
Abstract
Background
Acute heart failure (AHF) is a frequent reason for hospitalization worldwide and effective treatment options are limited. It is known that AHF is a condition characterized by impaired vasorelaxation, together with reduced nitric oxide (NO) bioavailability, an endogenous vasodilatory compound. Supplementation of inorganic sodium nitrate (NaNO3) is an indirect dietary source of NO, through bioconversion. It is proposed that oral sodium nitrate will favorably affect levels of circulating NO precursors (nitrate and nitrite) in AHF patients, resulting in reduced systemic vascular resistance, without significant hypotension.
Methods and outcomes
We propose a single center, randomized, double-blind, placebo-controlled pilot trial, evaluating the feasibility of sodium nitrate as a treatment for AHF. The primary hypothesis that sodium nitrate treatment will result in increased systemic levels of nitric oxide pre-cursors (nitrate and nitrite) in plasma, in parallel with improved vasorelaxation, as assessed by non-invasively derived systemic vascular resistance index. Additional surrogate measures relevant to the known pathophysiology of AHF will be obtained in order to assess clinical effect on dyspnea and renal function.
Discussion
The results of this study will provide evidence of the feasibility of this novel approach and will be of interest to the heart failure community. This trial may inform a larger study.
Similar content being viewed by others


Background and rationale
Acute heart failure: incidence and current treatment
In the United States, acute heart failure (AHF), also known as acute decompensated heart failure, is responsible for over 1 million hospital admissions annually [1] and is the leading reason for hospitalization among patients over 65 years of age. AHF refers to the heterogeneous syndrome of the gradual or rapid onset of worsening signs and/or symptoms of heart failure [2, 3], attributed to fluid overload, raised ventricular filling pressures and/or pulmonary congestion [4].
Although contemporary medical and device-based therapies (including non-invasive ventilation and ventricular assist devices) have substantially improved the outlook for patients with chronic heart failure, the lack of established effective treatment strategies for patients presenting with AHF has been identified as a major need [5]. This, together with (i) the re-emerging role of vasodilators in AHF, (ii) evidence for acutely impaired nitric oxide (NO) bioavailability and (iii) the potential therapeutic benefit of inorganic sodium nitrate, will be discussed as background to this protocol.
Vascular resistance in the hemodynamics of AHF
A discussion of typical hemodynamic profiles in the spectrum of AHF presentations is crucial to understand the re-emerging role of vasodilators in this context. Patients with AHF generally present with an elevated pulmonary capillary wedge pressure (PCWP), resulting in congestion (interstitial and/or alveolar pulmonary edema), present in the majority of cases of AHF. On the other hand, cardiac output/cardiac index (CI) can vary substantially within the AHF population. The majority of AHF patients (69%) actually present with a CI in the normal range, as illustrated by the work of Nohria et al. [6]. Furthermore, the majority of patients with AHF are known to present with hypertension (50%), rather than normotension (47%) or hypotension (3%) [7]. By inference, given that such patients are typically not hyperdynamic, systemic vascular resistance (SVR) is likely to be generally and markedly elevated at the time of acute presentations in this group. Elevated SVR was found to be the most consistent hemodynamic abnormality in a cross-sectional study employing non-invasive cardiac output measurements in a large cohort of AHF patients [8].
Acute elevation of SVR has several implications in the context of AHF. Increased SVR would generally be attributed to impaired vasorelaxation (or failure of vasodilation) at an arteriolar level, as a net result of neuroendocrine and paracrine signals, in addition to the influence of vasoactive drugs. Importantly, activation of the sympathetic nervous system, with arteriolar vasoconstriction and reduced venous capacitance, is thought to promote the centralization of blood volume, in the genesis of pulmonary congestion [9, 10]. The effects of high SVR on afterload are also likely to be crucial in the pathophysiology of AHF, which has been conceptualized as a condition of afterload mismatch, as reviewed by Cotter et al. [8, 11]. In respect to afterload, it should be noted that there are pulsatile and non-pulsatile elements of afterload and both are known to be abnormal in AHF [12, 13]. All of the above considerations are components of an emerging vasculocentric model of AHF [14].
