
Abstract
The concept of cancer as a cell-autonomous disease has been challenged by the wealth of knowledge gathered in the past decades on the importance of tumor microenvironment (TM) in cancer progression and metastasis. The significance of endothelial cells (ECs) in this scenario was initially attributed to their role in vasculogenesis and angiogenesis that is critical for tumor initiation and growth. Nevertheless, the identification of endothelial-derived angiocrine factors illustrated an alternative non-angiogenic function of ECs contributing to both physiological and pathological tissue development. Gene expression profiling studies have demonstrated distinctive expression patterns in tumor-associated endothelial cells that imply a bilateral crosstalk between tumor and its endothelium. Recently, some of the molecular determinants of this reciprocal interaction have been identified which are considered as potential targets for developing novel anti-angiocrine therapeutic strategies.
Similar content being viewed by others

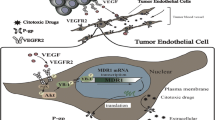
Introduction
Metastatic disease accounts for about 90% of patient mortality. The difficulty in controlling and eradicating metastasis might be related to the heterotypic interaction of tumor and its microenvironment [1]. The role of the tumor type specific microenvironment (TM) has been widely described in tumor initiation, maintenance and progression [2,3,4]. However effective targeting of TM in cancer therapy is not fully appreciated.
The vascular system comprises a vast network of capillary endothelial cells (ECs) that interconnect arteries and veins and create heterogenous vascular beds throughout body. The formation of these vascular networks and their interaction with the surrounding cells is the prerequisite of organ development and tissue regeneration [5, 6]. Similarly, neo-angiogenesis is considered as a critical process for establishing tumor vasculature that ensures tumor survival and expansion. Folkman originally proposed the term “angiogenic switch” to imply the essential role of blood vessels in initiation of tumor growth and stated that in the absence of such angiogenesis, tumors cannot expand their mass or display a metastatic phenotype [7]. Based on this theory, many investigators assumed tumor endothelium as the ultimate target for tumor therapy through prevention of neo-vascularization. However, strategies aimed at targeting tumor angiogenesis have mostly failed at clinical trial stage [8], indicating an alternative function for ECs in regulation of tumor progression.
The contribution of tumor endothelium to many hallmarks of cancer has already been reviewed by Hanahan and Coussens [2] and is beyond the scope of this review. In summary, the ECs infer a proliferative advantage to cancer cells in many ways such as neo-angiogenesis and provision of blood supply and nutrients, production of paracrine factors, metabolic regulation, modulation of tumor invasiveness, and resisting cellular apoptosis [5, 9,10,11,12,13]. This review will describe the emerging role of endothelial transmembrane and secretory growth factors and trophogens, referred to as angiocrine factors, as essential players in physiological processes and tumor growth [5]. We will describe the concept of angiocrine switch referring to the secreted and membrane bound endothelial factors that participate to the cross talk with cancer cells in association with their role in nutrients and oxygen delivery (angiogenic switch). We will review the data demonstrating that endothelial angiocrine repertoire is heterogenous and organ or tumor type specific. Our aim is to describe the ECs’ angiocrine role in developmental processes and illustrate how these mechanisms are hijacked by tumor cells. We will review the role of endothelial contexture in drug resistance, immune response, and tumor metabolic regulation. Finally, we will discuss new therapeutic approaches disrupting angiocrine endothelium.
Normal versus tumor endothelium: is there any angiocrine switch?
The importance of angiogenesis in initiation and development of tumors was postulated by Folkman in the context of an angiogenic switch with neovascularization [14]. While tumor vessels displayed an abnormal structure, the existence of a tumor specific endothelium compared to normal endothelium was revealed by gene expression alterations in tumor-associated ECs [15,16,17,18,19]. In addition to genetic alterations, inflammation and hypoxia, in the vicinity of ECs trigger the production of endothelial cytokines promoting tissue repair and tumor growth [20, 21]. Tumor endothelial cells usually display an activated state (discussed below) associated to a change of membrane bound and secretory elements referred here as angiocrine switch. Through the angiocrine switch that occurs early in tumor development, ECs establish a pro-tumoral niche that serves an alternative duty than the previously described angiogenic switch. Hence, the activated tumor ECs play a distinctive role in enhancement of tumorigenicity independent of its function as oxygen and nutrient transporters.
In a recent review by Friedl and Alexander, a model has been proposed in which tumor and its microenvironment induces reciprocal changes in their phenotypes and functions (spatiotemporal plasticity) by processing signals they receive from their environment (reciprocity) [22,23,24,25]. The interactions occurring in TM are similar to normal tissue development and remodeling processes and mediated by soluble factors or cell-to-cell contacts [3, 4, 26, 27] (Fig. 1). For instance, tumor-derived inflammatory cytokines stimulate the expression of notch ligands Jagged1 and Jagged2 on ECs [28] resulting in notch-dependent tumor growth [29]. In the leukemia model, cancer cell-secreted factors such as VEGF activate ECs in a paracrine fashion to support leukemia expansion by releasing leukemic cell trophogens or by increasing leukemic cell survival through HSP90-mediated induction of Bcl-2 expression and apoptosis inhibition [30,31,32]. In this model the activation of ECs by neighboring leukemia cells also triggers production of stem cell promoting angiocrine factors such as epidermal growth factors (EGFs) and fibroblast growth factors (FGFs) [33] enriching leukemia stem cells [34]. Similar findings by Cao et al. [35] showed participation of lymphoma-derived FGF4 in priming of a Jagged1 vascular niche that reciprocally reinforced lymphoma propagation and chemoresistance. Jagged1-dependent tumor cells stimulated a transient mesenchymal shift in a subset of tumor ECs in a notch and TGFβ-mediated manner [15]. The mesenchymal ECs (ECsMes) maintain their endothelial identity while acquiring improved angiocrine and angiogenic properties. Although different from endothelial-to-mesenchymal transition (EndMT) phenomenon implicated in the enrichment of cancer-associated fibroblasts (CAFs) [36], this might potentially represent selective activation of tumor endothelium leading to the generation of a pro-tumoral angiocrine endothelium.

The angiocrine and angiogenic switch during tumor progression. Vascular endothelial cells are capable of enhancing tumor growth and migration by secreting several cytokines and growth factors or activating the so-called angiocrine switch (bottom). This switch is activated by signals directed to ECs from their surrounding tumor cells. As a result, the activated ECs increase the production of angiocrine factors that confer many growth advantages to tumor cells. The angiocrine factors may be in the form of transmembrane ligands, secretory cytokines, or microparticle-packaged factors. There is evidence that activated ECs trough their interaction with tumor cells demonstrate some degree of plasticity toward a mesenchymal phenotype with enhanced tumor promoting potential
EC angiocrine role in organ development versus tumor progression
In addition to the primary role of ECs in vasculogenesis and angiogenesis, accumulating evidence shows that ECs contribute to complex patterning occurring during organogenesis [7]. The participation of vascular ECs in organ formation and regeneration has been described in a recent review by our group [6]. The involvement of angiocrine factors in many physiological body processes and their role in stem and progenitor cells regulation suggested their potential involvement in tumor progression and phenotypic plasticity [37,38,39,40,41,42] (Fig. 2). Here we provide an overview of the ECs angiocrine role in organ development in light of the angiocrine role in tumor progression mediated by secretion/expression of endothelial-specific angiocrine factors described above (Table 1).

The role of angiocrine endothelium in promoting tumorigenicity. ECs were initially known for their role in tumor angiogenesis. Emerging evidence shows an angiocrine role for tumor endothelium in inducing tumor growth and survival advantages through production of angiocrine factors. Several angiocrine factors such as VEGF, bFGF, IL-6, IL-8, TGFβ, PDGFβ, Jag1, and Endothelin, have been shown to enhance tumor cell proliferation. Also, a role for angiocrine factors is implicated in increased proliferation of cancer stem and progenitor cells in brain tumor (BDNF, PEDF, BMP2, and BMP4), head and neck cancer (EGF), and breast cancer (Jag1). Recent evidence demonstrates the participation of angiocrine endothelium in tumor immune tolerance. Angiocrine molecules such as ICAM-1, VCAM, and Selectin regulate the transmigration of lymphocytes through the lymphatic vessels. Endothelial bFGF plays a role in stimulation of leukocyte infiltration. Some secretory chemokine (CCL2, CCL3, CCL4, CCL5, CXCL9, and CXCL10) affect T cell infiltration to the tumor site. ECs are also capable of over-expressing specific molecules such as ETBR to provide a barrier for tumor-infiltrating lymphocytes. Additional mechanisms include escape from immune response regulated by Tim-3 or FasL. A recent angiocrine function for endothelial-derived thrombospondin-1 is defined in induction of tumor dormancy. Besides, endothelial FGF2 is capable of triggering resistance of anti-angiogenic drugs through activation of FGF2/FGFR1 loop. Quite notably, some angiocrine elements may have multiple functions in tumor expansion. Table 1 provides distinct functional information for the viewers
Brain
Endothelial cells from dorsal aorta secrete BMPs inducing mesenchymal SDF1 and neuregulin-1 expression, resulting in the attraction of sympatho-adrenal progenitors [43]. BMP signaling from ECs also governs progenitor’s segregation through neuregulin-ErbB signaling, forming the adrenal medulla and sympathetic ganglions. Brain capillaries’ ECs activate quiescent Neural stem cells (NSC) through jagged-1 and EphrinB2 pathways [6]. When activated, NSCs proliferates under the influence of pigment epithelium-derived factor (PEDF) [44], VEGF-C [45], SDF-1 [46] and placental growth factor-2 (PIGF-2) [47] secreted by ECs METTRE LES REF A LA FIN DE LA PHRASE POUR LECTURE PLUS FACILE. Finally, the differentiation of NSCs into neuroblast occurs under the influence of ECs through BDNF [48]. Cleaver et al. [49] have shown a reciprocal paracrine communication between ECs and the neighboring astrocytes mediated by VEGF as an essential factor in tissue differentiation and organ formation. Additional evidence including organization of peripheral nerves along the vascular system, endothelial secretion of neurogenic growth factors like artemin and neurotrophin-3, and neuronal secretion of endothelial stimulating factors illustrate the neuronal-endothelial cross-talk [50, 51]. ECs angiocrine factor have also a role in the regulation of brain stem/progenitor cells physiology. They induce quiescence through activation of BMP/Smad signaling instructing the neural stem cells to exit the cell cycle in the absence of differentiation. They maintain dormancy through EphrinB2 and Jagged-1 expression or stimulation of NO secretion by production of neurotrophin-3 [52,53,54,55,56]. EC in the neurogenic regions of the adult brain stimulate the self-renewal of neurons through PEDF signaling in the adult neural stem cell niche [57, 58]. Endothelial-associated increase in neuronal migration and survival has been attributed to BDNF since blocking this endothelial stimulating factor resulted in complete abrogation of ECs neurotrophic effects [48]. Graded deposition of BDNF and stromal-cell derived factor 1 (SDF-1) by ECs in the mouse brain induce proliferation and migration of brain cells to the olfactory bulb [59].
