
Abstract
Members in Motin family, or Angiomotins (AMOTs), are adaptor proteins that localize in the membranous, cytoplasmic or nuclear fraction in a cell context-dependent manner. They control the bioprocesses such as migration, tight junction formation, cell polarity, and angiogenesis. Emerging evidences have demonstrated that AMOTs participate in cancer initiation and progression. Many of the previous studies have focused on the involvement of AMOTs in Hippo-YAP1 pathway. However, it has been controversial for years that AMOTs serve as either positive or negative growth regulators in different cancer types because of the various cellular origins. The molecular mechanisms of these opposite roles of AMOTs remain elusive. This review comprehensively summarized how AMOTs function physiologically and how their dysregulation promotes or inhibits tumorigenesis. Better understanding the functional roles of AMOTs in cancers may lead to an improvement of clinical interventions as well as development of novel therapeutic strategies for cancer patients.
Similar content being viewed by others
Background
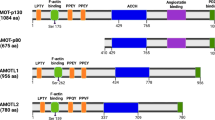
Motin family, also known as AMOTs, consist of three members: Angiomotin (AMOT), Angiomotin-like 1 (AMOTL1) and Angiomotin-like 2 (AMOTL2). AMOT was first identified as an angiostatin binding protein that regulated endothelial cell migration by yeast two-hybrid screening [1]. AMOTL1 and AMOTL2 are two human protein sequences in Motin family, which are similar to AMOT [2]. These proteins exhibit significant sequence homology and share several structural characteristics [2]. A later study identified two isoforms of AMOT, -p80 and -p130 [2, 3]. AMOT-p130 arises from alternative splicing of the AMOT gene between exons 2 and 3 with an extended 409 amino acids at N-terminus. Through alternative splicing, AMOT gene also produces AMOT-p80 which lacks the N-terminal 409 amino acids [4]. AMOT-p130, AMOTL1, and AMOTL2 share the same N-terminus, which is composed of conserved glutamine-rich domains, LPTY motif and PPXY motifs (AMOT-p130: 239PPEY242 and 284PPEY287; AMOTL1: 310PPEY313 and 367PPEY370; AMOTL2: 210PPQY213). PPXY motifs specifically interact with YAP1/TAZ via the WW domain of YAP1/TAZ (Fig. 1) [5,6,7]. In addition, a GST pull-down assay demonstrated that deletion of a single LPTY motif abolished the binding with YAP1, whereas deletion of both PPEY motifs still retained some ability to interact to YAP1. These results suggested that the LPTY is also critical for YAP1 interaction [8]. LPTY motifs were well reserved in AMOTL1 and AMOTL2 [8]. In the C-terminal region, they (AMOT-p80, AMOT-p130, AMOTL1, and AMOTL2) all compose of the conversed Bin/Amphiphysin/Rvs (BAR) domain and the C-terminal PDZ-binding domain [9]. Although there is high similarity among AMOT family members, their various functions are not fully understood. Molecular diversity within the AMOTs have also been revealed [10]. Functionally, AMOTs is involved in Hippo signaling pathway through interacting with multiple core proteins on this pathway, such as Merlin, MST1/2, and YAP1 (Fig. 2). Recent studies have demonstrated several features of AMOTs in numerous pathways, which are directly linked to the initiation and progression of cancers. Particularly, these members function oppositely as oncogenes or tumor suppressive genes depending on different cellular context. For example, most of studies suggested oncogenic functions of AMOT-p80, such as in hemangioendothelioma, head and neck squamous cell carcinoma, and prostate cancer. While for AMOT-p130, it could exert both its oncogenic functions and tumor suppressive functions according to literatures. Currently, AMOTL1 was mainly reported as oncogene in breast cancer and cervical cancer. AMOTL2 is also a promoter of breast cancer progression while it suppressed glioblastoma carcinogenesis (Table 1). However, specific molecular mechanisms behind this situation are not clearly demonstrated. In this review, we will introduce the physiological role of the Motin family members, their controversial indications in cancer, and the mechanisms toward Hippo or non-Hippo pathways.

Schematic illustrations of the domain structures and motifs of the Motin protein family and YAP1-2γ/TAZ. Because of the alternative splicing, AMOT-p80 is an N-terminal truncated protein of AMOT-p130. The N-terminal domain of AMOT-p130 contains LPTY motif and two PPEY motifs, which are required for YAP1-binding. Except for the Angiostatin-binding domain, AMOTL1 and AMOTL2 share sequence identity with AMOT-p130. In addition, all members of Motin possess a Bin/Amphiphysin/Rvs (BAR) domain. Prominent regions of YAP1-2γ/TAZ include WW domain(s), TEAD transcription factor-binding domain, transcriptional activation domain (TAD) and post synaptic density protein (PSD95) binding domain (PDZ BD). WW domains are required by AMOTs binding

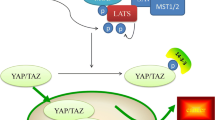
Schematic models of AMOTs interplayed with Hippo-YAP1 cascade. In this model, AMOTs mainly inhibits YAP1 by regulating its localization and promoting Hippo-mediated phosphorylation of YAP1. a AMOTs and YAP1 were co-translocated to the nuclear to promote the transcription of YAP1-target genes. b AMOTs bind with Merlin at the tight junctions and phosphorylate MST1/2 and LATS1/2. Phosphorylated LATS1/2 inactives YAP1 through phosphorylation, resulting in its degradation. c AMOTs physically interact with YAP1 to maintain it cytoplasmic retention. d Actin and YAP1 compete for binding with AMOTs in the tight junctions. TJ tight junction
The physiological roles of AMOTs
AMOTs are differently expressed across different tissues and they show variable spatiotemporal and expressional patterns. The highest expression of AMOT was reported to be in testis, followed by brain and thyroid. AMOTL1 is also highly expressed in skeletal muscle and the lowest levels are found in blood [11]. As for AMOTL2, breast tissue exhibits the highest expression among all the tissues [11]. According to previous researches, endogenous AMOTs expressions have been evaluated in many cell lines, such as in endothelial, epithelial, neural cells and fibroblasts. AMOT can be detected in most of epithelial, endothelial, fibroblast and neural cell lines. Nevertheless, AMOTL2 could not be measured in most cell types of these four cell lines. For AMOTL1, it is presented in a large proportion of endothelial and fibroblast cells [11].
Functional role of AMOTs in embryonic development
During the preimplantation stage, mouse embryos establish two cell lineages by the time of early blastocyst formation: the trophectoderm (TE) and the inner cell mass (ICM) [12]. AMOT, AMOTL1, and AMOTL2 behave variously in embryo development. In early mammalian embryo, AMOT is differentially expressed in the progenitors of the pluripotent ICM and the differentiated TE precursors [13, 14]. The appropriate formation of TE and ICM lineages is dependent on the differential activity of the Hippo-signaling pathway between the outer- and inner-cell populations [15]. The inner cells show high levels of Hippo pathway while outer cells of the embryo show low Hippo pathway [13, 14]. These different activities of Hippo pathway was at least partly regulated by AMOTs. At the adherent junctions of the inner cells, AMOT recruits and activates LATS1/2 to promote Hippo pathway [13, 14]. In apical membrane of the outer cells, F-actin inhibits the ability of AMOT to activate Hippo signaling [13, 14]. AMOT phosphorylation disrupts AMOT—F-actin interaction and leads to reduced F-actin stress fibers and focal adhesions. Also, this phosphorylation was reported to suppress endothelial cell migration in vitro and angiogenesis in zebrafish embryos in vivo [16]. The localization of AMOT is regulated by Rho-associated kinase (Rock) to regulate appropriate apical-basolateral polarization in outer cells [17]. In AMOTL2 knockdown (KD) embryos, they exhibit a normal YAP1 distribution and Hippo pathway activation. However, with AMOTL2 KD in AMOT-null mutants, these embryos exhibit strong nuclear localization of YAP1 in the inner cells, suggesting that some Motin family proteins are essential for the activation of Hippo pathway in preimplantation embryos [18]. During the peri-implantation period, AMOT, AMOTL1, and AMOTL2 are differentially expressed in uterine cells [19]. There was evidence showing a transitional expression pattern of AMOT during the embryonic development: from the anterior visceral endoderm (AVE) to the visceral endoderm (VE) subsequently, and the latter was associated with extra-embryonic ectoderm. In subregions of VE, AMOT regulated morphogenetic movements that were required for embryo viability [20]. AMOT-null mutant embryos failed to exclude YAP1 from the nuclei of the inner cells until 32-cell stage [18]. The embryos survived until the postimplantation stages [18].