Current therapeutics in AHF
The primary therapeutic objective during AHF hospitalization is decongestion, whilst avoiding the complications of hypotension and worsening renal function (WRF). Currently, loop diuretic is universally recommended in international guidelines, with the directed use of noninvasive ventilation, nitrovasodilators and inotropic support, in more severe cases [3, 15]. With the exception of noninvasive ventilation, this basic therapeutic tool kit has been consistent for over 30 years. Unfortunately, despite substantial effort, no novel pharmacological strategy has thus far been shown to be superior. Positive inotropes have been associated with increased mortality [16]. In regards to high dose intravenous organic nitrates (e.g. isosorbide dinitrate, glyceryltrinitrate), despite their availability and apparent benefit in several relatively small phase II studies [17,18,19], use is hampered by the development of tolerance [20], as well as the generation of reactive oxygen species [21] and impaired biotransformation in heart failure patients [22].
Several novel neurohumoral therapeutic strategies, including recombinant B-type natriuretic peptide infusion, adenosine receptor antagonism and vasopressin receptor antagonism, have not demonstrated an advantage over standard therapy [23,24,25]. Mechanical diuresis via ultrafiltration has likewise not demonstrated superiority to standard medical diuresis, in fact demonstrating an increased incidence of adverse events [26]. More recently, serelaxin, a recombinant vasodilating peptide, has shown some promise in a study by Teerlink et al. [27]. Whilst there was a significant benefit seen in dyspnea relief during treatment and biomarker endpoints, the 30-day readmission rate was not reduced. Intriguingly, however, the mortality at 6 months was improved over placebo. This finding has focused interest on vasodilation in AHF, as well as informed a large phase III study [28].
Nitric oxide bioavailability in AHF
The normal physiology of vascular relaxation involves the generation of NO via endothelial nitric oxide synthase, from l-arginine; NO freely diffuses to adjacent smooth muscle cells to activate soluble guanylate cyclase (sCG), which induces vasorelaxation via a second messenger cyclic guanylate monophosphate. Heart failure is associated with decreased NO bioavailability and dysfunction of endothelial NOS [29]. In acute decompensation, there is decreased overall NO production, as reflected by a 38% decrease in systemic nitrate and nitrite [30]. An additional mechanism by which NOS function may be acutely impaired is a substantial increase in levels of circulating asymmetric dimethylarginine (ADMA), an endogenous inhibitor of NOS, which was measured simultaneously with reduced nitrate and nitrite levels, by Saitoh and colleges [30]. Thus, in the AHF context, impaired NOS function may be at the nexus of poor NO biosynthesis/bioavailability and elevated SVR. Methods to therapeutically circumvent this deficiency may result in therapeutic benefit.
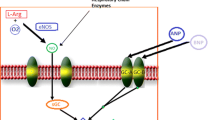
The concept of a circulating pool of inorganic nitrite (\( {\text{NO}}_{2}^{ - } \)) and nitrate (\( {\text{NO}}_{3}^{ - } \)) as an effective NO donor, independent of endothelial NOS function, has received increasing attention over the past decade [31]. Nitrite levels can be increased via direct administration (e.g. intravenous or inhaled), or by oral intake of nitrate (typically in foods or as a pure compound, with a half-life of 5–6 h), which is subsequently reduced in vivo, increasing plasma concentrations of nitrite [32] (see Fig. 1). The therapeutic potential of a nitrate/nitrite supplementation strategy in cardiovascular disease is underscored, when three additional facts are considered: (i) circulating nitrite may selectively donate NO to hypoxic vascular beds [33], (ii) repeat administration does not seem to induce tolerance [34], unlike the situation with organic nitrates and (iii) it is very well tolerated with modest effects on systemic blood pressure (e.g. dietary supplementation of inorganic nitrate produced an average blood pressure lowering of 8.1/3.8 mmHg [35]).