Brain tumors such as glioblastoma are highly aggressive tumors relying on important angiogenesis. However clinical trials assessing anti-angiogenic therapies such as bevacizumab or sorafenib did not result in the expected results and anti-angiogenic therapies are only considered as second line options. Multiple studies in the literature demonstrate how cancer cells use similar angiocrine cues that during development. Jagged1 ligand through notch activation increases neuroblastoma cells proliferation [60]. In glioblastoma, ECs are able to create a stem cell niche providing NOTCH ligands [61, 62], jagged-1 being essential for the glioma stem cell maintenance and self-renewal [63, 64]. Hence jagged-1 overexpression is linked to poor outcome in [65,66,67]. EphrinB1 is able to promote oncogenic signaling in medulloblastoma [68]. EphrinB2 expression is associated to the development of glioma and to glioblastoma patient’s prognosis [69]. Endothelial secretion of neuronal growth factors such as VEGF-C, BDNF, BMPs, and PEDF are implicated in brain tumor stem cell enrichment. Neurotrophin signaling through TrkB and TrkC receptors can stimulate brain tumor-initiating cell proliferation [70]. BMP/SMAD pathway is crucial in the development of childhood medulloblastoma [71]. In medulloblastoma, Placental growth factor is responsible for tumor growth and metastasis [72]. Finally the SDF-1/CXCR4 pathway have been widely study in the context of brain tumor (see [73] for review) and is now considered as a potential therapeutic target [74].
Liver
The development of the hepatic endoderm is under ECs control though Wnt and Notch signaling. ECs provide critical signals to hepatocytes leading to the establishment of the sinusoidal liver architecture [75] concomittent to biliary differentiation and morphogenesis regulated ECs derived Notch pathway [76]. ECs regulation of liver formation and growth is also mediated through bone morphogenetic protein 4 (BMP-4), fibroblast growth factor 8 (FGF8), and hepatocyte growth factor (HGF) [77,78,79]. The liver is an organ displaying important plasticity and regenerative ability, Ding et al. demonstrated that a subgroup of liver sinusoidal ECs were able to induce liver regeneration through HGF, Wnt2, and heparin-binding EGF (HB-EGF) activation regardless of liver perfusion [78, 80]. Most importantly EC do play a major role in the balance between fibrosis and regeneration. Fibrosis-free recovery requires CXCR7activation and up-regulation of apelin and follistatin-like-1 angiocrine factors. In contrast chronic liver injury results in activation of CXCR4 and a shift to secretion of angiocrine pro-fibrotic TGFβ1 and BMP2 [81].
In liver cancers (primary hepatocellular carcinoma, cholangiocarcinoma, and fibrolamellar hepatocellular carcinoma) ECs promotes tumor cells through Wnt/β-catenin and Notch pathways activation [82,83,84,85]. For example, Notch3 and Notch4 are highly expressed in primary liver cancer while they rarely present in regular liver [86]. Notch4 expression is limited to vascular ECs [87] hence Targeting Notch (especially 3 and 4) signaling could be an anti-angiocrine strategy in liver cancer [88, 89]. ECs derived BMP-4 induce proliferation [90], migration [91] and metastasis [92] in hepatocellular carcinoma and his overexpression is associated with poor prognosis [93]. Secreted FGF8 supports hepatocellular malignancy development and progression during neoangiogenesis [94]. In contact with ECs, cancer cells are stimulated by HGF and overexpress cell adhesion molecules, such as the α2β1 integrin, facilitating migration [95]. CXCR7 and CXCR4 essential for liver regeneration are regulating invasion and tumor growth of human hepatocellular carcinoma cells [96, 97].
Lung
The interaction between ECs and epithelial lung cells is essential for lung physiology [98]. ECs secretion of VEGF pathways are primordial in lung morphogenesis and cell proliferation [99]. For example, VEGF and FGF signaling increase metalloproteinase 14 (MMP14) secretion leading to EGF activation in alveolar epithelial cells and bronchioalveolar stem cells inducing their expansion as well as the proliferation of alveolar epithelium [100, 101]. During lung stem cell differentiation, BMP4 production triggers calcineurin/NFATc1-dependent TSP1 expression in ECs, promoting alveolar differentiation and repair [102]. The bilateral crosstalk between pulmonary ECs and lung progenitor cells derives the differentiation of lung epithelial stem and progenitor cells. VEGF, TGF-β1, FGF-2, and PDGF-B secreted by ECs are involved in development and progression of human small cell lung cancer progression and angiogenesis [103, 104]. Consequently, high levels of VEGF and low levels of TSP-1 are associated with a poor prognosis in non-small cell lung cancer [105]. MMP-14 increased expression is associated with poor prognosis in non-small cell lung cancer by increasing metastatic spread [106].
Pancreas
Endothelial cells induced endocrine pancreatic differentiation. ECs derived Sphingosine-1-phosphate (S1P) induces budding of pancreatic endoderm which is associated to VEGF pathway activation responsible for the differentiation of insulin-expressing cells [107, 108]. ECs’ laminins interact with β1-integrin and induce both insulin expression and proliferation of beta cells [109]. Kao et al. [110] also demonstrated that ECs play a stage-dependent role in pancreatic development by maintaining pancreatic progenitor cells self-renewal while impairing their differentiation into hormone-expressing cells through secretion of Epidermal growth factor-like domain 7 (EGFL7). Blood-free aortic ECs grown IS1-1-positive mesenchymal cells in vitro were able to selectively influence their differentiation to pancreatic cells thus illustrating the participation of ECs in pancreatic ectoderm differentiation independent of circulating factors [111].
In pancreatic cancer, Sp1 overexpression regulates VEGF expression and angiogenesis [112, 113]. Blockade of Sp1 has demonstrated a significant antitumor activity in pancreatic cancer [114]. Laminins induce pro-survival signal [115]. EGFL7 induce cell invasion and angiogenesis in pancreatic carcinoma and its expression is associated to poor prognosis [116, 117]. The role and expression of VEGF in pancreatic tumor have been widely studied (see [118] for review). VEGF is involved in metastasis process and its expression is associated to higher recurrence rate and poor prognosis [119,120,121].
Kidney
The development of kidney glomeruli is under the close control of ECs. Endothelial VEGF coordinates renal glomerulogenesis and tubulogenesis [122]. While podocytes recruit ECs through VEGF expression and release semaphorins (Sema 3a and Sema 3c) governing kidney vascular morphogenesis [123, 124]. ECs in return secrete MMP2 [125] and platelet-derived growth B (PDGF-B) leading to NNNNNNNNN < [125, 126]. ECs play also a role in renal function through the secretion of glomerular basement membrane formed by extracellular matrix proteins such as Laminin-α5β2γ1 (LM521) forming the glomerular basement membrane [127].
In tumor context, VEGF plays a primordial role in tumor angiogenesis [128]. PDGF-B and VEGF both promotes angiogenesis and cell proliferation in both primary and metastatic clear cell renal cell cancer [129]. MMP2 is involved in invasion and metastasis of clear cell renal cell carcinoma [130, 131]. Laminin-α4 is highly expressed in tumor blood vessels of renal cell carcinoma and is also associated with a poor prognosis [132].
Breast
The mammary gland consist of branched tubular structures resulting from the remodeling of epithelial or endothelial sheaths into multicellular tubular networks [133]. Epithelial-to-mesenchymal transition (EMT) is fundamental in breast morphogenesis [134]. ECs contribute to regulation of branching epithelial morphogenesis [135]. The specific pathways implicated in branching morphogenesis and EMT in the breast gland by ECs have not been yet clearly described.
In breast cancer ECs play several roles that have been widely studied from the development of primary tumors to metastatic evolution. ECs increase cancer stem (or initiating) cell (CSC) through contact mediated Jagged1/notch activation [136]. The CSCs participate in cancer progression and recurrence by their increased dormancy, chemoresistance and tumor-initiation potency [137, 138]. In addition, angiocrine-dependent CSCs may contribute to several tumor hallmark capabilities including apoptosis resistance, drug resistance, EMT, and invasion/metastasis [136, 139,140,141]. We have also gathered evidence on the EC contact- and microparticle-mediated regulation of breast tumor metastasis by showing the up-regulation of EMT markers CA SERAIT BIEN D EN CITER UN PEU in cancer cells once they were pre-exposed to ECs [142]. Our team also demonstrated that the uptake of mitochondria by breast cancer cells from ECs increase their chemoresistance through NNNNNN [143]. Recently Mina Bissel group (A VERIFIER) showed that the secretion of endothelial-derived growth factors influence tumor dormancy [144]. In the context of distant metastasis in the endothelial niche, thrombospondin-1 was shown to induce sustained tumor quiescence, thereby inhibiting tumor expansion and spreading. This suppressive state was inactivated in sprouting neo-vasculature by endothelial-derived tumor-promoting factors such as TGFβ1 and periostin.
Ovary
Of all the different organs, the ovary is the organ with the strongest physiological angiogenesis because of its cyclic corpus luteum [145]. Notch based interactions of ovarian cortical and medullar cells with ECs are primordial for ovarian morphogenesis and angiogenesis [146]. ECs in the ovary express Jagged 1, Dll1, and Dll4 ligands while ovarian cells express Notch 1, Notch 2, Notch 3, Notch 4, and Jagged 2. Notch pathway is regulating the VEGF-mediated microvascular growth essential to follicular maturation [147].
In the ovarian cancer, Dll4 and Notch 1 were associated with VEGFR1 and VEGFR2 expression respectively, and impacted microvessel density [148]. When comparing the gene expression in ECs from 10 invasive epithelial ovarian cancers and from five normal ovaries, Lu et al. [16] demonstrated that Jagged 1 was over expressed in invasive epithelial ovarian cancers compared to normal ovary. Dll4, Notch 1, Notch 3 or Jagged 1 are overexpressed in ovarian cancer suggesting that Notch signaling is primordial in ovarian cancer [146]. Finally, we demonstrated the role of angiocrine endothelium in expanding tumor cell population in a contact-dependent as well as microparticle-mediated paracrine fashion [142, 149, 150]. We showed that a co-culture between ovarian cancer cells and ECs induced an increase of ovarian cancer cell proliferation both in vitro and in vivo [151]. This co-culture induced an increase of Notch receptors expression on ovarian cancer cell surface and an increase of Jagged 1 expression on EC surface and activation of survival pathways leading to chemoresistance by ECs.