On the other hand, AMOTL2 is expressed maternally and in restricted cell types zygotically [21]. Silencing AMOTL2 expression impaired convergence and extension movement [21]. In mosaic embryos, cells with AMOTL2 knockdown failed to migrate properly [21]. AMOTL2 partially co-localized with RhoB- or EEA1- positive endosomes and tyrosine kinase c-Src, which in turn regulated the membrane architecture [21]. Thus, AMOTL2 is essential for cell movements in vertebrate embryos [21]. In zebrafish, AMOTL2 regulates embryonic development through inhibiting Wnt/β-catenin signaling [22]. It can interact with and trap β-catenin in the Rab11-positive recycling endosomes to reduce the amount of β-catenin in both the cytosol and nucleus [22].
Roles of AMOTs in cell migration
AMOT was first identified to mediate angiostatin inhibition of endothelial migration and tube formation in vitro [1]. In zebrafish, knocking down AMOT was shown to decrease the number of filopodia of endothelial tip cells [23]. AMOT is also required for endothelial polarization during migration in that AMOT regulates Rac1 activity in endothelial and epithelial cells [23]. When transgenic mice expressed C-terminal deletion mutant AMOT through endothelial cell-specific receptors tyrosine kinase (TIE) promoter, migration of endothelial cells into the neuroectoderm and intersomitic regions was inhibited [24]. AMOT also interacts with tissue factor pathway inhibitor-1 (TFPI-1), which leads to YAP1 activation and further increases the genes expression relating to proliferation and migration [25]. In Madin–Darby canine kidney (MDCK) cells, overexpression of AMOT induces relocalization of polarity proteins and loss of transepithelial electrical resistance [26]. And the interaction between AMOT and Rich1 (RhoGAP interacting with Cdc42-interacting protein four homologues protein 1) is necessary for maintaining TJ integrity [26]. MUPP1, which has an MRE domain and 13 PDZ domains, is located at TJs. The interaction between MUPP1 and AMOT has been identified in epithelial cells by yeast two-hybrid screening [27]. Patj is a close relative of MUPP1. Possessing a similar structure, Patj interacts with all AMOT family members to regulate the formation of TJ and epithelial polarity [27]. Pdlim2, a member of actin-associated LIM proteins subfamily, is expressed exclusively by podocytes in kidney. AMOTL1 interacts with Pdlim2 and governs the dynamics of the actin cytoskeleton in foot processes [28].
Roles of AMOTs in angiogenesis
Seventy five percentage of AMOT knockout mice died between embryonic day E11 and E11.5 [23]. Most of them exhibited severe vascular insufficiency in the intersomitic region and dilated vessels in the brain [23]. In addition, anti-AMOT antibody significantly inhibits endothelial migration and decreases the number of endothelial filopodia of tip cells during retinal angiogenesis [29]. Moreover, AMOT is a pivotal adaptor protein in the intersection between trafficking, cell junctions and cell migration, which plays a role in directional migration and angiogenesis [30]. Pals1-associated TJ protein (Patj), Lin Seven 1 (Pals1), and Par-3 are crucial for cell polarity. Together with Patj and synectin-binding GEF (Syx), AMOT forms a ternary complex. In vivo, AMOT plays an additive role with Syx in directing endothelial sprouts [31]. AMOT-Patj/multi-PDZ-domain protein 1 (Mupp1)-Syx govern the directional migration of capillaries in the embryo signaling through controlling RhoA GTPase activity to the leading front of migrating cells [32]. AMOT expression is up-regulated in dermal mesenchymal stem cells (DMSCs) in psoriasis, suggesting its involvement in the excessive angiogenesis and vasodilation [33].
AMOT distributes on cell surface and co-localizes with tight junction (zonula occludens) protein (ZO-1) in cell–cell contracts in endothelial cells in vitro and in angiogenic blood vessels of the postnatal mouse’s retina in vivo [3]. AMOT-p130 recruits ZO-1 to actin stress fibers, which is responsible for its localization to actin and tight junctions (TJs) [3]. Furthermore, AMOT-p130 coprecipitates with MAGI-1b, a component of endothelial TJs. Therefore, AMOT contributes to the assembly of endothelial cell–cell junctions [3]. AMOT-p80 and AMOT-p130 play coordinating roles in tube formation by affecting cell migration and cell shape respectively [4]. They were suggested to be differentially expressed in specific stages of mouse retina vascularization [34]. During retinal angiogenesis in vivo, AMOT-p80 was found to be expressed in the migratory phase [34]. In contrast, AMOT-p130 was detected during the period of blood vessel stabilization and maturation [34]. They have distinct functions, AMOT-p80 stimulated endothelial cell migration and angiogenesis while AMOT-p130 stabilized and matured vessels [34]. The rate of AMOT-p80 and AMOT-p130 expression served as an indicator for migration or stabilization of endothelial cells: increased AMOT-p80/-p130 ratio directly reflected the enhanced angiogenic ability of skeletal muscle in response to exercise training [35].
Similar to AMOT, AMOTL1 was also indicated to play a role in endothelial migration and TJ formation in vitro [36]. AMOTL1 mainly affected the stability of cell–cell junctions in stalk cells during sprouting angiogenesis in vivo [36]. It increased the velocity of migration and decreased the persistence of migration directionality. AMOTL1 interacted with AMOT-p80 to form a complex and induced the remodeling of actin cytoskeleton [37]. Motin family has also involved in controlling stability and permeability of endothelial cell junction. HECW2 (HECT, C2 and WW domain containing E3 ubiquitin protein ligase 2), the endothelial cell ubiquitin E3 ligase, physically interacts with AMOTL1 and enhances its stability [38]. In normal retinal, AMOTL1 is essential for normal establishment of vascular networks in the post-natal mouse retina. It forms a complex with N-cadherin to mediate endothelial junctional complex [39].
AMOTL2, accordingly, was detected in blood vessel cells, suggesting its critical roles in regulating multiple behaviors of endothelial cells during angiogenesis. In zebrafish transgenic embryos, knockdown of AMOTL2 impaired the intersegmental vessel growth and suppressed proliferation of endothelial cells [40]. AMOTL2 knockdown also inhibited cell proliferation and migration and disrupted cell polarity of cultured human umbilical vein endothelial cells [40]. It was required for MAPK/ERK activation during angiogenesis [40]. AMOTL2 also participates in aortic vessel lumen expansion through linking VE-cadherin to contractile actin fibers, which has been verified in zebrafish, mouse, and endothelial cell culture systems [41].
