novel tumor suppressing gene, ARHGAP9, is an independent prognostic biomarker for bladder cancer. Oncol Lett. 2020;19:476–86.
Li JP, Liu Y, Yin YH. ARHGAP1 overexpression inhibits proliferation, migration and invasion of C-33A and SiHa cell lines. Onco Targets Ther. 2017;10:691–701.
Hu N, Pang Y, Zhao H, Si C, Ding H, Chen L, Wang C, Qin T, Li Q, Han Y, et al. High expression of DOCK2 indicates good prognosis in acute myeloid leukemia. J Cancer. 2019;10:6088–94.
Du W, He J, Zhou W, Shu S, Li J, Liu W, Deng Y, Lu C, Lin S, Ma Y, et al. High IL2RA mRNA expression is an independent adverse prognostic biomarker in core binding factor and intermediate-risk acute myeloid leukemia. J Transl Med. 2019;17:191.
Song Y, Zhang W, He X, Liu X, Yang P, Wang J, Hu K, Liu W, Zhang X, Jing H, Yuan X. High NCALD expression predicts poor prognosis of cytogenetic normal acute myeloid leukemia. J Transl Med. 2019;17:166.
Zhou JD, Zhang TJ, Xu ZJ, Gu Y, Ma JC, Li XX, Guo H, Wen XM, Zhang W, Yang L, et al. BCL2 overexpression: clinical implication and biological insights in acute myeloid leukemia. Diagn Pathol. 2019;14:68.
Costa C, Germena G, Martin-Conte EL, Molineris I, Bosco E, Marengo S, Azzolino O, Altruda F, Ranieri VM, Hirsch E. The RacGAP ArhGAP15 is a master negative regulator of neutrophil functions. Blood. 2011;118:1099–108.
Csépányi-Kömi R, Sirokmány G, Geiszt M, Ligeti E. ARHGAP25, a novel Rac GTPase-activating protein, regulates phagocytosis in human neutrophilic granulocytes. Blood. 2012;119:573–82.
Wang LD, Ficarro SB, Hutchinson JN, Csepanyi-Komi R, Nguyen PT, Wisniewski E, Sullivan J, Hofmann O, Ligeti E, Marto JA, Wagers AJ. Phosphoproteomic profiling of mouse primary HSPCs reveals new regulators of HSPC mobilization. Blood. 2016;128:1465–74.
Csépányi-Kömi R, Wisniewski É, Bartos B, Lévai P, Németh T, Balázs B, Kurz AR, Bierschenk S, Sperandio M, Ligeti E. Rac GTPase Activating Protein ARHGAP25 Regulates Leukocyte Transendothelial Migration in Mice. J Immunol. 2016;197:2807–15.
Jimenez JJ, Chale RS, Abad AC, Schally AV. Acute promyelocytic leukemia (APL): a review of the literature. Oncotarget. 2020;11:992–1003.
Biro M, Munoz MA, Weninger W. Targeting Rho-GTPases in immune cell migration and inflammation. Br J Pharmacol. 2014;171:5491–506.
Pernis AB. Rho GTPase-mediated pathways in mature CD4+ T cells. Autoimmun Rev. 2009;8:199–203.
van Helden SF, Anthony EC, Dee R, Hordijk PL. Rho GTPase expression in human myeloid cells. PLoS ONE. 2012;7:e42563.
Csépányi-Kömi R, Pásztor M, Bartos B, Ligeti E. The neglected terminators: Rho family GAPs in neutrophils. Eur J Clin Invest. 2018;48(Suppl 2):e12993.
Marceaux C, Petit D, Bertoglio J, David MD. Phosphorylation of ARHGAP19 by CDK1 and ROCK regulates its subcellular localization and function during mitosis. J Cell Sci. 2018;131:jcs208397.
Ouadda A, He Y, Calabrese V, Ishii H, Chidiac R, Gratton JP, Roux PP, Lamarche-Vane N. CdGAP/ARHGAP31 is regulated by RSK phosphorylation and binding to 14–3–3β adaptor protein. Oncotarget. 2018;9:11646–64.
Luo W, Janoštiak R, Tolde O, Ryzhova LM, Koudelková L, Dibus M, Brábek J, Hanks SK, Rosel D. ARHGAP42 is activated by Src-mediated tyrosine phosphorylation to promote cell motility. J Cell Sci. 2017;130:2382–93.
Acknowledgements
We would like to thank the CCLE, GEPIA, GEO, HPA, LinkedOmics, TCGA, UALCAN, databases for open access to the AML sequencing datasets.
Funding
This work was supported by grants “Major New Drug Development Project” from Ministry of Science and Technology of China (2019ZX09201-002-003), State Key Program of National Natural Science of China (82030076), National Natural Science Foundation of China (82070161, 81970151, and 81870134), Beijing Natural Science Foundation (7202186), Natural Science Foundation of Shenzhen University General Hospital (SUGH2020QD008).
Author information
Authors and Affiliations
Contributions
CH wrote the manuscript and prepared tables and figures. SH, RW, and WH analyzed the data. QQ and XM performed statistical analyses. XG revised the manuscript critically. LY and YL contributed to study design and editing. All authors contributed to the article. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The written informed consent of all patients in this study was consistent with the Helsinki Declaration.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Additional tables.
Additional file 2:
Figure S1. The prognostic values of ARHGAP9 with different factors on the survival of AML patients without FAB M3 from the TCGA cohort. a OS and b EFS of CN-AML without FAB M3. c OS and d EFS in AML patients undergoing chemotherapy without FAB M3. e OS and f EFS in patients treating with auto/allo-HSCT without FAB M3. g OS and h EFS of patients without FAB M3 in ARHGAP9high group.OCX 178 KB).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Han, C., He, S., Wang, R. et al. The role of ARHGAP9: clinical implication and potential function in acute myeloid leukemia. J Transl Med 19, 65 (2021). https://doi.org/10.1186/s12967-021-02733-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-021-02733-5