Blood
Hematopoietic stem cell (HSC) give rise to both myeloid and lymphoid lineages. Several lines of evidence confirm a role for angiocrine endothelium in HSC growth and differentiation. (VEGFR-3+, VEGFR-2+, VE-cadherin+, and CD31+ Sinusoidal ECs (VEGFR-3+, VEGFR-2+, VE-cadherin+, and CD31+) are actively involved in self-renewal, maintenance, and regeneration of HSCs by producing membrane-bound or soluble angiocrine factors such as BMP2, BMP4, insulin growth factor binding protein 2 (IGFBP2), SDF-1, dessert hedgehog (DHH) protein, Notch ligands, wingless-type MMTV integration site 5a (Wnt5a)5a, and kit ligand. Our data shows that angiocrine, but not angiogenic function of ECs determine the lineage-specific differentiation of HSCs through differential secretion of granulocyte macrophage colony-stimulating factor (GM-CSF), interleukin 6 (IL-6), IL-8, granulocyte colony-stimulating factor (G-CSF), IL-1, and tumor necrosis factor (TNF). In Akt-activated endothelium, these angiocrine factors support the expansion of HSCs; whereas co-activation of Akt and p42/44 MAPK in ECs shifts the balance towards maintenance and differentiation of HSCs and progenitor cells [152].
Several reports have suggested the participation of tumor endothelium in neoplastic growth in solid and liquid hematological tumors. Dias et al. [32] demonstrated a non-angiogenic role for VEGF in promoting leukemic cell survival through activation of Hsp90 and induction of Bcl-2 expression. Furthermore, VEGF-C promoted leukemic cell survival and proliferation through interaction with VEGFR-3 (FLT-4) independent of its role in angiogenesis [153]. Other reports showed the angiocrine participation of VEGF signaling in modulation of cell growth in acute myeloid leukemia (AML) by induction of nitric oxide (NO) synthesis [154]. VEGF-mediated angiocrine activation of ECs also plays a role in supporting leukemic progression and resistance to chemotherapy treatment [155]. Inhibition of angiocrine VEGF by blocking VEGFR-2 increase the susceptibility of leukemic cells to chemotherapy. The involvement of vascular ECs in invasiveness and chemoresistance was recently reported in other non-solid hematological tumors [35]. Cao et al. showed that production of FGF4 by B-cell lymphoma and its interaction with FGFR1 receptor upregulates the Notch ligand Jagged 1 on neighboring ECs that reciprocally induces an autocrine loop of Notch2-Hey1 signaling in B cell lymphoma. This crosstalk enforces agressive CD44/IGF1R/CSF1R-positive phenotypes in lymphoma cells with enhanced extranodal invasiveness and chemoresistance.
The complex emerging angiocrine role of ECs may explain the relative failure of many anti-angiogenic therapies. The dichotomous role of ECs in angiogenesis and angiocrine-associated tumor development introduces new therapeutic targets, and instructs us on how to overcome the inefficiency of treatments caused by either redundant or substitutive functions of ECs.
Angiocrine endothelium and cancer therapy
Currently, cancer is not regarded as a single disease, but as a multi-faceted malignancy manifesting multiple capabilities to sustain proliferation and replicative immortality, evade growth suppression, avoid immune response, inducing angiogenesis, activating invasion and metastasis, promoting genome instability and mutation, resisting apoptosis, and deregulating cellular metabolism [3]. To remain within the scope of this paper, we provide a summary of the main anti-angiogenic strategies used to treat cancer and discuss their advantages and disadvantages. Then, we focus on the newly emerged angiocrine targeting of tumor endothelium and its potential application as adjuvant therapy in combination with other chemotherapeutic agents. The anti-angiogenic therapy originated from the evidence showing the importance of angiogenesis in non-neoplastic diseases as a route for delivery of inflammatory cells to the disease site [156]. The production of pro-angiogenic factors by tumor cells provided additional evidence to propose a role for angiogenesis in cancer development. The importance of VEGF family of pro-angiogenic factors in angiogenesis and tumor neovascularization leaded to the development of several anti-VEGF inhibitors [157,158,159]. VEGF is secreted by tumor cells and binds to its receptor VEGFR2 and neuropilin on ECs. Bevacizumab (Avastin) is a blocking antibody to VEGF is approved in combination with chemotherapy to treat metastatic colorectal cancer, non-small-cell lung cancer, and breast cancer [160]. Similarly, Ziv-aflibercept (VEGF trap) that functions as a VEGFR decoy is used to treat metastatic colorectal cancer in combination with chemotherapy AUTRE INDICATIONS. Also, an alternative therapy approach benefited from our understanding of the molecular mechanism of vessel stabilization and branching by tumor Angiopoietin1 (Ang-1) and Ang-2 and their tyrosine kinase receptor (Tie-2) on neighboring ECs [161]. Ang-2 antagonizes Ang-1 function in angiogenesis, but while acting in concert with VEGF they can both stabilize and mature new capillaries [162]. Several other angiogenesis inhibitors were approved for cancer therapy (refer to Table 2 for details) that either target receptor tyrosine kinases [163, 164] or reduce VEGF production [165,166,167]. Although angiogenesis inhibitors display obvious benefits including increased progression-free survival (PFS), the majority of them fail to produce prolonged response impacting overall survival (OS). The transient effectiveness of anti-angiogenesis drugs is reportedly related to two modes of resistance: (1) evasion of anti-angiogenic therapy and (2) indifference to therapy. In an extensive review, Bergers and Hanahan [8] have proposed several strategies that are exploited by each resistance mode including persistence of tumor angiogenesis, tumor blood vessel protection by pericytes, invasion of hypoxic conditions caused by therapeutic agents, and redundancy in angiogenic signals.
Recent discoveries of angiocrine factors produced by tumor ECs and their critical role in tumor growth and expansion can suggest new targets for drug development (Fig. 2). The angiocrine endothelium could also interfere with anti-angiogenic cancer treatment strategies. A recent work by our group showed a role for Akt-activated angiocrine endothelium in increased resistance to anti-angiogenic drugs such as bevacizumab through stimulation of the autocrine FGF2/FGFR1 loop [150]. The perivascular niche is also implicated in gaining resistance to radiation in medulloblastoma [168]. Hambardzumyan et al. showed that radiation activates the Akt/mTOR pathway in the perivascular nestin-expressing stem cells resulting in their re-entery to cell cycle and restoration of tumor bulk cells leading to tumor recurrence. They further confirmed their finding by demonstrating that inhibition of radiation-induced Akt phosphorylation sensitizes perivascular niche to radiation-induced apoptosis. Similarly the contribution of perivascular niche in promoting chemoresistance has been shown in non-solid tumors. Dias et al. [153] have demonstrated that leukemic cells stimulate ECs through secretion of pro-angiogenic and pro-inflammatory cytokines such as bFGF and IL-1 to VEGF-C interacting with its -3 receptor VEGFR on leukemic cells to induce survival and chemoresistance. Interestingly, Lim et al. [169] showed that inhibition of VEGFR3 on ovarian cancer cells was associated with improved response to chemotherapy through down-regulation of BRCA1 and BRCA2. Cao et al. [35] have also defined a role for vascular niche in inducing chemoresistance in B cell lymphoma. They have demonstrated that interaction of lymphoma cells with tumor ECs results in up-regulation of endothelial Jagged1 and induction of Notch2/Hey1 pathway in lymphoma cells resulting in their improved chemoresistance. Concordant with these findings, our group has shown the participation of tumor endothelial-derived microvesicles in shuttling endothelial angiocrine factors to breast and ovarian cancer cells resulting in enhanced chemoresistance [142, 149]. Taken together, targeting endothelial-specific angiocrine factors might represent several advantages. Targeting those involved in tumor initiation in an adjuvant setting may inhibit tumor recurrence and prevent residual disease; inactivating those that are involved in tumor cell self-renewal and expansion might block tumor growth and metastasis and combinatorial use of those which confer drug resistance might improve chemotherapy outcome.
Angiocrine endothelium and immune invasion
Most tumor cells express antigens that can mediate recognition by host CD8+ T cells. Thus, cancers that are detected clinically must have evaded antitumor immune responses to grow progressively. In solid tumors, it has been shown that the components of TM influence the host immune response (Fig. 2). It has long been recognized that tumor stimulation of an immune response is exceedingly inefficient when tumor is surrounded by microenvironment components. Analysis of the TM in patients with a variety of tumors has revealed that a major subset of tumors shows evidence of a T-cell-infiltrated phenotype. However, the infiltrated T-cells are consequently functionally inhibited [170,171,172]. The expression of immune checkpoint genes such as programmed cell death 1 ligand 1 (PDL1 or B7-H1), programmed cell death 1 (PD1), and cytotoxic T-lymphocyte-associated protein 4 (CTLA4) in stromal components plays an important role in down-regulation of the immune response to tumor by preventing the activation of T-cells. LaGier et al. have recently shown that the expression of PDL1 on human ECs was associated with reduced T-cell responses, a phenomenon that is also used by tumor cells to escape direct immune destruction [173,174,175]. Besides, Demaria et al. showed that antitumor efficacy of endothelial-derived interferon response was enhanced by PD1 and CTLA4 blockade [176]. More recently, the checkpoint blockade therapies targeting T-cell inhibitory pathways have produced promising results in many tumor setting [177,178,179,180]. Another interesting aspect of stromal regulation of antitumor immune response is associated with the production of angiocrine factors. Some of human tumors that represent poor lymphocyte infiltration appear to have higher expression of several angiogenic factors such as VEGF. VEGF-A that can be produced by both tumor and endothelial cells, is reportedly involved in creation of an immunosuppressive microenvironment through enhancing the production of inhibitory checkpoints molecules such as PD1 [181]. Also, the ineffectiveness of cancer vaccines may be related to the role of tumor endothelium in regulating T-cell homing to tumor location, a process that plays a significant role in optimal anti-tumor immunity and tumor elimination [182,183,184]. In order for the lymphocytes to enter the TM, they require to transmigrate across lymphatic vessels, a process regulated by endothelial adhesion molecule 1 (ICAM-1), vascular adhesion molecule (VCAM), and Selectin (CD62) as well as local production of specific chemokines [185,186,187]. Initial report on the participation of tumor endothelium in leukocyte infiltration reported a role for ICAM-1. In their work, Griffioen et al. [188] suggested a regulatory role for angiocrine factor bFGF in stimulating ICAM-1 expression on ECs followed by improved leukocyte infiltration to tumor site. Buckanovich et al. [189] have also shown that the tumor endothelium provides a barrier for tumor-infiltrating lymphocytes by overexpression of endothelin B receptor (ETBR). They showed that the overexpression of ETBR was correlated with the absence of tumor-infiltrating lymphocytes and poor patient survival. This might be the underlying reason for ineffectiveness of tumor vaccines despite being highly immunogenic. Additionally, Harlin et al. [190] have shown the importance of chemokines such as CCL2, CCL3, CCL4, CCL5, CXCL9, and CXCL10 in T cell infiltration into TM. Although these chemokines are produced by tumor cells, they can also be secreted by stromal cells in the TM and contribute to the recruitment of immune effector cells [191].