The role of AMOTs in Hippo-YAP1 pathway
AMOTs have been reported in kinds of cancer types (Fig. 3 and Table 1). According to GENT database, AMOT expression showed up-regulated in colon cancer and lung cancer while it is down-regulated in breast cancer, kidney cancer, head and neck tumor when comparing to correspondingly normal control tissues (Fig. 3a). Decreased AMOTL1 expression was observed in breast cancer and cervical cancer (Fig. 3b). Interestingly, suppressed and over-expressed AMOTL2 was verified in breast cancer and brain cancer respectively (Fig. 3c). Similar with their different expressional patterns, functional roles of AMOTs are distinct in various cancer types. For example, AMOT, AMOTL1, as well as AMOTL2 exert both oncogene or tumor suppressive gene in different cancer types (Table 1). In this review, we mainly focused on the functions of AMOTs in Hippo-YAP1 pathway. Meanwhile, different mechanisms were summarized based on their involvement in Hippo-YAP1 signaling and other pathways.

Expression pattern of Motin family members in various cancer types which have been reported previously. a According to GENT database, there is discrepancy of expression regarding to AMOT in different tumors. When compared to correspondingly normal control, its expression level is significantly lower in breast cancer (P < 0.001), kidney cancer (P = 0.002), as well as head and neck tumor (P = 0.008); however, AMOT is overexpressed in colon cancer (P = 0.003) and lung cancer (P = 0.018). The difference of AMOT expression between prostate cancer and the corresponding normal is insignificant (P = 0.230). b AMOTL1 exhibits downregulation in both breast cancer (P < 0.001) and cervical cancer (P < 0.001). c Expression of AMOTL2 is only down-regulated in breast cancer (P < 0.001), rather than brain cancer (P = 0.353)
Functional roles of AMOTs in Hippo-YAP1 pathway
Role of AMOTs in Hippo-YAP1 pathway is not clearly proven, even still highly controversial. Some laboratories have shown they inhibit cell proliferation, where others demonstrated the promotion of cell proliferation or tumor growth. Hippo pathway functions as an organ size controller, mainly through its ability to regulate cell proliferation and apoptosis [42, 43]. The core kinase cascade in mammals consists of MST1/2 kinases, WW45, LATS1/2 and Mob1. MST1/2 interacts with WW45 and phosphorylates Mob1 and LATS1/2, resulting in their activation [44]. Activated LATS1/2 phosphorylates YAP1/TAZ. YAP1 and TAZ are the nuclear effectors of Hippo pathway and they belong to the paralogous multifunctional co-activators [45]. AMOTs correlate with YAP1 through the binging of their PPXY and LPXY motifs with the WW domains of YAP1 and TAZ [5,6,7]. YAP1 is a transcriptional co-activator and a key component in Hippo pathway. Therefore, Motin family proteins have been also identified as novel components in Hippo-YAP1 pathway [6, 46].
Merlin functions upstream of the core Hippo pathway kinases LATS1/2 and MST1/2. An AMOT-dependent complex comprised of AMOT, YAP and Merlin has been described [47]. Merlin is a kind of TJ associated protein and interacts directly with AMOT through their mutual coiled-coil domains. In epithelial cells, AMOT retains Merlin at mature TJs. In addition, Merlin functions through AMOT and Rich1 to inhibit Rac1 and Ras-MAPK signaling. Interaction of AMOT and Merlin regulates Rac signaling via Cdc42/Rac1 GAP and Rich1. Therefore, AMOT is considered as a potential player in Merlin-related cancers [6, 48]. An elongated α-helix-rich region of AMOT (residues 404–728 of AMOT-p130) robustly bound to the entire C-terminal part of Merlin to form a stable complex [49]. This interaction releases the auto-inhibition and promotes Merlin’s binding to LATS1/2 to activate Hippo pathway [49].
AMOT was first identified as a partner of YAP1 via large screening of multi-protein complexes that were assembled on de-ubiquitinating enzymes [50]. In MDCK cells, AMOT inhibits YAP1-induced transformation and loss of cell contact inhibition. Knockdown AMOTL2 induces MDCK cell transformation in a YAP1/TAZ dependent manner [6]. AMOTL1, YAP1 and ZO-2 form a tripartite complex to regulate the function of YAP1 in HEK293 cells: AMOTL1 inhibits proapoptotic function of YAP1, while ZO-2 enhance it [51]. AMOTL1 was also reported to be stabilized by being directly phosphorylated by AMPK, which contributing to YAP1 inhibition [52]. On the other hand, AMOTL2 interacts with YAP1 and the Wnt/β-catenin effector Lef1 to control tissue size [53]. Moreover, mono-ubiquitination in turn mediates the function of AMOTL2 itself [54]. Moreover, Hippo-refractory-TAZ mutant (S89A) was reported to be negatively regulated by AMOT and AMOTL1. These results suggest a mechanism of Hippo pathway-independent restriction of TAZ and YAP1 by AMOTs, indicating that the function of AMOTs compensate for the absence of LATS1/2 kinases [55]. As for mammalian cells, AMOT could also regulate YAP1 activity through competitively interacting with endosomal integral membrane protein endotubin (EDTB) [56]. Overexpression of AMOT and AMOTL1 leads to the cytoplasmic retention of TAZ and downregulation of CTGF and Cyr61 expression. Cytoplasmic YAP1 recruits c-Abl to protect AMOTL1 against Nedd4.2-mediated degradation. Therefore, cytoplasmic YAP1 involves in the protecting of AMOTL1 [57].