(Source: Allen et al. [52]. (Reproduced with permission from J.D Allen))
Nitrate–nitrite–nitric oxide formation/recycle pathways. In the presence of oxygen, endothelial nitric oxide synthase (NOS) catalyzes the oxidation l-arginine to nitric oxide (NO). NO is oxidized to nitrite (\( {\text{NO}}_{2}^{ - } \)) and nitrate (\( {\text{NO}}_{3}^{ - } \)). Dietary intake of inorganic nitrate (leafy green vegetables, beetroot) has been shown to increase plasma nitrate, which is concentrated in saliva where is it is reduced to nitrite by commensal oral bacteria. Nitrite can then be absorbed and further reduced to NO via several mechanisms, under hypoxic conditions. The pools of circulating nitrate and nitrite are subject to renal excretion, but are also able to be recycled back to NO [52]
Furthermore, recent experience in stable heart failure patients suggests that inorganic nitrate/nitrite supplementation will be effective as a decongestive and vasorelaxant therapy in AHF. In stable compensated heart failure (NB. with preserved ejection fraction), Zamani et al. demonstrated in 17 patients following an acute dose of beetroot juice (containing 8.4 mmol inorganic nitrate), an increase in aerobic work capacity in a maximal supine cycle ergometer test, which was accompanied by a reduction in SVR, and an increase in cardiac output [36]. In advanced but compensated heart failure patients, the Frenneaux group recently reported on the central hemodynamic effects of a 5-min high dose infusion of sodium nitrite (NaNO2). At an infusion rate of 50 μg/kg/min, a 12% reduction in SVR was observed, together with a 13% increase in cardiac output and substantial venodilation, with only a 4 mmHg drop in systolic blood pressure [37]. Whilst these short infusions did not produce any methemoglobinemia, a study of prolonged infusions (up to 48 h) in healthy volunteers by Pluta et al. of ~5 μg/kg/min sodium nitrite did result in methemoglobinemia [38]. Thus, whilst intravenous sodium nitrite holds therapeutic promise for hemodynamic optimization in advanced HF, due to the limited number of studies, the safety of sustained treatment in an AHF episode currently remains a concern.
On the other hand, oral administration of inorganic sodium nitrate has the advantage of having been more widely studied. For example, supplementation of sodium nitrate in the range of 5.1–45 mmol/day (321–2790 mg) was demonstrated to be safe in a meta-analysis of studies in hypertensive patients [39] and may therefore be a safe means of delivering the hemodynamic benefits of NO in AHF. Furthermore, no clinically significant methemoglobinemia (defined by levels ≥5%) has been demonstrated to occur at this dose [40, 41].
Methods and design
This study is considered a pilot and feasibility study [42]. It is a single-center, double blind, placebo-controlled, randomized trial evaluating inorganic sodium nitrate versus placebo. The trial will be conducted at two hospital campuses (Footscray Hospital and Sunshine Hospital) within the Western Health hospital network, in metropolitan Melbourne, Australia. The trial is funded by a grant from the University of Melbourne. The protocol conforms to the SPIRIT 2013 statement, and the CONSORT-EHEALTH checklist. An overview of the trial design is presented in Fig. 2. The trial is registered with the Australian New Zealand Clinical Trials Registry (ACTRN12616000951459).

Overview of trial design
Aims and hypotheses
The aim of this study is to evaluate the feasibility of inorganic sodium nitrate administration, using a single dose (8.4 mmol) and to gain an indication of its effects as a vasoactive treatment for AHF, via bioconversion to NO precursors.
Primary hypothesis
Supplemental sodium nitrate in comparison to placebo will result in (a) increased plasma nitrite levels (as a surrogate for a circulating potential NO pool) in parallel with (b) improved vasorelaxation, as assessed by SVR.
Secondary hypotheses
Supplemental sodium nitrate in comparison to placebo will result in:
-
reduced arterial stiffness, as assessed by augmentation pressure (AP) and augmentation index (AI);
-
preserved/improved renal function, as assessed by serial plasma cystatin C levels;
-
no clinically significant elevation of systemic methemoglobin levels, i.e. not exceeding 5%;
-
greater dyspnea relief as per the Likert scale;
-
greater systemic oxygenation as determined by arterial oxygen saturation as assessed by pulse oximetry;
-
improved plasma troponin and B-type natriuretic peptide (BNP) concentrations;
-
reduced hospital length of stay (LOS) and incidence of clinical deterioration and complications of treatment, including WRF, progression to respiratory failure/ventilatory support, hemodynamic compromise, stroke, and death.
Subject recruitment
Inclusion criteria
Eligible subjects will be ≥18 years who present to hospital within the previous 24 h with AHF and underlying HF with reduced ejection fraction (HFrEF), diagnosed on the basis of at least one symptom (dyspnea, orthopnea or edema) and one sign (chest crackles, peripheral edema, ascites) and/or radiological confirmation of pulmonary vascular congestion on a chest X-ray (CXR). Both the Boston criteria [43] and plasma BNP concentrations (>100 pg/mL) will be used to independently confirm (or refute) the AHF diagnosis. The diagnosis of HFrEF will be based on the European Society of Cardiology Guidelines [3] and adjudicated by the study cardiologist (CN). In order to minimize the risk of serious hypotension in this study, it will be required that the patient must have a baseline systolic blood pressure of ≥110 mmHg at enrolment.