As discussed earlier, ECs demonstrate high degrees of plasticity based on their contexture. Emerging data is now revealing the role of tumor cells in activating immune response escape mechanisms in ECs. For instance, the expression of T-cell immunoglobulin and mucin domain-containing molecule 3 (Tim-3) in lymphoma endothelium greatly facilitates the growth and dissemination of lymphoma cells by suppressing CD4+ T-cell activation through the stimulation of the IL-6/STAT3 pathway and providing protective immunity [192]. Furthermore, the mechanism by which the tumor vasculature regulates tumor endothelial death barrier and T-cell infiltration in solid tumors has been lately described in establishing immune tolerance [193]. Tumor-derived VEGF-A, IL-10 and prostaglandin E2 were shown to cooperatively induce FasL expression in ECs enabling them to kill effector CD8+ T-cells, but not regulatory T-cells.
The above findings highlight the importance of the angiocrine endothelium in regulation of tumor response to immunotherapy and demonstrate that tumor cells are capable of hijacking a physiological anti-inflammatory mechanism of normalizing immune response from angiocrine endothelium to suppress immune reaction against tumor progression.
The endothelium metabolic regulation of tumor progression
The strong dependency of tumor cells on nutrients and oxygen is the driving force for stimulating blood vessel sprouting. Blood perfusion provides the requirements for cancer progression. Therefore, understanding the connection between tumor nourishment and blood vessel budding may introduce new mechanisms for preventing cancer growth. Recent evidence shows that in addition to tumor-induced angiogenic signals, the EC metabolic requirements are an essential regulators of angiogenesis [194, 195]. Cancer and stromal cells adapt to harsh conditions of TM such as hypoxia and lack of nutrients by switching their metabolism from oxidative phosphorylation to glycolysis [196,197,198]. The question is when this metabolic conversion happens and how it is regulated? Carmeliet’s group proposed that at the time of vessel sprouting (angiogenic switch), quiescent ECs undergo activation (angiocrine switch) and adapt their metabolism to generate additional energy and biomass for growth and division. This step is referred to as “metabolic switch” that occurs in parallel with angiogenic and angiocrine switches [199]. Interestingly, the mechanism of metabolic switch is partly regulated by signals from pro-angiogenic factors produced by tumor cells. De Bock et al. [194, 200] have documented that tumor cells activate the endothelium metabolic switch through VEGF production leading to enhancement of glycolysis by up-regulation of phosphofructokinase-2/fructose-2,6-bisphosphatase 3 (PFKFB3). As a result, a novel hypothesis describes an anti-glycolytic therapy that targets the VEGF/PFKFB3-stimulated glycolysis in vessel sprouting [195]. Inhibition of PFKFB3 resulted in reduced vessel sprouting in endothelial spheroids in zebrafish embryo and postnatal mouse retina as a result of partial and transient reduction in glycolysis. As vessels sprouts by a migratory tip cell and the elongating stalk cells, blocking PFKFB3 will possibly impair the activity of both cell types leading to inhibition of vessel budding. Therefore, inhibition of a key metabolic regulator of ECs might efficiently block neovascularization despite the presence of new pro-angiogenic factors. Furthermore, current anti-angiogenic drugs induce tumor hypoxia and nutrient deprivation that adversely activate the tumor non-oxidative metabolism (glycolysis) contributing to the enrichment of resistant tumor colonies [201]. Hence, identification of angiocrine factors that mediate endothelial metabolism might have implications in developing novel approaches for targeting the concomitant angiocrine, angiogenic, and metabolically activated ECs. Besides, it is possible that anti-metabolism drugs could increase tumor response to therapy by impairing the tumor resistance strategies induced by angiogenic and angiocrine factors. The metabolic regulation of tumor growth introduces a promising area for designing effective anti-metabolism treatments. However, we first need to establish a metabolomic map of the entire metabolites generated in different EC subtypes during different stages of tumor growth and vascular branching to characterize how these compounds are involved in various metabolic pathways [200]. Moreover, tumor and ECs possess some similarities and differences in their metabolic processes. Both cell types are highly glycolytic and use glycolysis for many cellular mechanisms such as proliferation and invasion. These similarities provide great opportunities for targeting both cell types. On the other hand, we must be aware of the differences between tumor and endothelial metabolic needs and mechanisms while applying anti-metabolic strategies. Some of these differences include ability of cancer cells to switch on alternative metabolic pathways, modify their metabolic substrate, and differences in reacting to metabolic stress [202,203,204].
Conclusions and future directions
Emerging evidence on the angiocrine role of ECs raises the possibility of designing novel therapy approaches. To ensure the success of anti-angiocrine drugs, precise understanding of all cellular mechanisms involved in the activation of angiocrine switch will be required. This knowledge will be beneficial in designing mechanism-based targeted therapies for interrupting angiocrine activation as well as for impairing the crosstalk between tumor and ECs. While the pathway-specific drugs potentially moderate therapy-induced toxicity, they will provide more effective targeting of endothelial cancer crosstalk. A systematic understanding of the role of angiocrine factors may also lead to functional targeting of ECs. For instance, targeting specific angiocrine factors that foster cancer stem cells may be beneficial in addressing drug resistance and/or tumor recurrence hurdles. However, we ought to learn from earlier angiogenic-targeting therapies and bear in mind that a successful treatment will not be achieved by solely aiming at individual molecules as it may reportedly lead to transient patient survival or development of adaptive patient resistance [2, 205,206,207]. To avoid succumbing to redundancy in stromal regulation of tumor development, we must first develop comprehensive knowledge of the stroma-tumor interaction to be able to design combinatorial strategies for targeting not only intracellular pathways in cancer cells, but also in cancer stromal cells in many ways [2, 208, 209]. The dichotomous role of ECs in angiogenesis and angiocrine-associated tumor development not only introduces new therapeutic targets, but also instructs us on how to overcome the inefficiency of treatments caused by either redundant or substitutive functions of ECs. This demonstrate that the so called “high jacked” of the surrounding cells by the tumors is in reality the use of regular pathway of the organ by the dedifferentiate cell that composed the tumor. Molecules expressed during embryogenesis and downregulated in adult tissue are re-expressed in tumors.
Availability of data and materials
Not applicable.
Abbreviations
- AML:
-
Acute myeloid leukemia
- bFGF:
-
Basic FGF
- BMP-4:
-
Endothelial-derived bone morphogenetic protein 4
- CSC:
-
Cancer stem cells
- CTLA4T:
-
Lymphocyte-associated protein 4
- DHH:
-
Dessert hedgehog
- EC:
-
Endothelial Cells
- EGFs:
-
Epidermal growth factors
- EGFL7:
-
Epidermal growth factor-like domain 7
- EMT:
-
Epithelial-to-mesenchymal transition
- FDA:
-
Food and Drug Administration
- FGFs:
-
Fibroblast growth factors
- FGF8:
-
Fibroblast growth factor 8
- GM-CSF:
-
Granulocyte macrophage colony-stimulating factor
- HGF:
-
Hepatocyte growth factor
- HSC:
-
Hematopoietic stem cell
- ICAM-1:
-
Endothelial adhesion molecule 1
- IGFBP2:
-
Insulin growth factor binding protein 2
- IL-6:
-
Interleukin 6
- LM521:
-
Laminin-α5β2γ1
- MMP14:
-
Matrix metalloproteinase 14
- NO:
-
Nitric oxide
- NSC:
-
Neural stem cells
- OS:
-
Overall survival
- PD1:
-
Programmed cell death 1
- PDGF-B:
-
Platelet-derived growth B
- PDL1:
-
Programmed cell death 1 ligand 1
- PFKFB3:
-
Phosphofructokinase-2/fructose-2,6-bisphosphatase 3
- PFS:
-
Progression-free survival
- SDF-1:
-
Stromal-cell derived factor
- S1P:
-
Sphingosine-1-phosphate
- TM:
-
Tumor microenvironment
- VCAM:
-
Vascular adhesion molecule
References
Hoarau-Vechot J, Rafii A, Touboul C, Pasquier J. Halfway between 2D and animal models: are 3D cultures the ideal tool to study cancer-microenvironment interactions? Int J Mol Sci. 2018;19(1):181.
Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–22.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Pasquier J, Rafii A. Role of the microenvironment in ovarian cancer stem cell maintenance. Biomed Res Int. 2013;2013:630782.
Butler JM, Kobayashi H, Rafii S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer. 2010;10(2):138–46.
Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature. 2016;529(7586):316–25.
Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. 1989;339(6219):58–61.
Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603.
Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97(6):512–23.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307.
Daenen LG, Shaked Y, Man S, Xu P, Voest EE, Hoffman RM, Chaplin DJ, Kerbel RS. Low-dose metronomic cyclophosphamide combined with vascular disrupting therapy induces potent antitumor activity in preclinical human tumor xenograft models. Mol Cancer Ther. 2009;8(10):2872–81.
Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219(4587):983–5.
Halama A, Guerrouahen BS, Pasquier J, Satheesh NJ, Suhre K, Rafii A. Nesting of colon and ovarian cancer cells in the endothelial niche is associated with alterations in glycan and lipid metabolism. Sci Rep. 2017;7:39999.
Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–64.
Ghiabi P, Jiang J, Pasquier J, Maleki M, Abu-Kaoud N, Halabi N, Guerrouahen BS, Rafii S, Rafii A. Breast cancer cells promote a notch-dependent mesenchymal phenotype in endothelial cells participating to a pro-tumoral niche. J Transl Med. 2015;13(1):27.
Lu C, Bonome T, Li Y, Kamat AA, Han LY, Schmandt R, Coleman RL, Gershenson DM, Jaffe RB, Birrer MJ, et al. Gene alterations identified by expression profiling in tumor-associated endothelial cells from invasive ovarian carcinoma. Cancer Res. 2007;67(4):1757–68.
Nagy JA, Chang SH, Shih SC, Dvorak AM, Dvorak HF. Heterogeneity of the tumor vasculature. Semin Thromb Hemost. 2010;36(3):321–31.
Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188(6):759–68.
Seaman S, Stevens J, Yang MY, Logsdon D, Graff-Cherry C, St Croix B. Genes that distinguish physiological and pathological angiogenesis. Cancer Cell. 2007;11(6):539–54.
Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho RA, Zeiher AM, Dimmeler S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Investig. 2005;115(9):2382–92.
Phung TL, Ziv K, Dabydeen D, Eyiah-Mensah G, Riveros M, Perruzzi C, Sun J, Monahan-Earley RA, Shiojima I, Nagy JA, et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell. 2006;10(2):159–70.
Bissell MJ, Hall HG, Parry G. How does the extracellular matrix direct gene expression? J Theor Biol. 1982;99(1):31–68.
Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147(5):992–1009.
Nelson CM, Khauv D, Bissell MJ, Radisky DC. Change in cell shape is required for matrix metalloproteinase-induced epithelial–mesenchymal transition of mammary epithelial cells. J Cell Biochem. 2008;105(1):25–33.
Xu R, Boudreau A, Bissell MJ. Tissue architecture and function: dynamic reciprocity via extra- and intra-cellular matrices. Cancer Metastasis Rev. 2009;28(1–2):167–76.
Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25(1):30–8.
Pasquier J, Gosset M, Geyl C, Hoarau-Vechot J, Chevrot A, Pocard M, Mirshahi M, Lis R, Rafii A, Touboul C. CCL2/CCL5 secreted by the stroma induce IL-6/PYK2 dependent chemoresistance in ovarian cancer. Mol Cancer. 2018;17(1):47.
Fernandez L, Rodriguez S, Huang H, Chora A, Fernandes J, Mumaw C, Cruz E, Pollok K, Cristina F, Price JE, et al. Tumor necrosis factor-alpha and endothelial cells modulate Notch signaling in the bone marrow microenvironment during inflammation. Exp Hematol. 2008;36(5):545–58.
Zeng Q, Li S, Chepeha DB, Giordano TJ, Li J, Zhang H, Polverini PJ, Nor J, Kitajewski J, Wang CY. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell. 2005;8(1):13–23.
Dias S, Hattori K, Heissig B, Zhu Z, Wu Y, Witte L, Hicklin DJ, Tateno M, Bohlen P, Moore MA, et al. Inhibition of both paracrine and autocrine VEGF/VEGFR-2 signaling pathways is essential to induce long-term remission of xenotransplanted human leukemias. Proc Natl Acad Sci USA. 2001;98(19):10857–62.
Dias S, Hattori K, Zhu Z, Heissig B, Choy M, Lane W, Wu Y, Chadburn A, Hyjek E, Gill M, et al. Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J Clin Investig. 2000;106(4):511–21.
Dias S, Shmelkov SV, Lam G, Rafii S. VEGF(165) promotes survival of leukemic cells by Hsp90-mediated induction of Bcl-2 expression and apoptosis inhibition. Blood. 2002;99(7):2532–40.
Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304(5675):1338–40.
Zhang Z, Dong Z, Lauxen IS, Filho MS, Nor JE. Endothelial cell-secreted EGF induces epithelial to mesenchymal transition and endows head and neck cancer cells with stem-like phenotype. Cancer Res. 2014;74(10):2869–81.
Cao Z, Ding BS, Guo P, Lee SB, Butler JM, Casey SC, Simons M, Tam W, Felsher DW, Shido K, et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell. 2014;25(3):350–65.
Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. 2008;99(9):1375–9.
Yoshida S, Sukeno M, Nabeshima Y. A vasculature-associated niche for undifferentiated spermatogonia in the mouse testis. Science. 2007;317(5845):1722–6.
Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B, Garcia-Verdugo JM, Doetsch F. A specialized vascular niche for adult neural stem cells. Cell Stem Cell. 2008;3(3):279–88.
Tang W, Zeve D, Suh JM, Bosnakovski D, Kyba M, Hammer RE, Tallquist MD, Graff JM. White fat progenitor cells reside in the adipose vasculature. Science. 2008;322(5901):583–6.
Shen Q, Wang Y, Kokovay E, Lin G, Chuang SM, Goderie SK, Roysam B, Temple S. Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell–cell interactions. Cell Stem Cell. 2008;3(3):289–300.
Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425(4):479–94.
Christov C, Chretien F, Abou-Khalil R, Bassez G, Vallet G, Authier FJ, Bassaglia Y, Shinin V, Tajbakhsh S, Chazaud B, et al. Muscle satellite cells and endothelial cells: close neighbors and privileged partners. Mol Biol Cell. 2007;18(4):1397–409.
Saito D, Takase Y, Murai H, Takahashi Y. The dorsal aorta initiates a molecular cascade that instructs sympatho-adrenal specification. Science. 2012;336(6088):1578–81.
Andreu-Agullo C, Morante-Redolat JM, Delgado AC, Farinas I. Vascular niche factor PEDF modulates Notch-dependent stemness in the adult subependymal zone. Nat Neurosci. 2009;12(12):1514–23.
Han J, Calvo CF, Kang TH, Baker KL, Park JH, Parras C, Levittas M, Birba U, Pibouin-Fragner L, Fragner P, et al. Vascular endothelial growth factor receptor 3 controls neural stem cell activation in mice and humans. Cell Rep. 2015;10(7):1158–72.
Kokovay E, Goderie S, Wang Y, Lotz S, Lin G, Sun Y, Roysam B, Shen Q, Temple S. Adult SVZ lineage cells home to and leave the vascular niche via differential responses to SDF1/CXCR4 signaling. Cell Stem Cell. 2010;7(2):163–73.
Crouch EE, Liu C, Silva-Vargas V, Doetsch F. Regional and stage-specific effects of prospectively purified vascular cells on the adult V-SVZ neural stem cell lineage. J Neurosci. 2015;35(11):4528–39.
Leventhal C, Rafii S, Rafii D, Shahar A, Goldman SA. Endothelial trophic support of neuronal production and recruitment from the adult mammalian subependyma. Mol Cell Neurosci. 1999;13(6):450–64.
Cleaver O, Melton DA. Endothelial signaling during development. Nat Med. 2003;9(6):661–8.
Mukouyama YS, Shin D, Britsch S, Taniguchi M, Anderson DJ. Sensory nerves determine the pattern of arterial differentiation and blood vessel branching in the skin. Cell. 2002;109(6):693–705.
Carmeliet P, Tessier-Lavigne M. Common mechanisms of nerve and blood vessel wiring. Nature. 2005;436(7048):193–200.
Mathieu C, Sii-Felice K, Fouchet P, Etienne O, Haton C, Mabondzo A, Boussin FD, Mouthon MA. Endothelial cell-derived bone morphogenetic proteins control proliferation of neural stem/progenitor cells. Mol Cell Neurosci. 2008;38(4):569–77.
Ottone C, Krusche B, Whitby A, Clements M, Quadrato G, Pitulescu ME, Adams RH, Parrinello S. Direct cell–cell contact with the vascular niche maintains quiescent neural stem cells. Nat Cell Biol. 2014;16(11):1045–56.
Ottone C, Parrinello S. Multifaceted control of adult SVZ neurogenesis by the vascular niche. Cell Cycle. 2015;14(14):2222–5.
Delgado AC, Ferron SR, Vicente D, Porlan E, Perez-Villalba A, Trujillo CM, D’Ocon P, Farinas I. Endothelial NT-3 delivered by vasculature and CSF promotes quiescence of subependymal neural stem cells through nitric oxide induction. Neuron. 2014;83(3):572–85.
Silva-Vargas V, Doetsch F. A new twist for neurotrophins: endothelial-derived NT-3 mediates adult neural stem cell quiescence. Neuron. 2014;83(3):507–9.
Pumiglia K, Temple S. PEDF: bridging neurovascular interactions in the stem cell niche. Nat Neurosci. 2006;9(3):299–300.
Ramirez-Castillejo C, Sanchez-Sanchez F, Andreu-Agullo C, Ferron SR, Aroca-Aguilar JD, Sanchez P, Mira H, Escribano J, Farinas I. Pigment epithelium-derived factor is a niche signal for neural stem cell renewal. Nat Neurosci. 2006;9(3):331–9.
Whitman MC, Fan W, Rela L, Rodriguez-Gil DJ, Greer CA. Blood vessels form a migratory scaffold in the rostral migratory stream. J Comp Neurol. 2009;516(2):94–104.
Ferrari-Toninelli G, Bonini SA, Uberti D, Buizza L, Bettinsoli P, Poliani PL, Facchetti F, Memo M. Targeting Notch pathway induces growth inhibition and differentiation of neuroblastoma cells. Neuro-Oncology. 2010;12(12):1231–43.
Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, He XB, Hervey-Jumper SL, Heth JA, Muraszko KM, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011;71(18):6061–72.
Nduom EKE, Hadjipanayis CG, Van Meir EG. Glioblastoma cancer stem-like cells implications for pathogenesis and treatment. Cancer J. 2012;18(1):100–6.
Purow BW, Haque RM, Noel MW, Su Q, Burdick MJ, Lee J, Sundaresan T, Pastorino S, Park JK, Mikolaenko I, et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005;65(6):2353–63.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11.
Qiu XX, Wang CH, You N, Chen BJ, Wang XF, Chen YP, Lin ZX. High Jagged1 expression is associated with poor outcome in primary glioblastoma. Med Oncol. 2015;32(1):341.
Fiaschetti G, Shalaby T, Baumgartner M, Grotzer MA. Notch ligands Jag1 and Jag2 control medulloblastoma cell survival and represent potential prognostic markers and therapeutic targets. Neuro-Oncology. 2014;16:74.
Yavropoulou MP, Maladaki A, Topouridou K, Kotoula V, Poulios C, Daskalaki E, Foroglou N, Karkavelas G, Yovos JG. Expression pattern of the Hedgehog signaling pathway in pituitary adenomas. Neurosci Lett. 2016;611:94–100.
McKinney N, Yuan L, Zhang H, Liu J, Cho YJ, Rushing E, Schniederjan M, MacDonald TJ. EphrinB1 expression is dysregulated and promotes oncogenic signaling in medulloblastoma. J Neurooncol. 2015;121(1):109–18.
Tu Y, He S, Fu J, Li G, Xu R, Lu H, Deng J. Expression of EphrinB2 and EphB4 in glioma tissues correlated to the progression of glioma and the prognosis of glioblastoma patients. Clin Transl Oncol. 2012;14(3):214–20.