Tumor suppressive functions of AMOTs
Tumor suppressive functions of AMOTs have been identified in different cancer types. Overexpression of YAP1 impairs the nuclear retention of SRSF1 (serine/arginine-rich splicing factor 1) and itself via AMOT [58]. Therefore, AMOT mediates translocation of SRSF1 through YAP1 in liver cancer cells [58]. In clinical lung cancer specimens, AMOT expression was found to be significantly decreased, which indicated its tumor suppressive role. In vivo, AMOT knockdown increased the growth and spread of Lewis lung carcinoma [59]. Besides, tankyrase inhibitor sensitized cancer cells to endothelial growth factor receptor (EGFR) inhibition through stabilizing AMOT and inhibiting YAP1 signaling in lung cancer cells [60]. Tankyrases inhibitors stabilize AMOT family proteins, disabling the oncogenic function of YAP1 [61, 62]. Through controlling the stability of the AMOT family proteins, E3 ligase ITCH, and the LATS kinases, DUB3 has the potential to act as a tumor suppressor by limiting YAP/TAZ activity [63]. LPTY and PPEY domains are required for full activity of AMOT. Unlike AMOT-p130, due to the lack of these motifs, AMOT-p80 fails to bind with YAP1. Thereby, it is AMOT-p130, instead of AMOT-p80, that promotes YAP1 phosphorylation and inhibits YAP1 transcriptional activity [8]. In addition, AMOT undergoes proteasomal degradation. Three members of Nedd4 (neural-precursor-cell-expressed developmentally down-regulated)-like ubiquitin E3 ligases, Nedd4, Nedd4-2 and Itch, mediate poly-ubiquitination of AMOT-p130 [8]. Nedd4-like ubiquitin E3 ligases competes with YAP1 to bind AMOT-p130 and subsequently targets AMOT-p130 for ubiquitin-dependent degradation [8]. Ubiquitin ligase atrophin-1 interacting protein 4 (AIP4) is a member of the Nedd4 family. AMOT-p130 is ubiquitinated by AIP4 at residue Lys-481 [64]. AMOT-p130 activates AIP4, resulting in the catalysis of ubiquitination of AIP4, AMOT-p130 and YAP1, meaning that AMOT-p130 works interdependently with AIP4 to reduce the stability of YAP1 and inhibit YAP1-dependent transcription as well as cell growth [64]. Hence, AMOT-p130 and AIP4 cooperatively mediate anti-growth partly through the inhibition of YAP1 [64]. AMOTs could also serve as direct activators of LATS2. AMOTL1, AMOTL2 and AMOT-p130 bind with LATS2 and YAP1 to promote LATS2 phosphorylating YAP1 [65]. Post-translational modifications of AMOT family proteins are critical for their context-dependent functions during carcinogenesis. The N-terminal domain of AMOT contains a consensus motif (HxRxxS) for phosphorylation by LATS1/2 [18]. The phosphorylation of AMOT at Serine 176 shifts localization of this complex to the plasma membrane, where it correlates with the tight-junction proteins Pals1/PATJ and E-cadherin. Therefore, phosphorylation of AMOT S175 is a critical post-translational modification that suppresses YAP1’s ability of promoting cell proliferation and tumorigenesis [47]. The phosphorylation of AMOT-p130 by LATS is a key feature that enables it to inhibit YAP1-dependent signaling and cell growth [66]. Phosphorylation of AMOT by LATS1/2 inhibits F-actin binding, cell migration and angiogenesis [16]. AMOT S175 is crucial for the actin-binding activity. Wild-type and non-phosphorylated AMOT (AMOT-S175A) interact with actin filaments, whereas phospho-mimic AMOT (AMOT-S175D) fails to be localized with actin [67]. S175A promotes cell proliferation, while AMOT and S175D inhibits it [67]. Motin family members are required for YAP1 cytoplasmic relocalization in response to the actin cytoskeleton perturbation [68]. F-actin and YAP1 compete to bind with AMOT-p130, accounting for how F-actin inhibits AMOT-p130-mediated cytoplasmic retention of YAP1 [68]. Furthermore, LATS synergizes with F-actin perturbations by phosphorylating free AMOT-p130 to keep it from interacting with F-actin [68]. Mutants’ defective of AMOTs for F-actin binding shows an enhanced ability to retain YAP1 in the cytoplasm. LATS2-phosphorylated AMOT-p130 inhibits the association of AMOT-p130 with F-actin, and AMOT-p130 binding to F-actin inhibited LATS phosphorylation. In short, F-actin and YAP1 competes to bind with AMOT-p130 [68]. Down-regulation of AMOTL2 promotes epithelial-mesenchymal transition (EMT), which was also observed when YAP1 is overexpressed [69].
Oncogenic functions of AMOTs
In some specific cancer types, oncogenic functions of AMOT have also been revealed. RNF146 (an E3 ubiquitin ligase that recognizes ADP-ribosylated substrates) and tankyrase stabilize the Crumbs complex through downregulation of AMOT proteins at the apical membrane [70]. In breast cancer, AMOT was observed to be highly expressed in cancer tissues comparing with adjacent non-cancerous tissues. Decreased AMOT inhibited cell proliferation and invasion. In addition, the expression of YAP1 and LATS1 was suppressed, especially the expression of YAP1, which was significantly decreased [71]. In renal cell carcinoma (RCC), AMOT maintained nuclear localization of YAP so as to increase cell proliferation [72]. Atypical protein kinase C1 (PKC1) was suggested to be an oncogene in lung and ovarian cancer. Knockdown PKC1 enhanced the binding between YAP1 and AMOT, which retained YAP1 in the cytoplasm. PKC1 directly phosphorylated AMOT at Thr750, whose phosphorylation decreases YAP1 binding. PKC1-AMOT-YAP1 signaling axis enhanced the growth of ovarian serous carcinoma tumor [73]. AMOT-p80 functions as a tumor promoter by enhancing PCa cell proliferation. AMOT-p80 promotes nuclear localization of YAP1 through the Hippo pathway, which resulting in an overexpression of YAP1 targeting protein BMP4 [74]. Oncogenic functions of AMOT-p130 have also been justified. Yi et al. [75] suggested that AMOT-p130 was required for YAP1-mediated hepatic epithelial cell proliferation and tumorigenesis. Mice with a liver-specific AMOT knockout presented reduced hepatic ‘oval cell’ proliferation and tumorigenesis in response to toxin-induced injury [75]. These researchers also revealed that AMOT-p130 interacts with YAP1 in both cytoplasm and nucleus. In the cytoplasm, AMOT-p130 prevented the phosphorylation of YAP1 by blocking the access of the WW domains to the kinase LATS1 [75]. In the nucleus, AMOT-p130 interacted with the transcriptional complex containing YAP1 and TEADs to induce the expression of downstream targets [75]. Based on the facts above, AMOT promoted YAP1 nuclear translocation and acted as a transcriptional co-factor of the YAP1-TEAD complex to facilitate proliferation of biliary epithelial cells and cancer development of the liver [75]. In glioblastoma, phosphorylation of AMOTL2 by the mTORC2 kinase enhances YAP1 signaling, leading to cancer growth and invasiveness [76].
AMOTs involved in other molecular mechanisms
In hemangioendothelioma invasion, AMOT promotes endothelial invasion by both stimulating invasion and stabilizing established tubes [77]. In human breast tumor tissues, AMOT was reported to be highly expressed, which was related to angiogenesis [78]. There was evidence that a DNA vaccine targeting AMOT overcame immune tolerance and hampered the progression of incipient tumors [79]. In addition, angiogenesis and tumor growth were inhibited in vivo with targeting AMOT DNA vaccination [80]. Silencing of AMOT prevented the activation and angiogenic effects [81]. In clear cell renal cell carcinoma (ccRCC), AMOT transcripts were associated with poor differentiation, venous invasion, which made it an independent prognostic factor for survival of ccRCC patients [82]. Overexpression of AMOT has also been detected in sinonasal inverted papilloma [83]. In head and neck squamous cell carcinoma (HNSCC), through regulating AMOT-p80 expression, different cell behaviors are induced. For instance, its high expression spured cells proliferate and migrate while its low level induces invasion or metastasis [84]. In prostate cancer, AMOT-p80 functioned as a component of the Cadl1 protein complex, which played a role in cell migration [85]. In colorectal cancer, upregulation of AMOT has been observed. Overexpressed AMOT promoted cell proliferation, cell invasion and migration, as well as apoptotic resistance to 5-fluorouracil. Moreover, AMOT abundance also led to the activation of ERK and AKT pathways [86]. In human osteosarcoma cells, elevated AMOT expression facilitated cell growth and migration [87], whereas its silencing decreased cell proliferation, migration and invasion [88]. A similar results was detected in breast cancer accordingly [89]. In human breast and colon cancer patients, AMOTL2 expression was associated with deficiency of tissue architecture. Hypoxic-stress-induced AMOTL2 activation promoted the loss of polarity [90]. AMOTL2 interacting with AKT and negatively regulating AKT have been identified recently [91]. In vivo, Liver-specific depletion of AMOTL2 enlarged mouse liver, which was correlated with the concomitant activation of YAP and AKT [91]. These observations suggested a dual tumor suppressive function of AMOTL2 through targeting both YAP and AKT [91].