Exclusion criteria
Patients will be excluded if they have a pre-existing and ongoing need for either inotrope therapy or organic nitrates. Patients will be ineligible for enrollment if they have an estimated glomerular filtration rate (eGFR) <20 mL/min/1.73 m2 or are receiving renal replacement therapy (dialysis). The following concomitant conditions will represent further exclusion criteria: anemia with a hemoglobin of less than 10.0 g/dL, critical aortic stenosis, acute severe ischaemia, isolated severe right sided heart failure, respiratory failure, cirrhosis of the liver (Child–Pugh class C) and uncontrolled heart rate (tachycardia or unaddressed bradycardia), advanced malignancy or advanced dementia. Women of child-bearing potential or nursing mothers will also be excluded. Finally, those with phosphodiesterase 5 inhibitor (PDE5I) use within the preceding 24 h will be excluded, on specific enquiry.
Randomization protocol and blinding of treatment allocation
Following informed consent and all baseline assessments, eligible patients will be randomized 1:1 to sodium nitrate or placebo. The randomization list will be computer-generated by an independent statistician. A stratified block permuted randomization will be used with hospital campus as the stratification variable. The block size will not be disclosed to ensure concealment. Randomization will be enabled through a web-based application using REDCap® electronic data capture tools hosted at the University of Melbourne. Patient enrollment and random assignment will be performed by trial investigators. Sodium nitrate and placebo will be packaged identically and labelled by the hospital pharmacy. Treatment assignment numbers will be logged with the hospital pharmacy. Aside from the trial’s hospital pharmacist, patients, nurses, treating clinicians, the trial investigators and the statistician will be blind to treatment allocation.
Preparation and administration of the investigational product
In addition to standard care, patients will be randomly allocated to receive either a single dose of oral sodium nitrate (8.4 mmol, or 714 mg) or matching oral placebo. Food-grade sodium nitrate, a crystalline powder, will be compounded locally into white gelatin capsules. An identical placebo capsule will also be provided, as per randomization, containing an inert substance (microcrystalline cellulose, Avicel®).
Techniques for outcome determination
Measurements for primary outcome determination
Measuring NO directly is inherently difficult, due to compound instability and short biological half-life. The majority of studies measure the concentration of plasma nitrite in order to evaluate the degree of bioconversion resulting from nitrate dosing. In this context, plasma nitrite is therefore a surrogate, reflecting the effective NO pool [44].
In order to assess sodium nitrate bioconversion and pharmacokinetics (including steady state concentrations), blood sampling will occur at time points based on known peak plasma \( {\text{NO}}_{3}^{ - } \) and \( {\text{NO}}_{2}^{ - } \) concentrations and half-lives of orally administered inorganic nitrate [44, 45]. Blood will be drawn at 0, 1, 2, 3 and 6 h, centrifuged for plasma, snap frozen in liquid nitrogen, and stored at −80 °C until analysis. Subsequent batched NO analysis (NOA) will be performed to determine plasma nitrate and nitrite levels. NOA will be performed by chemical reduction, followed by chemiluminescence (Sievers NO analyzer) as previously described [44, 46]. An overview of the trial timeline is presented in Fig. 3.

Overview of trial timeline in hours
In order to assess the vasomotor effects of supplemental nitrate, SVR (indexed to body surface area) will be serially assessed non-invasively. In order to obtain this, bioimpedance cardiography cardiac output measurement will be performed utilizing the PhysioFlow® device, together with simultaneous blood pressure [47] at 0, 3 and 6 h.
Measurements for secondary outcome determination
-
Indices of arterial stiffness (AP and AI) will be derived utilizing the SphygmoCor® device as previously described [48, 49] at 0, 3 and 6 h.
-
Venous methemoglobin will be assayed from blood drawn at time points 0, 3 and 6 utilizing a standard blood gas analyzer.
-
Blood tests: plasma cystatin C, troponin and BNP will be determined at 0 and 6 h, and pre-discharge.
-
Clinical measurements: subjective dyspnea and systemic oxygenation will be prospectively measured via the Likert scale and finger plethysmography, respectively at 0, 3 and 6 h; length of stay and the incidence of complications (WRF, progression to respiratory failure, hypotension/hemodynamic compromise, stroke, and death) will be obtained from the medical record at the time of patient discharge; WRF, or acute kidney injury, will be specifically quantified according to the Kidney Disease Improving Global Outcomes (KDIGO) classification.