Lawn S, Krishna N, Pisklakova A, Qu X, Fenstermacher DA, Fournier M, Vrionis FD, Tran N, Chan JA, Kenchappa RS, et al. Neurotrophin signaling via TrkB and TrkC receptors promotes the growth of brain tumor-initiating cells. J Biol Chem. 2015;290(6):3814–24.
Fiaschetti G, Castelletti D, Zoller S, Schramm A, Schroeder C, Nagaishi M, Stearns D, Mittelbronn M, Eggert A, Westermann F, et al. Bone morphogenetic protein-7 is a MYC target with prosurvival functions in childhood medulloblastoma. Oncogene. 2011;30(25):2823–35.
Dewerchin M, Carmeliet P. Placental growth factor in cancer. Expert Opin Ther Targets. 2014;18(11):1339–54.
Jiang Z, Zhou W, Guan S, Wang J, Liang Y. Contribution of SDF-1alpha/CXCR4 signaling to brain development and glioma progression. Neurosignals. 2013;21(3–4):240–58.
Terasaki M, Sugita Y, Arakawa F, Okada Y, Ohshima K, Shigemori M. CXCL12/CXCR4 signaling in malignant brain tumors: a potential pharmacological therapeutic target. Brain Tumor Pathol. 2011;28(2):89–97.
Matsumoto K, Yoshitomi H, Rossant J, Zaret KS. Liver organogenesis promoted by endothelial cells prior to vascular function. Science. 2001;294(5542):559–63.
Zong Y, Panikkar A, Xu J, Antoniou A, Raynaud P, Lemaigre F, Stanger BZ. Notch signaling controls liver development by regulating biliary differentiation. Development. 2009;136(10):1727–39.
Jung J, Zheng M, Goldfarb M, Zaret KS. Initiation of mammalian liver development from endoderm by fibroblast growth factors. Science. 1999;284(5422):1998–2003.
LeCouter J, Moritz DR, Li B, Phillips GL, Liang XH, Gerber HP, Hillan KJ, Ferrara N. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science. 2003;299(5608):890–3.
Rossi JM, Dunn NR, Hogan BL, Zaret KS. Distinct mesodermal signals, including BMPs from the septum transversum mesenchyme, are required in combination for hepatogenesis from the endoderm. Genes Dev. 2001;15(15):1998–2009.
Ding BS, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, Mittal V, Kobayashi H, Shido K, Lyden D, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature. 2010;468(7321):310–5.
Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, Penfold ME, Shido K, Rabbany SY, Rafii S. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505(7481):97–102.
Kim W, Khan SK, Yang YZ. Interacting network of Hippo, Wnt/beta-catenin and Notch signaling represses liver tumor formation. BMB Rep. 2017;50(1):1–2.
Robson AJ, Guest RV, Boulter L, Iredale JP, Forbes SJ. The role of notch signalling within the tumour-stromal microenvironment in cholangiocarcinoma. Br J Surg. 2012;99:3.
Robson AJ, Samuel K, Pellicoro A, Garden OJ, Wigmore SJ, Iredale JP, Forbes SJ. The role of the bone marrow and notch signalling in the stromal microenvironment in cholangiocarcinoma. Br J Surg. 2011;98:21.
Malouf GG, Job S, Paradis V, Fabre M, Brugieres L, Saintigny P, Vescovo L, Belghiti J, Branchereau S, Faivre S, et al. Transcriptional profiling of pure fibrolamellar hepatocellular carcinoma reveals an endocrine signature. Hepatology. 2014;59(6):2228–37.
Geisler F, Nagl F, Mazur PK, Lee M, Zimber-Strobl U, Strobl LJ, Radtke F, Schmid RM, Siveke JT. Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology. 2008;48(2):607–16.
Chen YX, Weng ZH, Zhang SL. Notch3 regulates the activation of hepatic stellate cells. World J Gastroenterol. 2012;18(12):1397–403.
Giovannini C, Bolondi L, Gramantieri L. Targeting Notch3 in hepatocellular carcinoma: molecular mechanisms and therapeutic perspectives. Int J Mol Sci. 2017;18(1):56.
Lu J, Xia YJ, Chen K, Zheng YY, Wang JR, Lu WX, Yin Q, Wang F, Zhou YQ, Guo CY. Oncogenic role of the Notch pathway in primary liver cancer (Review). Oncol Lett. 2016;12(1):3–10.
Maegdefrau U, Amann T, Winklmeier A, Braig S, Schubert T, Weiss TS, Schardt K, Warnecke C, Hellerbrand C, Bosserhoff AK. Bone morphogenetic protein 4 is induced in hepatocellular carcinoma by hypoxia and promotes tumour progression. J Pathol. 2009;218(4):520–9.
Chiu CY, Kuo KK, Kuo TL, Lee KT, Cheng KH. The activation of MEK/ERK signaling pathway by bone morphogenetic protein 4 to increase hepatocellular carcinoma cell proliferation and migration. Mol Cancer Res. 2012;10(3):415–27.
Zeng S, Zhang Y, Ma J, Deng G, Qu Y, Guo C, Han Y, Yin L, Cai C, Li Y, et al. BMP4 promotes metastasis of hepatocellular carcinoma by an induction of epithelial-mesenchymal transition via upregulating ID2. Cancer Lett. 2017;390:67–76.
Guo X, Xiong L, Zou L, Zhao J. Upregulation of bone morphogenetic protein 4 is associated with poor prognosis in patients with hepatocellular carcinoma. Pathol Oncol Res. 2012;18(3):635–40.
Gauglhofer C, Sagmeister S, Schrottmaier W, Fischer C, Rodgarkia-Dara C, Mohr T, Stattner S, Bichler C, Kandioler D, Wrba F, et al. Up-regulation of the fibroblast growth factor 8 subfamily in human hepatocellular carcinoma for cell survival and neoangiogenesis. Hepatology. 2011;53(3):854–64.
Kawakami-Kimura N, Narita T, Ohmori K, Yoneda T, Matsumoto K, Nakamura T, Kannagi R. Involvement of hepatocyte growth factor in increased integrin expression on HepG2 cells triggered by adhesion to endothelial cells. Br J Cancer. 1997;75(1):47–53.
Zheng K, Li HY, Su XL, Wang XY, Tian T, Li F, Ren GS. Chemokine receptor CXCR7 regulates the invasion, angiogenesis and tumor growth of human hepatocellular carcinoma cells. J Exp Clin Cancer Res. 2010;29:31.
Furusato B, Mohamed A, Uhlen M, Rhim JS. CXCR4 and cancer. Pathol Int. 2010;60(7):497–505.
DeLisser HM, Helmke BP, Cao G, Egan PM, Taichman D, Fehrenbach M, Zaman A, Cui Z, Mohan GS, Baldwin HS, et al. Loss of PECAM-1 function impairs alveolarization. J Biol Chem. 2006;281(13):8724–31.
Jakkula M, Le Cras TD, Gebb S, Hirth KP, Tuder RM, Voelkel NF, Abman SH. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol. 2000;279(3):L600–7.
Ramasamy SK, Kusumbe AP, Adams RH. Regulation of tissue morphogenesis by endothelial cell-derived signals. Trends Cell Biol. 2015;25(3):148–57.
Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z, Crystal RG, Simons M, Sato TN, Worgall S, et al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell. 2011;147(3):539–53.
Lee JH, Bhang DH, Beede A, Huang TL, Stripp BR, Bloch KD, Wagers AJ, Tseng YH, Ryeom S, Kim CF. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell. 2014;156(3):440–55.
Ao Z, Yu S, Qian P, Gao W, Guo R, Dong X, Xu J, Zhang R, Jiang C, Ji F, et al. Tumor angiogenesis of SCLC inhibited by decreased expression of FMOD via downregulating angiogenic factors of endothelial cells. Biomed Pharmacother. 2017;87:539–47.
Tsuchida R, Osawa T, Wang F, Nishii R, Das B, Tsuchida S, Muramatsu M, Takahashi T, Inoue T, Wada Y, et al. BMP4/Thrombospondin-1 loop paracrinically inhibits tumor angiogenesis and suppresses the growth of solid tumors. Oncogene. 2014;33(29):3803–11.
Fleitas T, Martinez-Sales V, Vila V, Reganon E, Mesado D, Martin M, Gomez-Codina J, Montalar J, Reynes G. VEGF and TSP1 levels correlate with prognosis in advanced non-small cell lung cancer. Clin Transl Oncol. 2013;15(11):897–902.
Wang YZ, Wu KP, Wu AB, Yang ZC, Li JM, Mo YL, Xu M, Wu B, Yang ZX. MMP-14 overexpression correlates with poor prognosis in non-small cell lung cancer. Tumor Biol. 2014;35(10):9815–21.
Edsbagge J, Johansson JK, Esni F, Luo Y, Radice GL, Semb H. Vascular function and sphingosine-1-phosphate regulate development of the dorsal pancreatic mesenchyme. Development. 2005;132(5):1085–92.
Lammert E, Cleaver O, Melton D. Induction of pancreatic differentiation by signals from blood vessels. Science. 2001;294(5542):564–7.
Nikolova G, Jabs N, Konstantinova I, Domogatskaya A, Tryggvason K, Sorokin L, Fassler R, Gu G, Gerber HP, Ferrara N, et al. The vascular basement membrane: a niche for insulin gene expression and Beta cell proliferation. Dev Cell. 2006;10(3):397–405.
Kao DI, Lacko LA, Ding BS, Huang C, Phung K, Gu G, Rafii S, Stuhlmann H, Chen S. Endothelial cells control pancreatic cell fate at defined stages through EGFL7 signaling. Stem Cell Rep. 2015;4(2):181–9.
Jacquemin P, Yoshitomi H, Kashima Y, Rousseau GG, Lemaigre FP, Zaret KS. An endothelial–mesenchymal relay pathway regulates early phases of pancreas development. Dev Biol. 2006;290(1):189–99.
Mukhopadhyay D, Datta K. Multiple regulatory pathways of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in tumors. Semin Cancer Biol. 2004;14(2):123–30.
Ellis LM, Takahashi Y, Fenoglio CJ, Cleary KR, Bucana CD, Evans DB. Vessel counts and vascular endothelial growth factor expression in pancreatic adenocarcinoma. Eur J Cancer. 1998;34(3):337–40.
Jia Z, Gao Y, Wang L, Li Q, Zhang J, Le X, Wei D, Yao JC, Chang DZ, Huang S, et al. Combined treatment of pancreatic cancer with mithramycin A and tolfenamic acid promotes Sp1 degradation and synergistic antitumor activity. Cancer Res. 2010;70(3):1111–9.
Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6(4):1186–97.