Other functional roles of Motin family
AMOTs have been identified to be essential among viruses as well. Based on previous studies, AMOT-p130 was a host protein required for efficient HIV-1 release, which bound with both NEDD4L and HIV-1 Gag to stimulate HIV-1 release. In addition, expression of either AMOTL1 or AMOTL2 also promoted HIV-1 release and infectivity when AMOT-p130 is absent [92]. Moreover, AMOTL1 has been suggested to play a role in paramyxovirus infection, since AMOTL1 deletion reduced the budding of parainfluenza virus 5 (PIV5) [93]. A raising expression of AMOT was observed among rheumatoid arthritis (RA) patients. However, there was no evidence suggesting a correlation between AMOT level and other clinical variables [94]. In rat models of incipient diabetic nephropathy, inhibition of AMOT reduced glomerular hypertrophy and periodic acid-Schiff positivity. Consequently, the progression of diabetic nephropathy was found to be inhibited [95]. In lung cells, interaction between AMOTL2 and TAZ inhibits surfactant proteins expression. Besides, abundant AMOTL2 inhibits TAZ nuclear distribution, which subsequently decreases the expression of target genes [96]. In mammalian skeletal muscle, AMOTL2 associates with synaptic podosomes in cultured myotubes and it regulates postsynaptic differentiation in muscle cells [97].
Conclusion
Taken together, the functional roles of AMOT-p80, AMOT-p130, AMOTL1, and AMOTL2 in different cancer types are controversial and they highly depend on cell context. In some cancer types, AMOTs promote cell proliferation and invasion, including breast cancer, renal cell carcinoma, colon cancer, prostate cancer, and cervical cancer. However in lung cancer and glioblastoma, AMOTs inhibit tumor growth. In addition, the functional roles of AMOTs in Hippo-YAP1 signaling are still elusive. AMOT promotes either YAP1 nuclear localization or cytoplasmic retention in different cancer types. Moreover, the protein expression of AMOT members varies depending on the cell density. For examples, AMOT protein expression is upregulated in dense-cell condition. In different cancer types, AMOTs play either oncogenic or tumor suppressive role. The mechanisms may rely on the following issues. Firstly, there are three family members of AMOT family. Their expression patterns are distinct, indicating the functional roles of AMOTs might be different. Secondly, the functional roles of different AMOT members are contradictory in different cancer types. In this review, the knowledge about AMOTs is summarized and hints are offered for further investigation.
Therefore, more studies are required to elucidate the controversial function of AMOTs in carcinogenesis. Based on our summary above, several issues need to be addressed in the future study. Firstly, which AMOT family member are predominantly expressed in some specific cancer cells or tissues? Secondly, what is the real function of AMOTs and what are the detailed molecular mechanisms determining the oncogenic or tumor-suppressive function of AMOTs? Thirdly, what are the crucial upstream regulators and downstream effectors of AMOTs in tumorigenesis? Elucidation of AMOTs’ real function and mechanisms may lead to novel therapeutic strategies and promote anti-cancer drug development.
Abbreviations
- AMOT:
-
Angiomotin
- AMOTL1:
-
Angiomotin-like 1
- AMOTL2:
-
Angiomotin-like 2
- BAR domain:
-
Bin/Amphiphysin/Rvs domain
- TE:
-
trophectoderm
- ICM:
-
inner cell mass
- Rock:
-
Rho-associated kinase
- KD:
-
knockdown
- AVE:
-
anterior visceral endoderm
- VE:
-
visceral endoderm
- TIE:
-
tyrosine kinase
- Patj:
-
Pals1-associated TJ protein
- Pals1:
-
Lin Seven 1
- Syx:
-
synectin-binding GEF
- DMSCs:
-
dermal mesenchymal stem cells
- TFPI-1:
-
tissue factor pathway inhibitor-1
- ZO-1:
-
zonula occludens protein 1
- TJs:
-
tight junctions
- HECW2:
-
HECT, C2 and WW domain containing E3 ubiquitin protein ligase 2
- MDCK:
-
Madin–Darby canine kidney
- Rich1:
-
RhoGAP interacting with Cdc42-interacting protein four homologues protein 1
- MUPP1:
-
multi-PDZ-domain protein 1
- Malat1:
-
metastasis-associated lung adenocarcinoma transcript 1
- SRSF1:
-
serine/arginine-rich splicing factor 1
- EGFR:
-
endothelial growth factor receptor
- RNF146:
-
an E3 ubiquitin ligase that recognizes ADP-ribosylated substrates
- RCC:
-
renal cell carcinoma
- PKC1:
-
protein kinase C1
- Nedd4:
-
neural-precursor-cell-expressed developmentally down-regulated
- AIP4:
-
atrophin-1 interacting protein 4
- EMT:
-
epithelial-mesenchymal transition
- HNSCC:
-
head and neck squamous cell carcinoma
- PIV5:
-
parainfluenza virus 5
- EDTB:
-
endosomal integral membrane protein endotubin
- RA:
-
rheumatoid arthritis
- GC:
-
gastric cancer
- TCGA:
-
The Cancer Genome Atlas
References
Troyanovsky B, Levchenko T, Månsson G, Matvijenko O, Holmgren L. Angiomotin an angiostatin binding protein that regulates endothelial cell migration and tube formation. J Cell Biol. 2001;152:1247–54.
Bratt A, Wilson WJ, Troyanovsky B, Aase K, Kessler R, Meir EGV, Holmgren L. Angiomotin belongs to a novel protein family with conserved coiled-coil and PDZ binding domains. Gene. 2002;298:69–77.
Bratt A, Birot O, Sinha I, Veitonmäki N, Aase K, Ernkvist M, Holmgren L. Angiomotin regulates endothelial cell-cell junctions and cell motility. J Biol Chem. 2005;280:34859–69.
Ernkvist M, Aase K, Ukomadu C, Wohlschlegel J, Blackman R, Veitonmäki N, Bratt A, Dutta A, Holmgren L. p130-Angiomotin associates to actin and controls endothelial cell shape. FEBS J. 2006;273:2000–11.
Webb C, Upadhyay A, Giuntini F, Eggleston I, Furutani-Seiki M, Ishima R, Bagby S. Structural features and ligand binding properties of tandem WW domains from YAP and TAZ, nuclear effectors of the Hippo pathway. Biochemistry. 2011;50:3300–9.
Zhao B, Li L, Lu Q, Wang LH, Liu C-Y, Lei Q, Guan K-L. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011;25:51–63.
Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. DOG 1.0: illustrator of protein domain structures. Cell Res. 2009;19:271–3.
Wang C, An J, Zhang P, Xu C, Gao K, Wu D, Wang D, Yu H, Liu JO, Yu L. The Nedd4-like ubiquitin E3 ligases target Angiomotin/p130 to ubiquitin-dependent degradation. Biochem J. 2012;444:279–89.
Liu W, Xie Y, Ma J, Luo X, Nie P, Zuo Z, Lahrmann U, Zhao Q, Zheng Y, Zhao Y. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics. 2015;31:3359–61.
Moreau J, Lord M, Boucher M, Belleau P, Fernandes MJG. Protein diversity is generated within the motin family of proteins by alternative pre-mRNA splicing. Gene. 2005;350:137–48.
Moleirinho S, Guerrant W, Kissil JL. The Angiomotins–from discovery to function. FEBS Lett. 2014;588:2693–703.
Marikawa Y, Alarcón VB. Establishment of trophectoderm and inner cell mass lineages in the mouse embryo. Mol Reprod Dev. 2009;76:1019–32.
Hirate Y, Sasaki H. The role of Angiomotin phosphorylation in the Hippo pathway during preimplantation mouse development. Tissue Barriers. 2014;2:1181–94.
Sasaki H. Position-and polarity-dependent Hippo signaling regulates cell fates in preimplantation mouse embryos. Amsterdam: Elsevier; 2015. p. 80–7.
Nishioka N, Inoue K-I, Adachi K, Kiyonari H, Ota M, Ralston A, Yabuta N, Hirahara S, Stephenson RO, Ogonuki N. The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Dev Cell. 2009;16:398–410.