Data collection and data management
Study data will be collected and managed using REDCap® electronic data capture tools hosted at the University of Melbourne [50]. REDCap® (Research Electronic Data Capture) is a secure, web-based application designed to support data capture for research studies, providing (1) an intuitive interface for validated data entry; (2) audit trails for tracking data manipulation and export procedures; (3) automated export procedures for seamless data downloads to common statistical packages; and (4) procedures for importing data from external sources. Training of those who collect, check and enter study data will facilitate high quality data, including regular data checks for inconsistency and missing data between and within measurements. Before the start of the statistical analysis, a check will be performed to evaluate the correctness of the randomization.
Statistical methods
In this pilot and feasibility study, the sample size calculation was limited by available resources. As there is currently no relevant literature available to inform the sample size calculation on the primary outcomes in this vulnerable patient population, a total of 40 acute heart failure patients was felt to be realistic and achievable. Twenty patients per treatment group will allow detection of a moderate effect size of 0.6 between sodium nitrate and placebo with a power of 85% using a t test to evaluate the change from baseline to a post-baseline time point at a two-sided significance level of 0.05, assuming a correlation of 0.8 between the baseline and post-baseline measurement. Based on the observed standard deviation for SVR of 296–416 (dyn s/cm5) at baseline in a total of 40 patients reported by Kim et al. [51] and assuming the standard deviation will be similar after sodium nitrate treatment, an effect size of 0.6 corresponds to an absolute treatment effect of about 178–250 (dyn s/cm5) in favour of sodium nitrate compared to placebo, which is considered a clinically relevant improvement.
All randomized patients will be included in the analysis of all outcomes. Descriptive statistics will be used to describe the characteristics of the sample and summarize the data by treatment group and overall. The primary outcomes plasma nitrite levels and SVR will be analyzed separately using a mixed effect repeated measures analysis, including in the model the factors treatment group, time point and treatment by time interaction, while also accounting for baseline plasma levels, baseline by time interaction and the factor hospital campus. The primary hypothesis will be evaluated by examining the main difference between sodium nitrate and placebo as well as the difference between both over time. Secondary outcomes measured will be analyzed similarly to the primary outcomes. If the underlying assumptions of the primary or secondary outcome data are violated, then either an appropriate transformation to allow for parametric methods or non-parametric (or bootstrapping) methods will be used. The number and percentage of patients with events of interest (e.g. complications) will be compared using the Fisher’s exact test. All tests will be two-sided.
Discussion
The combined aims of this project seek to determine the effects of a single dose of orally administered inorganic nitrate in patients with AHF. While previous research indicates that administration of organic nitrates, such as glyceryltrinitrate, can be effective at improving the acute hemodynamic and clinical status of patients with AHF, the induction of tolerance limits usefulness in acute practice. In contrast, we have outlined the therapeutic potential of inorganic nitrate for the clinical context of AHF, which can be given orally and is not subject to the pharmacological tolerance, noting that there have been no recorded studies examining either the bioconversion of inorganic nitrate, or its vascular effects, in this context. If oral inorganic nitrate supplementation is shown to enhance NO bioavailability and produce beneficial changes in peripheral vascular function in AHF, appropriately powered studies may be undertaken. This mechanistic translational study, therefore, has the potential to provide the foundation of a new strategy in AHF, where therapeutic options are acknowledged to be limited.
Abbreviations
- ADMA:
-
asymmetric dimethylarginine
- AHF:
-
acute heart failure
- AI:
-
augmentation index
- AP:
-
augmentation pressure
- BNP:
-
B-type natriuretic peptide
- CI:
-
cardiac index
- CXR:
-
chest X-ray
- eGFR:
-
estimated glomerular filtration rate
- G6PD:
-
glucose-6-phosphate dehydrogenase
- Hb:
-
hemoglobin
- HR:
-
heart rate
- LOS:
-
length of stay
- NaNO2 :
-
sodium nitrite
- NaNO3 :
-
sodium nitrate
- NO:
-
nitric oxide
- \( {\text{NO}}_{2}^{ - } \) :
-
nitrite
- \( {\text{NO}}_{3}^{ - } \) :
-
nitrate
- PCWP:
-
pulmonary capillary wedge pressure
- PDE5I:
-
phosphodiesterase 5 inhibitor
- SVR:
-
systemic vascular resistance
- WRF:
-
worsening renal function
References
Friedewald VE, Gheorghiade M, Yancy CW, Young JB, Roberts WC. The editor’s roundtable: acute decompensated heart failure. Am J Cardiol. 2007;99:1560–7.