Shen X, Han Y, Xue X, Li W, Guo X, Li P, Wang Y, Li D, Zhou J, Zhi Q. Epidermal growth factor-like domain 7 promotes cell invasion and angiogenesis in pancreatic carcinoma. Biomed Pharmacother. 2016;77:167–75.
Zhou L, Li J, Zhao YP, Guo JC, Cui QC, Zhou WX, Zhang TP, Wu WM, You L, Shu H. Prognostic significance of epidermal growth factor-like domain 7 in pancreatic cancer. Hepatobiliary Pancreat Dis Int. 2014;13(5):523–8.
Costache MI, Ioana M, Iordache S, Ene D, Costache CA, Saftoiu A. VEGF expression in pancreatic cancer and other malignancies: a review of the literature. Romanian J Intern Med. 2015;53(3):199–208.
Korc M. Pathways for aberrant angiogenesis in pancreatic cancer. Mol Cancer. 2003;2:8.
Ikeda N, Adachi M, Taki T, Huang C, Hashida H, Takabayashi A, Sho M, Nakajima Y, Kanehiro H, Hisanaga M, et al. Prognostic significance of angiogenesis in human pancreatic cancer. Br J Cancer. 1999;79(9–10):1553–63.
Niedergethmann M, Hildenbrand R, Wostbrock B, Hartel M, Sturm JW, Richter A, Post S. High expression of vascular endothelial growth factor predicts early recurrence and poor prognosis after curative resection for ductal adenocarcinoma of the pancreas. Pancreas. 2002;25(2):122–9.
Tufro A, Norwood VF, Carey RM, Gomez RA. Vascular endothelial growth factor induces nephrogenesis and vasculogenesis. J Am Soc Nephrol. 1999;10(10):2125–34.
Reidy K, Tufro A. Semaphorins in kidney development and disease: modulators of ureteric bud branching, vascular morphogenesis, and podocyte-endothelial crosstalk. Pediatr Nephrol. 2011;26(9):1407–12.
Eremina V, Cui S, Gerber H, Ferrara N, Haigh J, Nagy A, Ema M, Rossant J, Jothy S, Miner JH, et al. Vascular endothelial growth factor a signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol. 2006;17(3):724–35.
Serluca FC, Drummond IA, Fishman MC. Endothelial signaling in kidney morphogenesis: a role for hemodynamic forces. Curr Biol. 2002;12(6):492–7.
Bjarnegard M, Enge M, Norlin J, Gustafsdottir S, Fredriksson S, Abramsson A, Takemoto M, Gustafsson E, Fassler R, Betsholtz C. Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development. 2004;131(8):1847–57.
Miner JH. The glomerular basement membrane. Exp Cell Res. 2012;318(9):973–8.
Qian CN, Huang D, Wondergem B, Teh BT. Complexity of tumor vasculature in clear cell renal cell carcinoma. Cancer. 2009;115(10 Suppl):2282–9.
Duran I, Lambea J, Maroto P, Gonzalez-Larriba JL, Flores L, Granados-Principal S, Graupera M, Saez B, Vivancos A, Casanovas O. Resistance to targeted therapies in renal cancer: the importance of changing the mechanism of action. Target Oncol. 2017;12(1):19–35.
Li HL, Han L, Chen HR, Meng F, Liu QH, Pan ZQ, Bai J, Zheng JN. PinX1 serves as a potential prognostic indicator for clear cell renal cell carcinoma and inhibits its invasion and metastasis by suppressing MMP-2 via NF-kappa B-dependent transcription. Oncotarget. 2015;6(25):21406–20.
Wang J, Ren Y, Guo X, Cheng H, Ye YP, Qi J, Yang CH, You HB. Alterations in enhancer of zeste homolog 2, matrix metalloproteinase-2 and tissue inhibitor of metalloproteinase-2 expression are associated with ex vivo and in vitro bone metastasis in renal cell carcinoma. Mol Med Rep. 2015;11(5):3585–92.
Wragg JW, Finnity JP, Anderson JA, Ferguson HJM, Porfiri E, Bhatt RI, Murray PG, Heath VL, Bicknell R. MCAM and LAMA4 are highly enriched in tumor blood vessels of renal cell carcinoma and predict patient outcome. Cancer Res. 2016;76(8):2314–26.
Hens JR, Wysolmerski JJ. Key stages in mammary gland development—molecular mechanisms involved in the formation of the embryonic mammary gland. Breast Cancer Res. 2005;7(5):220–4.
Pasquier J, Abu-Kaoud N, Al Thani H, Rafii A. Epithelial to mesenchymal transition in a clinical perspective. J Oncol. 2015;2015:792182.
Ingthorsson S, Briem E, Bergthorsson JT, Gudjonsson T. Epithelial plasticity during human breast morphogenesis and cancer progression. J Mammary Gland Biol. 2016;21(3–4):139–48.
Ghiabi P, Jiang J, Pasquier J, Maleki M, Abu-Kaoud N, Rafii S, Rafii A. Endothelial cells provide a notch-dependent pro-tumoral niche for enhancing breast cancer survival, stemness and pro-metastatic properties. PLoS ONE. 2014;9(11):e112424.
Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, Butler PD, Yang GP, Joshua B, Kaplan MJ, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466(7302):133–7.
Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456(7222):593–8.
Buck E, Eyzaguirre A, Barr S, Thompson S, Sennello R, Young D, Iwata KK, Gibson NW, Cagnoni P, Haley JD. Loss of homotypic cell adhesion by epithelial–mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol Cancer Ther. 2007;6(2):532–41.
Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA. 2009;106(33):13820–5.
Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–51.
Pasquier J, Thawadi HA, Ghiabi P, Abu-Kaoud N, Maleki M, Guerrouahen BS, Vidal F, Courderc B, Ferron G, Martinez A, et al. Microparticles mediated cross-talk between tumoral and endothelial cells promote the constitution of a pro-metastatic vascular niche through Arf6 up regulation. Cancer Microenviron. 2014;7(1–2):41–59.
Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S, et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med. 2013;11:94.
Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, Koller A, Hajjar KA, Stainier DY, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013;15(7):807–17.
Augustin HG. Vascular morphogenesis in the ovary. Baillieres Best Pract Res Clin Obstet Gynaecol. 2000;14(6):867–82.
Xie Q, Cheng Z, Chen X, Lobe CG, Liu J. The role of Notch signalling in ovarian angiogenesis. J Ovarian Res. 2017;10(1):13.
Hellstrom M, Phng LK, Gerhardt H. VEGF and Notch signaling: the yin and yang of angiogenic sprouting. Cell Adhes Migr. 2007;1(3):133–6.
Wang H, Huang X, Zhang J, Shao N, Chen LO, Ma D, Ji C. The expression of VEGF and Dll4/Notch pathway molecules in ovarian cancer. Clin Chim Acta. 2014;436:243–8.
Al Thawadi H, Abu-Kaoud N, Al Farsi H, Hoarau-Vechot J, Rafii S, Rafii A, Pasquier J. VE-cadherin cleavage by ovarian cancer microparticles induces beta-catenin phosphorylation in endothelial cells. Oncotarget. 2016;7(5):5289–305.
Guerrouahen BS, Pasquier J, Abu Kaoud N, Maleki M, Beauchamp MC, Yasmeen A, Ghiabi P, Lis R, Vidal F, Saleh A, et al. Akt-activated endothelium constitute the niche for residual disease and resistance to bevacizumab in ovarian cancer. Mol Cancer Ther. 2014;13:3123–36.
Hoarau-Vechot J, Touboul C, Halabi N, Blot-Dupin M, Lis R, Abi Khalil C, Rafii S, Rafii A, Pasquier J. Akt-activated endothelium promotes ovarian cancer proliferation through notch activation. J Transl Med. 2019;17(1):194.
Kobayashi H, Butler JM, O’Donnell R, Kobayashi M, Ding BS, Bonner B, Chiu VK, Nolan DJ, Shido K, Benjamin L, et al. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol. 2010;12(11):1046–56.
Dias S, Choy M, Alitalo K, Rafii S. Vascular endothelial growth factor (VEGF)-C signaling through FLT-4 (VEGFR-3) mediates leukemic cell proliferation, survival, and resistance to chemotherapy. Blood. 2002;99(6):2179–84.
Koistinen P, Siitonen T, Mantymaa P, Saily M, Kinnula V, Savolainen ER, Soini Y. Regulation of the acute myeloid leukemia cell line OCI/AML-2 by endothelial nitric oxide synthase under the control of a vascular endothelial growth factor signaling system. Leukemia. 2001;15(9):1433–41.
Poulos MG, Gars EJ, Gutkin MC, Kloss CC, Ginsberg M, Scandura JM, Rafii S, Butler JM. Activation of the vascular niche supports leukemic progression and resistance to chemotherapy. Exp Hematol. 2014;42(11):976–986.e971–973.
Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6(4):273–86.
Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438(7070):967–74.
Ferrara N, Mass RD, Campa C, Kim R. Targeting VEGF-A to treat cancer and age-related macular degeneration. Annu Rev Med. 2007;58:491–504.
Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8(8):579–91.
Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. 2005;333(2):328–35.
Eklund L, Saharinen P. Angiopoietin signaling in the vasculature. Exp Cell Res. 2013;319(9):1271–80.
Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358(19):2039–49.
Zhong H, Bowen JP. Recent advances in small molecule inhibitors of VEGFR and EGFR signaling pathways. Curr Top Med Chem. 2011;11(12):1571–90.
Bhargava P, Robinson MO. Development of second-generation VEGFR tyrosine kinase inhibitors: current status. Curr Oncol Rep. 2011;13(2):103–11.
Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10(11):868–80.
Zaytseva YY, Valentino JD, Gulhati P, Evers BM. mTOR inhibitors in cancer therapy. Cancer Lett. 2012;319(1):1–7.
Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P, Miglarese M, Epstein DM, McDonald DM. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res. 2011;71(5):1573–83.
Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008;22(4):436–48.
Lim JJ, Yang K, Taylor-Harding B, Wiedemeyer WR, Buckanovich RJ. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia. 2014;16(4):343–353.e341–342.
Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5(200):200ra116.
Chen L, Ashe S, Brady WA, Hellstrom I, Hellstrom KE, Ledbetter JA, McGowan P, Linsley PS. Costimulation of antitumor immunity by the B7 counterreceptor for the T lymphocyte molecules CD28 and CTLA-4. Cell. 1992;71(7):1093–102.
Brown IE, Blank C, Kline J, Kacha AK, Gajewski TF. Homeostatic proliferation as an isolated variable reverses CD8+ T cell anergy and promotes tumor rejection. J Immunol. 2006;177(7):4521–9.
LaGier AJ, Pober JS. Immune accessory functions of human endothelial cells are modulated by overexpression of B7-H1 (PDL1). Hum Immunol. 2006;67(8):568–78.
Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. 2002;99(19):12293–7.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800.
Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci USA. 2015;112(50):15408–13.
Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–94.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–44.
Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, Latreche S, Bergaya S, Benhamouda N, Tanchot C, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212(2):139–48.
Boissonnas A, Fetler L, Zeelenberg IS, Hugues S, Amigorena S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J Exp Med. 2007;204(2):345–56.
Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010;70(15):6171–80.
Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, De Palma A, Mauri P, Monegal A, Rescigno M, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. 2011;208(10):1949–62.
Mukai S, Kagamu H, Shu S, Plautz GE. Critical role of CD11a (LFA-1) in therapeutic efficacy of systemically transferred antitumor effector T cells. Cell Immunol. 1999;192(2):122–32.
Strasly M, Cavallo F, Geuna M, Mitola S, Colombo MP, Forni G, Bussolino F. IL-12 inhibition of endothelial cell functions and angiogenesis depends on lymphocyte-endothelial cell cross-talk. J Immunol. 2001;166(6):3890–9.
Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med. 2006;203(12):2763–77.
Griffioen AW, Damen CA, Martinotti S, Blijham GH, Groenewegen G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: the role of angiogenic factors. Cancer Res. 1996;56(5):1111–7.
Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, Katsaros D, O’Brien-Jenkins A, Gimotty PA, Coukos G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14(1):28–36.
Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69(7):3077–85.
Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–22.
Huang X, Bai X, Cao Y, Wu J, Huang M, Tang D, Tao S, Zhu T, Liu Y, Yang Y, et al. Lymphoma endothelium preferentially expresses Tim-3 and facilitates the progression of lymphoma by mediating immune evasion. J Exp Med. 2010;207(3):505–20.
Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20(6):607–15.
De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154(3):651–63.
Schoors S, De Bock K, Cantelmo AR, Georgiadou M, Ghesquiere B, Cauwenberghs S, Kuchnio A, Wong BW, Quaegebeur A, Goveia J, et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014;19(1):37–48.
Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab. 2014;2(1):3.
Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Investig. 2013;123(9):3664–71.
Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14.
Verdegem D, Moens S, Stapor P, Carmeliet P. Endothelial cell metabolism: parallels and divergences with cancer cell metabolism. Cancer Metab. 2014;2:19.
De Bock K, Georgiadou M, Carmeliet P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013;18(5):634–47.
Quintieri L, Selmy M, Indraccolo S. Metabolic effects of antiangiogenic drugs in tumors: therapeutic implications. Biochem Pharmacol. 2014;89(2):162–70.
DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell. 2012;148(6):1132–44.
Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13(4):227–32.
Schoors S, Cantelmo AR, Georgiadou M, Stapor P, Wang X, Quaegebeur A, Cauwenberghs S, Wong BW, Bifari F, Decimo I, et al. Incomplete and transitory decrease of glycolysis: a new paradigm for anti-angiogenic therapy? Cell Cycle. 2014;13(1):16–22.
Casanovas O. The adaptive stroma joining the antiangiogenic resistance front. J Clin Investig. 2011;121(4):1244–7.
Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL, Johnson M, Lusis AJ, Cohen DA, Iruela-Arispe ML, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115(7):1461–71.
Shojaei F. Anti-angiogenesis therapy in cancer: current challenges and future perspectives. Cancer Lett. 2012;320(2):130–7.
Pasquier J, Abu-Kaoud N, Abdesselem H, Madani A, Hoarau-Vechot J, Thawadi HA, Vidal F, Couderc B, Favre G, Rafii A. SDF-1alpha concentration dependent modulation of RhoA and Rac1 modifies breast cancer and stromal cells interaction. BMC Cancer. 2015;15:569.
Pasquier J, Vidal F, Hoarau-Vechot J, Bonneau C, Darai E, Touboul C, Rafii A. Surgical peritoneal stress creates a pro-metastatic niche promoting resistance to apoptosis via IL-8. J Transl Med. 2018;16(1):271.
Pirtskhalaishvili G, Nelson JB. Endothelium-derived factors as paracrine mediators of prostate cancer progression. Prostate. 2000;44(1):77–87.
Lu J, Ye X, Fan F, Xia L, Bhattacharya R, Bellister S, Tozzi F, Sceusi E, Zhou Y, Tachibana I, et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell. 2013;23(2):171–85.
Lammert E, Cleaver O, Melton D. Role of endothelial cells in early pancreas and liver development. Mech Dev. 2003;120(1):59–64.
Nikolova G, Strilic B, Lammert E. The vascular niche and its basement membrane. Trends Cell Biol. 2007;17(1):19–25.
Dengel LT, Norrod AG, Gregory BL, Clancy-Thompson E, Burdick MD, Strieter RM, Slingluff CL Jr, Mullins DW. Interferons induce CXCR3-cognate chemokine production by human metastatic melanoma. J Immunother. 2010;33(9):965–74.
Kunz M, Toksoy A, Goebeler M, Engelhardt E, Brocker E, Gillitzer R. Strong expression of the lymphoattractant C-X-C chemokine Mig is associated with heavy infiltration of T cells in human malignant melanoma. J Pathol. 1999;189(4):552–8.
von Minckwitz G, Eidtmann H, Rezai M, Fasching PA, Tesch H, Eggemann H, Schrader I, Kittel K, Hanusch C, Kreienberg R, et al. Neoadjuvant chemotherapy and bevacizumab for HER2-negative breast cancer. N Engl J Med. 2012;366(4):299–309.
Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, Erdkamp FL, Vos AH, van Groeningen CJ, Sinnige HA, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360(6):563–72.
Allegra CJ, Yothers G, O’Connell MJ, Sharif S, Petrelli NJ, Colangelo LH, Atkins JN, Seay TE, Fehrenbacher L, Goldberg RM, et al. Phase III trial assessing bevacizumab in stages II and III carcinoma of the colon: results of NSABP protocol C-08. J Clin Oncol. 2011;29(1):11–6.
Bennouna J, Sastre J, Arnold D, Osterlund P, Greil R, Van Cutsem E, von Moos R, Vieitez JM, Bouche O, Borg C, et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol. 2013;14(1):29–37.
Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G, Carey MS, Beale P, Cervantes A, Kurzeder C, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–96.
Zhu AX, Finn RS, Mulcahy M, Gurtler J, Sun W, Schwartz JD, Dalal RP, Joshi A, Hozak RR, Xu Y, et al. A phase II and biomarker study of ramucirumab, a human monoclonal antibody targeting the VEGF receptor-2, as first-line monotherapy in patients with advanced hepatocellular cancer. Clin Cancer Res. 2013;19(23):6614–23.
Garcia JA, Hudes GR, Choueiri TK, Stadler WM, Wood LS, Gurtler J, Bhatia S, Joshi A, Hozak RR, Xu Y, et al. A phase 2, single-arm study of ramucirumab in patients with metastatic renal cell carcinoma with disease progression on or intolerance to tyrosine kinase inhibitor therapy. Cancer. 2014;120(11):1647–55.
Garcia-Carbonero R, Rivera F, Maurel J, Ayoub JP, Moore MJ, Cervantes A, Asmis TR, Schwartz JD, Nasroulah F, Ballal S, et al. An open-label phase II study evaluating the safety and efficacy of ramucirumab combined with mFOLFOX-6 as first-line therapy for metastatic colorectal cancer. Oncologist. 2014;19(4):350–1.
Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausova J, Macarulla T, Ruff P, van Hazel GA, Moiseyenko V, Ferry D, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol. 2012;30(28):3499–506.
Rapisarda A, Melillo G. Overcoming disappointing results with antiangiogenic therapy by targeting hypoxia. Nat Rev Clin Oncol. 2012;9(7):378–90.
Ramlau R, Gorbunova V, Ciuleanu TE, Novello S, Ozguroglu M, Goksel T, Baldotto C, Bennouna J, Shepherd FA, Le-Guennec S, et al. Aflibercept and docetaxel versus docetaxel alone after platinum failure in patients with advanced or metastatic non-small-cell lung cancer: a randomized, controlled phase III trial. J Clin Oncol. 2012;30(29):3640–7.
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, Negrier S, Chevreau C, Desai AA, Rolland F, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol. 2009;27(20):3312–8.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90.
Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, Negrier S, Szczylik C, Pili R, Bjarnason GA, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27(22):3584–90.
Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JV, Booth BP, Verbois SL, Morse DE, Liang CY, et al. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007;13(5):1367–73.
Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501–13.
Sternberg CN, Hawkins RE, Wagstaff J, Salman P, Mardiak J, Barrios CH, Zarba JJ, Gladkov OA, Lee E, Szczylik C, et al. A randomised, double-blind phase III study of pazopanib in patients with advanced and/or metastatic renal cell carcinoma: final overall survival results and safety update. Eur J Cancer. 2013;49(6):1287–96.
van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, Schoffski P, Aglietta M, Staddon AP, Beppu Y, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879–86.
Herbst RS, Sun Y, Eberhardt WE, Germonpre P, Saijo N, Zhou C, Wang J, Li L, Kabbinavar F, Ichinose Y, et al. Vandetanib plus docetaxel versus docetaxel as second-line treatment for patients with advanced non-small-cell lung cancer (ZODIAC): a double-blind, randomised, phase 3 trial. Lancet Oncol. 2010;11(7):619–26.
Wells SA Jr, Gosnell JE, Gagel RF, Moley J, Pfister D, Sosa JA, Skinner M, Krebs A, Vasselli J, Schlumberger M. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol. 2010;28(5):767–72.
Schmoll HJ, Cunningham D, Sobrero A, Karapetis CS, Rougier P, Koski SL, Kocakova I, Bondarenko I, Bodoky G, Mainwaring P, et al. Cediranib with mFOLFOX6 versus bevacizumab with mFOLFOX6 as first-line treatment for patients with advanced colorectal cancer: a double-blind, randomized phase III study (HORIZON III). J Clin Oncol. 2012;30(29):3588–95.
Motzer RJ, Escudier B, Tomczak P, Hutson TE, Michaelson MD, Negrier S, Oudard S, Gore ME, Tarazi J, Hariharan S, et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013;14(6):552–62.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
JP, PG, LC, KR, SR and AR. Conception and design are made by AR and SR. Paper preparation was done by JP, PG, and AR. JP and PG wrote the paper. Paper reviewing is done by JP, LC, KR, and AR. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pasquier, J., Ghiabi, P., Chouchane, L. et al. Angiocrine endothelium: from physiology to cancer. J Transl Med 18, 52 (2020). https://doi.org/10.1186/s12967-020-02244-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-020-02244-9