Dai X, She P, Chi F, Feng Y, Liu H, Jin D, Zhao Y, Guo X, Jiang D, Guan K-L. Phosphorylation of Angiomotin by Lats1/2 kinases inhibits F-actin binding, cell migration, and angiogenesis. J Biol Chem. 2013;288:34041–51.
Mihajlović AI, Bruce AW. Rho-associated protein kinase regulates subcellular localisation of Angiomotin and Hippo-signalling during preimplantation mouse embryo development. Reprod BioMed Online. 2016;33:381–90.
Hirate Y, Hirahara S, Inoue K-I, Suzuki A, Alarcon VB, Akimoto K, Hirai T, Hara T, Adachi M, Chida K. Polarity-dependent distribution of angiomotin localizes Hippo signaling in preimplantation embryos. Curr Biol. 2013;23:1181–94.
Matsumoto H, Fukui E, Yoshizawa M, Sato E, Daikoku T. Differential expression of the motin family in the peri-implantation mouse uterus and their hormonal regulation. J Reprod Dev. 2012;58:649–53.
Shimono A, Behringer RR. Angiomotin regulates visceral endoderm movements during mouse embryogenesis. Curr Biol. 2003;13:613–7.
Huang H, Lu F-I, Jia S, Meng S, Cao Y, Wang Y, Ma W, Yin K, Wen Z, Peng J. Amotl2 is essential for cell movements in zebrafish embryo and regulates c-Src translocation. Development. 2007;134:979–88.
Li Z, Wang Y, Zhang M, Xu P, Huang H, Wu D, Meng A. The Amotl2 gene inhibits Wnt/β-catenin signaling and regulates embryonic development in zebrafish. J Biol Chem. 2012;287:13005–15.
Aase K, Ernkvist M, Ebarasi L, Jakobsson L, Majumdar A, Yi C, Birot O, Ming Y, Kvanta A, Edholm D. Angiomotin regulates endothelial cell migration during embryonic angiogenesis. Genes Dev. 2007;21:2055–68.
Levchenko T, Aase K, Troyanovsky B, Bratt A, Holmgren L. Loss of responsiveness to chemotactic factors by deletion of the C-terminal protein interaction site of Angiomotin. J Cell Sci. 2003;116:3803–10.
Xiao J, Jin K, Wang J, Ma J, Zhang J, Jiang N, Wang H, Luo X, Fei J, Wang Z. Conditional knockout of TFPI-1 in VSMCs of mice accelerates atherosclerosis by enhancing AMOT/YAP pathway. Int J Cardiol. 2017;228:605–14.
Wells CD, Fawcett JP, Traweger A, Yamanaka Y, Goudreault M, Elder K, Kulkarni S, Gish G, Virag C, Lim C. A Rich1/Amot complex regulates the Cdc42 GTPase and apical-polarity proteins in epithelial cells. Cell. 2006;125:535–48.
Sugihara-Mizuno Y, Adachi M, Kobayashi Y, Hamazaki Y, Nishimura M, Imai T, Furuse M, Tsukita S. Molecular characterization of Angiomotin/JEAP family proteins: interaction with MUPP1/Patj and their endogenous properties. Genes Cells. 2007;12:473–86.
Sistani L, Dunér F, Udumala S, Hultenby K, Uhlen M, Betsholtz C, Tryggvason K, Wernerson A, Patrakka J. Pdlim2 is a novel actin-regulating protein of podocyte foot processes. Kidney Int. 2011;80:1045–54.
Levchenko T, Veitonmäki N, Lundkvist A, Gerhardt H, Ming Y, Berggren K, Kvanta A, Carlsson R, Holmgren L. Therapeutic antibodies targeting Angiomotin inhibit angiogenesis in vivo. FASEB J. 2008;22:880–9.
Wu C, Agrawal S, Vasanji A, Drazba J, Sarkaria S, Xie J, Welch CM, Liu M, Anand-Apte B, Horowitz A. Rab13-dependent trafficking of RhoA is required for directional migration and angiogenesis. J Biol Chem. 2011;286:23511–20.
Garnaas MK, Moodie KL, Liu ML, Samant GV, Li K, Marx R, Baraban JM, Horowitz A, Ramchandran R. Syx, a RhoA guanine exchange factor, is essential for angiogenesis in vivo. Circ Res. 2008;103:710–6.
Ernkvist M, Persson NL, Audebert S, Lecine P, Sinha I, Liu M, Schlueter M, Horowitz A, Aase K, Weide T. The Amot/Patj/Syx signaling complex spatially controls RhoA GTPase activity in migrating endothelial cells. Blood. 2009;113:244–53.
Niu X, Chang W, Liu R, Hou R, Li J, Wang C, Li X, Zhang K. mRNA and protein expression of theangiogenesis-related genes EDIL3, AMOT and ECM1 in mesenchymal stem cells in psoriatic dermis. ClinExp Dermatol. 2016;41:533–40.
Ernkvist M, Birot O, Sinha I, Veitonmaki N, Nyström S, Aase K, Holmgren L. Differential roles of p80-and p130-Angiomotin in the switch between migration and stabilization of endothelial cells. Biochimica et Biophysica Acta (BBA)-Mol Cell Res. 2008;1783:429–37.
Roudier E, Chapados N, Decary S, Gineste C, Le Bel C, Lavoie JM, Bergeron R, Birot O. Angiomotin p80/p130 ratio: a new indicator of exercise-induced angiogenic activity in skeletal muscles from obese and non-obese rats? J Physiol. 2009;587:4105–19.
Zheng Y, Vertuani S, Nyström S, Audebert S, Meijer I, Tegnebratt T, Borg J-P, Uhlén P, Majumdar A, Holmgren L. Angiomotin-like protein 1 controls endothelial polarity and junction stability during sprouting angiogenesis. Circ Res. 2009;105:260–70.
Gagné V, Moreau J, Plourde M, Lapointe M, Lord M, Gagnon É, Fernandes MJG. Human Angiomotin-like 1 associates with an Angiomotin protein complex through its coiled-coil domain and induces the remodeling of the actin cytoskeleton. Cell Motil Cytoskelet. 2009;66:754–68.
Choi K-S, Choi H-J, Lee J-K, Im S, Zhang H, Jeong Y, Park JA, Lee I-K, Kim Y-M, Kwon Y-G. The endothelial E3 ligase HECW2 promotes endothelial cell junctions by increasing AMOTL1 protein stability via K63-linked ubiquitination. Cell Signal. 2016;28:1642–51.
Zheng Y, Zhang Y, Barutello G, Chiu K, Arigoni M, Giampietro C, Cavallo F, Holmgren L. Angiomotin like-1 is a novel component of the N-cadherin complex affecting endothelial/pericyte interaction in normal and tumor angiogenesis. Sci Rep. 2016;6:30622.
Wang Y, Li Z, Xu P, Huang L, Tong J, Huang H, Meng A. Angiomotin-like2 gene (amotl2) is required for migration and proliferation of endothelial cells during angiogenesis. J Biol Chem. 2011;286:41095–104.
Hultin S, Zheng Y, Mojallal M, Vertuani S, Gentili C, Balland M, Milloud R, Belting H-G, Affolter M, Helker CSM. AmotL2 links VE-cadherin to contractile actin fibres necessary for aortic lumen expansion. Nat Commun. 2014;5:3743.
Zhao B, Tumaneng K, Guan K-L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. 2011;13:877–83.
Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505.
Edgar BA. From cell structure to transcription: hippo forges a new path. Cell. 2006;124:267–73.
Chen L, Loh PG, Song H. Structural and functional insights into the TEAD-YAP complex in the Hippo signaling pathway. Protein Cell. 2010;1:1073–83.