Ponikowski P, Jankowska EA. Pathogenesis and clinical presentation of acute heart failure. Rev Esp Cardiol. 2015;68:331–7.
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, Gonzalez-Juanatey JR, Harjola VP, Jankowska EA, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37:2129–200.
Parrinello G, Torres D, Paterna S, di Pasquale P, Licata G. The pathophysiology of acute heart failure: the key role of fluid accumulation. Am Heart J. 2008;156:e19.
Metra M, Felker GM, Zaca V, Bugatti S, Lombardi C, Bettari L, Voors AA, Gheorghiade M, Dei Cas L. Acute heart failure: multiple clinical profiles and mechanisms require tailored therapy. Int J Cardiol. 2010;144:175–9.
Nohria A, Tsang SW, Fang JC, Lewis EF, Jarcho JA, Mudge GH, Stevenson LW. Clinical assessment identifies hemodynamic profiles that predict outcomes in patients admitted with heart failure. J Am Coll Cardiol. 2003;41:1797–804.
Fonarow GC, Corday E, Committee ASA. Overview of acutely decompensated congestive heart failure (ADHF): a report from the ADHERE registry. Heart Fail Rev. 2004;9:179–85.
Cotter G, Moshkovitz Y, Kaluski E, Milo O, Nobikov Y, Schneeweiss A, Krakover R, Vered Z. The role of cardiac power and systemic vascular resistance in the pathophysiology and diagnosis of patients with acute congestive heart failure. Eur J Heart Fail. 2003;5:443–51.
Burchell AE, Sobotka PA, Hart EC, Nightingale AK, Dunlap ME. Chemohypersensitivity and autonomic modulation of venous capacitance in the pathophysiology of acute decompensated heart failure. Curr Heart Fail Rep. 2013;10:139–46.
Isbister JP. Physiology and pathophysiology of blood volume regulation. Transfus Sci. 1997;18:409–23.
Cotter G, Metra M, Milo-Cotter O, Dittrich HC, Gheorghiade M. Fluid overload in acute heart failure—re-distribution and other mechanisms beyond fluid accumulation. Eur J Heart Fail. 2008;10:165–9.
London GM, Marchais SJ, Guerin AP, Pannier B. Arterial stiffness: pathophysiology and clinical impact. Clin Exp Hypertens. 2004;26:689–99.
Marti CN, Gheorghiade M, Kalogeropoulos AP, Georgiopoulou VV, Quyyumi AA, Butler J. Endothelial dysfunction, arterial stiffness, and heart failure. J Am Coll Cardiol. 2012;60:1455–69.
Colombo PC, Onat D, Sabbah HN. Acute heart failure as “acute endothelitis”—interaction of fluid overload and endothelial dysfunction. Eur J Heart Fail. 2008;10:170–5.
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147–239.
Abraham WT, Adams KF, Fonarow GC, Costanzo MR, Berkowitz RL, LeJemtel TH, Cheng ML, Wynne J, Committee ASA and Investigators, Group AS. In-hospital mortality in patients with acute decompensated heart failure requiring intravenous vasoactive medications: an analysis from the Acute Decompensated Heart Failure National Registry (ADHERE). J Am Coll Cardiol. 2005;46:57–64.
Cotter G, Metzkor E, Kaluski E, Faigenberg Z, Miller R, Simovitz A, Shaham O, Marghitay D, Koren M, Blatt A, et al. Randomised trial of high-dose isosorbide dinitrate plus low-dose furosemide versus high-dose furosemide plus low-dose isosorbide dinitrate in severe pulmonary oedema. Lancet. 1998;351:389–93.
Sharon A, Shpirer I, Kaluski E, Moshkovitz Y, Milovanov O, Polak R, Blatt A, Simovitz A, Shaham O, Faigenberg Z, et al. High-dose intravenous isosorbide-dinitrate is safer and better than Bi-PAP ventilation combined with conventional treatment for severe pulmonary edema. J Am Coll Cardiol. 2000;36:832–7.
Beltrame JF, Zeitz CJ, Unger SA, Brennan RJ, Hunt A, Moran JL, Horowitz JD. Nitrate therapy is an alternative to furosemide/morphine therapy in the management of acute cardiogenic pulmonary edema. J Card Fail. 1998;4:271–9.