Bossuyt W, Chen CL, Chen Q, Sudol M, McNeill H, Pan D, Kopp A, Halder G. An evolutionary shift in the regulation of the Hippo pathway between mice and flies. Oncogene. 2014;33:1218–28.
Moleirinho S, Hoxha S, Mandati V, Curtale G, Troutman S, Ehmer U, Kissil JL. Regulation of localization and function of the transcriptional co-activator YAP by Angiomotin. Elife. 2017;6:1–23.
Yi C, Troutman S, Fera D, Stemmer-Rachamimov A, Avila JL, Christian N, Persson NL, Shimono A, Speicher DW, Marmorstein R. A tight junction-associated Merlin-Angiomotin complex mediates Merlin’s regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell. 2011;19:527–40.
Li Y, Zhou H, Li F, Chan SW, Lin Z, Wei Z, Yang Z, Guo F, Lim CJ, Xing W. Angiomotin binding-induced activation of Merlin/NF2 in the Hippo pathway. Cell Res. 2015;25:801–17.
Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403.
Oka T, Schmitt AP, Sudol M. Opposing roles of Angiomotin-like-1 and zona occludens-2 on pro-apoptotic function of YAP. Oncogene. 2012;31:128–34.
DeRan M, Yang J, Shen C-H, Peters EC, Fitamant J, Chan P, Hsieh M, Zhu S, Asara JM, Zheng B. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of Angiomotin-like 1 protein. Cell Rep. 2014;9:495–503.
Agarwala S, Duquesne S, Liu K, Boehm A, Grimm L, Link S, König S, Eimer S, Ronneberger O, Lecaudey V. Amotl2a interacts with the Hippo effector Yap1 and the Wnt/β-catenin effector Lef1 to control tissue size in zebrafish. Elife. 2015;4:e08201.
Kim M, Kim M, Park SJ, Lee C, Lim DS. Role of Angiomotin-like 2 mono-ubiquitination on YAP inhibition. EMBO Rep. 2016;17:64–78.
Chan SW, Lim CJ, Chong YF, Pobbati AV, Huang C, Hong W. Hippo pathway-independent restriction of TAZ and YAP by Angiomotin. J Biol Chem. 2011;286:7018–26.
Cox CM, Mandell EK, Stewart L, Lu R, Johnson DL, McCarter SD, Tavares A, Runyan R, Ghosh S, Wilson JM. Endosomal regulation of contact inhibition through the AMOT: YAP pathway. Mol Biol Cell. 2015;26:2673–84.
Skouloudaki K, Walz G. YAP1 recruits c-Abl to protect Angiomotin-like 1 from Nedd4-mediated degradation. PLoS ONE. 2012;7:e35735.
Wang J, Wang H, Zhang Y, Zhen N, Zhang L, Qiao Y, Weng W, Liu X, Ma L, Xiao W. Mutual inhibition between YAP and SRSF1 maintains long non-coding RNA, Malat1-induced tumourigenesis in liver cancer. Cell Signal. 2014;26:1048–59.
Hsu YL, Hung JY, Chou SH, Huang MS, Tsai MJ, Lin YS, Chiang SY, Ho YW, Wu CY, Kuo PL. Angiomotin decreases lung cancer progression by sequestering oncogenic YAP/TAZ and decreasing Cyr61 expression. Oncogene. 2015;34:4056–68.
Wang H, Lu B, Castillo J, Zhang Y, Yang Z, McAllister G, Lindeman A, Reece-Hoyes J, Tallarico J, Russ C. Tankyrase inhibitor sensitizes lung cancer cells to endothelial growth factor receptor (EGFR) inhibition via stabilizing Angiomotins and inhibiting YAP signaling. J Biol Chem. 2016;291:15256–66.
Wang W, Li N, Li X, Tran MK, Han X, Chen J. Tankyrase inhibitors target YAP by stabilizing Angiomotin family proteins. Cell Rep. 2015;13:524–32.
Troilo A, Benson EK, Esposito D, Garibsingh RA, Reddy EP, Mungamuri SK, Aaronson SA. Angiomotin stabilization by tankyrase inhibitors antagonizes constitutive TEAD-dependent transcription and proliferation of human tumor cells with Hippo pathway core component mutations. Oncotarget. 2016;7:28765–82.
Nguyen HT, Kugler J-M, Cohen SM. DUB3 deubiquitylating enzymes regulate Hippo pathway activity by regulating the stability of ITCH, LATS and AMOT proteins. PloS ONE. 2017;12:e0169587.
Adler JJ, Heller BL, Bringman LR, Ranahan WP, Cocklin RR, Goebl MG, Oh M, Lim H-S, Ingham RJ, Wells CD. Amot130 adapts atrophin-1 interacting protein 4 to inhibit yes-associated protein signaling and cell growth. J Biol Chem. 2013;288:15181–93.
Paramasivam M, Sarkeshik A, Yates JR, Fernandes MJG, McCollum D. Angiomotin family proteins are novel activators of the LATS2 kinase tumor suppressor. Mol Biol Cell. 2011;22:3725–33.
Adler JJ, Johnson DE, Heller BL, Bringman LR, Ranahan WP, Conwell MD, Sun Y, Hudmon A, Wells CD. Serum deprivation inhibits the transcriptional co-activator YAP and cell growth via phosphorylation of the 130-kDa isoform of Angiomotin by the LATS1/2 protein kinases. Proc Natl Acad Sci. 2013;110:17368–73.
Chan SW, Lim CJ, Guo F, Tan I, Leung T, Hong W. Actin-binding and cell proliferation activities of Angiomotin family members are regulated by Hippo pathway-mediated phosphorylation. J Biol Chem. 2013;288:37296–307.
Mana-Capelli S, Paramasivam M, Dutta S, McCollum D. Angiomotins link F-actin architecture to Hippo pathway signaling. Mol Biol Cell. 2014;25:1676–85.
Wang W, Huang J, Chen J. Angiomotin-like proteins associate with and negatively regulate YAP1. J Biol Chem. 2011;286:4364–70.
Campbell CI, Samavarchi-Tehrani P, Barrios-Rodiles M, Datti A, Gingras A-C, Wrana JL. The RNF146 and tankyrase pathway maintains the junctional Crumbs complex through regulation of Angiomotin. J Cell Sci. 2016;129:3396–411.
Lv M, Lv M, Chen L, Qin T, Zhang X, Liu P, Yang J. Angiomotin promotes breast cancer cell proliferation and invasion. Oncol Rep. 2015;33:1938–46.
Lv M, Li S, Luo C, Zhang X, Shen Y, Sui Y, Wang F, Wang X, Yang J, Liu P. Angiomotin promotes renal epithelial and carcinoma cell proliferation by retaining the nuclear YAP. Oncotarget. 2016;7:12393–403.
Wang Y, Justilien V, Brennan KI, Jamieson L, Murray NR, Fields AP. PKCι regulates nuclear YAP1 localization and ovarian cancer tumorigenesis. Oncogene. 2017;36:534–45.
Zeng H, Ortiz A, Shen P-F, Cheng C-J, Lee Y-C, Yu G, Lin S-C, Creighton CJ, Yu-Lee L-Y, Lin S-H. Angiomotin regulates prostate cancer cell proliferation by signaling through the Hippo-YAP pathway. Oncotarget. 2017;8:10145.
Yi C, Shen Z, Stemmer-Rachamimov A, Dawany N, Troutman S, Showe LC, Liu Q, Shimono A, Sudol M, Holmgren L. The p130 isoform of Angiomotin is required for Yap-mediated hepatic epithelial cell proliferation and tumorigenesis. Sci Signal. 2013;6:ra77.
Artinian N, Cloninger C, Holmes B, Benavides-Serrato A, Bashir T, Gera J. Phosphorylation of the Hippo pathway component AMOTL2 by the mTORC2 kinase promotes YAP signaling, resulting in enhanced glioblastoma growth and invasiveness. J Biol Chem. 2015;290:19387–401.
Levchenko T, Bratt A, Arbiser JL, Holmgren L. Angiomotin expression promotes hemangioendothelioma invasion. Oncogene. 2004;23:1469–73.
Jiang WG, Watkins G, Douglas-Jones A, Holmgren L, Mansel RE. Angiomotin and Angiomotin like proteins, their expression and correlation with angiogenesis and clinical outcome in human breast cancer. BMC Cancer. 2006;6:1.
Arigoni M, Barutello G, Lanzardo S, Longo D, Aime S, Curcio C, Iezzi M, Zheng Y, Barkefors I, Holmgren L. A vaccine targeting Angiomotin induces an antibody response which alters tumor vessel permeability and hampers the growth of established tumors. Angiogenesis. 2012;15:305–16.
Holmgren L, Ambrosino E, Birot O, Tullus C, Veitonmäki N, Levchenko T, Carlson L-M, Musiani P, Iezzi M, Curcio C. A DNA vaccine targeting Angiomotin inhibits angiogenesis and suppresses tumor growth. Proc Natl Acad Sci. 2006;103:9208–13.
Stalin J, Harhouri K, Hubert L, Subrini C, Lafitte D, Lissitzky J-C, Elganfoud N, Robert S, Foucault-Bertaud A, Kaspi E. Soluble melanoma cell adhesion molecule (sMCAM/sCD146) promotes angiogenic effects on endothelial progenitor cells through Angiomotin. J Biol Chem. 2013;288:8991–9000.
Yang J, Liu P, Tian M, Li Y, Chen W, Li X. Proteomic identification of Angiomotin by ProteomeLab PF-2D and correlation with clinical outcome in human clear cell renal cell carcinoma. Int J Oncol. 2013;42:2078–86.
Byun JY, Lee SH, Shin JM, Baek BJ, Lee JY. Overexpression of Angiomotin in sinonasal inverted papilloma. Int Forum Allergy Rhinol. 2014;4(6):512–6.
Hakami F, Darda L, Stafford P, Woll P, Lambert DW, Hunter KD. The roles of HOXD10 in the development and progression of head and neck squamous cell carcinoma (HNSCC). Br J Cancer. 2014;111:807–16.
Ortiz A, Lee Y-C, Yu G, Liu H-C, Lin S-C, Bilen MA, Cho H, Yu-Lee L-Y, Lin S-H. Angiomotin is a novel component of cadherin-11/β-catenin/p120 complex and is critical for cadherin-11-mediated cell migration. FASEB J. 2015;29:1080–91.
Zhang Y, Yuan J, Zhang X, Yan F, Huang M, Wang T, Zheng X, Zhang M. Angiomotin promotes the malignant potential of colon cancer cells by activating the YAP-ERK/PI3K-AKT signaling pathway. Oncol Rep. 2016;36:3619–26.
Ruan W, Wang P, Feng S, Xue Y, Li Y. Long non-coding RNA small nucleolar RNA host gene 12 (SNHG12) promotes cell proliferation and migration by upregulating Angiomotin gene expression in human osteosarcoma cells. Tumor Biol. 2016;37:4065–73.
Ruan W-D, Wang P, Feng S, Xue Y, Zhang B. MicroRNA-497 inhibits cell proliferation, migration, and invasion by targeting AMOT in human osteosarcoma cells. OncoTargets Ther. 2016;9:303.
Zhang H, Fan Q. MicroRNA-205 inhibits the proliferation and invasion of breast cancer by regulating AMOT expression. Oncol Rep. 2015;34:2163–70.
Mojallal M, Zheng Y, Hultin S, Audebert S, van Harn T, Johnsson P, Lenander C, Fritz N, Mieth C, Corcoran M. AmotL2 disrupts apical–basal cell polarity and promotes tumour invasion. Nat Commun. 2014;5:4557.
Han H, Yang B, Wang W. Angiomotin-like 2 interacts with and negatively regulates AKT. Oncogene. 2017;36(32):4662.
Mercenne G, Alam SL, Arii J, Lalonde MS, Sundquist WI. Angiomotin functions in HIV-1 assembly and budding. Elife. 2015;4:e03778.
Pei Z, Bai Y, Schmitt AP. PIV5 M protein interaction with host protein Angiomotin-like 1. Virology. 2010;397:155–66.
Park MC, Chung SJ, Park YB, Lee SK. Relationship of angiogenic factors to disease activity and radiographic damage in rheumatoid arthritis. Clin Exp Rheumatol. 2008;26:881.
Celec P, Hodosy J, Gardlík R, Behuliak M, Pálffy R, Pribula M, Jáni P, Turňa J, Šebeková K. The effects of anti-inflammatory and anti-angiogenic DNA vaccination on diabetic nephropathy in rats. Hum Gene Ther. 2011;23:158–66.
Lucci V, Di Palma T, D’Ambrosio C, Scaloni A, Zannini M. AMOTL2 interaction with TAZ causes the inhibition of surfactant proteins expression in lung cells. Gene. 2013;529:300–6.
Proszynski TJ, Sanes JR. Amotl2 interacts with LL5β, localizes to podosomes and regulates postsynaptic differentiation in muscle. J Cell Sci. 2013;126:2225–35.
Nguyen HT, Andrejeva D, Gupta R, Choudhary C, Hong X, Eichhorn PJA, Loya AC, Cohen SM. Deubiquitylating enzyme USP9x regulates Hippo pathway activity by controlling Angiomotin protein turnover. Cell Discov. 2016;2:16001.
Couderc C, Boin A, Fuhrmann L, Vincent-Salomon A, Mandati V, Kieffer Y, Mechta-Grigoriou F, Del Maestro L, Chavrier P, Vallerand D. AMOTL1 promotes breast cancer progression and is antagonized by merlin. Neoplasia. 2016;18:10–24.
Wan H-Y, Li Q-Q, Zhang Y, Tian W, Li Y-N, Liu M, Li X, Tang H. MiR-124 represses vasculogenic mimicry and cell motility by targeting amotL1 in cervical cancer cells. Cancer Lett. 2014;355:148–58.
Authors’ contributions
TH, YZ and JZ conducted the literature search and selected articles for review and wrote the first draft. ASLC, JY, KFT and WK revised manuscript. All authors read and approved the final manuscript.
Acknowledgements
We acknowledge the TCGA research Network (http://cancergenome.nih.gov/), The UCSC Cancer Genomics Browser (https://genome-cancer.ucsc.edu/), and NCI Center for Cancer Genomics Office (http://gdc.nci.nih.gov/) for providing the gastric cancer data set and analysis. We thank the Core Utilities of Cancer Genomics and Pathobiology (CUHK) for providing the facilities and assistance in support of this review.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
No animals have been used in this study and therefore, no requirement of Institutional animal ethical clearance required.
Funding
This study is supported by General Research Fund (RGC Reference Nos. CUHK14114414, 14110016, 14104415, and 14138016) from The Research Grants Council of Hong Kong.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Huang, T., Zhou, Y., Zhang, J. et al. The physiological role of Motin family and its dysregulation in tumorigenesis. J Transl Med 16, 98 (2018). https://doi.org/10.1186/s12967-018-1466-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-018-1466-y