Mayer B, Beretta M. The enigma of nitroglycerin bioactivation and nitrate tolerance: news, views and troubles. Br J Pharmacol. 2008;155:170–84.
Munzel T, Steven S, Daiber A. Organic nitrates: update on mechanisms underlying vasodilation, tolerance and endothelial dysfunction. Vascul Pharmacol. 2014;63:105–13.
Petersson M, Rundqvist B, Bennett BM, Adams MA, Friberg P. Impaired nitroglycerin biotransformation in patients with chronic heart failure. Clin Physiol Funct Imaging. 2008;28:229–34.
Konstam MA, Gheorghiade M, Burnett JC Jr, Grinfeld L, Maggioni AP, Swedberg K, Udelson JE, Zannad F, Cook T, Ouyang J, et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297:1319–31.
Massie BM, O’Connor CM, Metra M, Ponikowski P, Teerlink JR, Cotter G, Weatherley BD, Cleland JG, Givertz MM, Voors A, et al. Rolofylline, an adenosine A1-receptor antagonist, in acute heart failure. N Engl J Med. 2010;363:1419–28.
O’Connor CM, Starling RC, Hernandez AF, Armstrong PW, Dickstein K, Hasselblad V, Heizer GM, Komajda M, Massie BM, McMurray JJ, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365:32–43.
Bart BA. Treatment of congestion in congestive heart failure: ultrafiltration is the only rational initial treatment of volume overload in decompensated heart failure. Circ Heart Fail. 2009;2:499–504.
Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams KF Jr, et al. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet. 2013;381:29–39.
Tietjens J, Teerlink JR. Serelaxin and acute heart failure. Heart. 2016;102:95–9.
Hermansen SE, Kalstad T, How OJ, Myrmel T. Inflammation and reduced endothelial function in the course of severe acute heart failure. Transl Res. 2011;157:117–27.
Saitoh M, Osanai T, Kamada T, Matsunaga T, Ishizaka H, Hanada H, Okumura K. High plasma level of asymmetric dimethylarginine in patients with acutely exacerbated congestive heart failure: role in reduction of plasma nitric oxide level. Heart Vessels. 2003;18:177–82.
Modin A, Bjorne H, Herulf M, Alving K, Weitzberg E, Lundberg JO. Nitrite-derived nitric oxide: a possible mediator of ‘acidic-metabolic’ vasodilation. Acta Physiol Scand. 2001;171:9–16.
Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–67.
Dejam A, Hunter CJ, Schechter AN, Gladwin MT. Emerging role of nitrite in human biology. Blood Cells Mol Dis. 2004;32:423–9.
Dejam A, Hunter CJ, Tremonti C, Pluta RM, Hon YY, Grimes G, Partovi K, Pelletier MM, Oldfield EH, Cannon RO 3rd, et al. Nitrite infusion in humans and nonhuman primates: endocrine effects, pharmacokinetics, and tolerance formation. Circulation. 2007;116:1821–31.
Kapil V, Khambata RS, Robertson A, Caulfield MJ, Ahluwalia A. Dietary nitrate provides sustained blood pressure lowering in hypertensive patients: a randomized, phase 2, double-blind, placebo-controlled study. Hypertension. 2015;65:320–7.
Zamani P, Rawat D, Shiva-Kumar P, Geraci S, Bhuva R, Konda P, Doulias PT, Ischiropoulos H, Townsend RR, Margulies KB, et al. Effect of inorganic nitrate on exercise capacity in heart failure with preserved ejection fraction. Circulation. 2015;131:371–80 (discussion 380).
Ormerod JO, Arif S, Mukadam M, Evans JD, Beadle R, Fernandez BO, Bonser RS, Feelisch M, Madhani M, Frenneaux MP. Short-term intravenous sodium nitrite infusion improves cardiac and pulmonary hemodynamics in heart failure patients. Circ Heart Fail. 2015;8:565–71.
Pluta RM, Oldfield EH, Bakhtian KD, Fathi AR, Smith RK, Devroom HL, Nahavandi M, Woo S, Figg WD, Lonser RR. Safety and feasibility of long-term intravenous sodium nitrite infusion in healthy volunteers. PLoS ONE. 2011;6:e14504.
Siervo M, Lara J, Ogbonmwan I, Mathers JC. Inorganic nitrate and beetroot juice supplementation reduces blood pressure in adults: a systematic review and meta-analysis. J Nutr. 2013;143:818–26.
Lambers AC, Koppeschaar HPF, van Isselt JW, Slob W, Schothorst RC, Mensinga TT, Meulenbelt J. The effect of nitrate on the thyroid gland function in healthy volunteers in a 4-week oral toxicity study. RIVM National Institute for Public Health and the Environment, Netherlands; 2000.
Nitrite (WHO Food Additive Series 35). http://www.inchem.org/documents/jecfa/jecmono/v35je13.htm. Accessed 07 July 2016.
Eldridge SM, Lancaster GA, Campbell MJ, Thabane L, Hopewell S, Coleman CL, Bond CM. Defining feasibility and pilot studies in preparation for randomised controlled trials: development of a conceptual framework. PLoS ONE. 2016;11:e0150205.
Marantz PR, Tobin JN, Wassertheil-Smoller S, Steingart RM, Wexler JP, Budner N, Lense L, Wachspress J. The relationship between left ventricular systolic function and congestive heart failure diagnosed by clinical criteria. Circulation. 1988;77:607–12.
Woessner M, Smoliga JM, Tarzia B, Stabler T, Van Bruggen M, Allen JD. A stepwise reduction in plasma and salivary nitrite with increasing strengths of mouthwash following a dietary nitrate load. Nitric Oxide. 2016;54:1–7.
Kapil V, Milsom AB, Okorie M, Maleki-Toyserkani S, Akram F, Rehman F, Arghandawi S, Pearl V, Benjamin N, Loukogeorgakis S, et al. Inorganic nitrate supplementation lowers blood pressure in humans: role for nitrite-derived NO. Hypertension. 2010;56:274–81.
Allen JD, Stabler T, Kenjale AA, Ham KL, Robbins JL, Duscha BD, Kraus WE, Annex BH. Diabetes status differentiates endothelial function and plasma nitrite response to exercise stress in peripheral arterial disease following supervised training. J Diabetes Complicat. 2014;28:219–25.
Tonelli AR, Alkukhun L, Arelli V, Ramos J, Newman J, McCarthy K, Pichurko B, Minai OA, Dweik RA. Value of impedance cardiography during 6-min walk test in pulmonary hypertension. Clin Transl Sci. 2013;6:474–80.
Chen CH, Nevo E, Fetics B, Pak PH, Yin FCP, Maughan WL, Kass DA. Estimation of central aortic pressure waveform by mathematical transformation of radial tonometry pressure—validation of generalized transfer function. Circulation. 1997;95:1827–36.
Allen JD, Robbins JL, Vanbruggen MD, Credeur DP, Johannsen NM, Earnest CP, Pieper CF, Johnson JL, Church TS, Ravussin E, et al. Unlocking the barriers to improved functional capacity in the elderly: rationale and design for the “Fit for Life trial”. Contemp Clin Trials. 2013;36:266–75.
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–81.
Kim BG, Cho SW, Lee HY, Kim DH, Byun YS, Goh CW, Rhee KJ, Kim BO. Reduced systemic vascular resistance is the underlying hemodynamic mechanism in nitrate-stimulated vasovagal syncope during head-up tilt-table test. J Arrhythm. 2015;31:196–200.
Allen JD, Giordano T, Kevil CG. Nitrite and nitric oxide metabolism in peripheral artery disease. Nitric Oxide. 2012;26:217–22.
Authors’ contributions
CN conception of research. CN, JDA, MS, JS, SB, RF design of research protocol. CN, MS, RF drafted the manuscript. CN, MS, JDA, RF, SB edited and revised the manuscript. MS, RF prepared figures. All authors read and approved the final manuscript.
Acknowledgements
This research will be conducted at Western Health Footscray and Sunshine Campuses, Melbourne, Victoria, Australia. This investigation is solely the responsibility of the authors and does not necessarily represent the official views of Western Health or the University of Melbourne.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable. This manuscript does not contain any data.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This protocol has been approved by the Melbourne Health Human Research Ethics Committee (HREC/15/MH/402 2015.214) on April 15th 2016. It is being supported by a research grant from the University of Melbourne, Melbourne, Australia, and is registered under The Australian New Zealand Clinical Trials Registry (ACTRN12616000951459).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Falls, R., Seman, M., Braat, S. et al. Inorganic nitrate as a treatment for acute heart failure: a protocol for a single center, randomized, double-blind, placebo-controlled pilot and feasibility study. J Transl Med 15, 172 (2017). https://doi.org/10.1186/s12967-017-1271-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-017-1271-z