
Abstract
Background
Despite the prevalence of CDH23 mutations in East Asians, its large size hinders investigation. The pathologic mutation p.P240L in CDH23 is common in East Asians. However, whether this mutation represents a common founder or a mutational hot spot is unclear. The prevalence of CDH23 mutations with prelingual severe-to-profound sporadic or autosomal recessive sensorineural hearing loss (arSNHL) is unknown in Koreans.
Methods
From September 2010 to October 2014, children with severe-to-profound sporadic or arSNHL without phenotypic markers, and their families, were tested for mutations in connexins GJB2, GJB6 and GJB3. Sanger sequencing of CDH23 p.P240L was performed on connexin-negative samples without enlarged vestibular aqueducts (EVA), followed by targeted resequencing of 129 deafness genes, including CDH23, unless p.P240L homozygotes were detected in the first screening. Four p.P240L-allele-linked STR markers were genotyped in 40 normal-hearing control subjects, and the p.P240L carriers in the hearing-impaired cohort, to identify the haplotypes.
Results
Four (3.1 %) of 128 children carried two CDH23 mutant alleles, and SLC26A4 and GJB2 accounted for 18.0 and 17.2 %, respectively. All four children showed profound nonsyndromic SNHL with minimal residual hearing. Interestingly, all had at least one p.P240L mutant allele. Analysis of p.P240L-linked STR markers in these children and other postlingual hearing-impaired adults carrying p.P240L revealed that p.P240L was mainly carried on a single haplotype.
Conclusions
p.P240L contributed significantly to Korean pediatric severe arSNHL with a strong founder effect, with implications for future phylogenetic studies. Screening for p.P240L as a first step in GJB2-negative arSNHL Koreans without EVA is recommended.
Similar content being viewed by others


Background
Many loci can cause genetic sensorineural hearing loss (SNHL), which accounts for more than 50 % of SNHL [1]. New molecular genetic techniques, such as whole-exome sequencing [2] and targeted resequencing (TRS) [3], have enabled more in-depth understanding of the molecular etiologies of SNHL. These next-generation sequencing (NGS) technologies have assisted in screening of large deafness genes.
CDH23 (NM_022124) is a prime example as this gene consists of 69 exons, 11,073 bases, and is located on chromosome 10q21–q22 [4–7]. It encodes cadherin 23, consisting of 3,354 amino acids and forms 27 extracellular domains (EC), a single transmembrane domain and a short cytoplasmic domain. This protein is reported to contribute to tip link formation by interacting with protocadherin 15 which is encoded by PCDH15 (NM_001142771). It thereby maintains the structural integrity of the tip link and the mechanoelectrical transducer currents of hair cells [8, 9]. Mutations in CDH23 lead to two types of phenotypic disorder or nonsense, splice-site, frameshift mutations, which are assumed to severely disrupt the functions of cadherin 23, usually associated with Usher syndrome type 1D (USH1D; MIM601067) [5]. In contrast, hypo-functional missense mutations result in nonsyndromic hearing loss, preserving retinal and vestibular functions (DFNB12; MIM601386) [5, 10]. When these hypofunctional (DFNB12 allele) and nonfunctional mutations (USH1D allele) are in trans configuration to each other, it is presumed that the residual function of the DFNB12 allele preserves vision and the vestibular function, and impacts the function of only hair cells in the inner ear, resulting in the DFNB12 phenotype [11, 12]. To date, more than 50 mutations have been identified in USH1D patients, and over 24 mutations in DFNB12 patients [13].
The advent of NGS rapidly led to identification of CDH23 as a cause of deafness, especially in East Asians. Indeed, CDH23 mutations contributed significantly to SNHL, with genetic loads that differed according to the ethnicity of the population [10, 14–16]. In East Asians, CDH23 mutations were the third-most frequent after GJB2 and SLC26A4. They accounted for 3.7 % of total hearing loss patients and 5.7 % of autosomal recessive SNHL (arSNHL) patients in Japan [13]. Recently, Park et al. [17] reported an association between CDH23 mutations in hearing loss in 3 (3.22 %) of 93 Korean cochlear implantees with varying degrees of onset, and with diverse inheritance patterns [17]. Another small Korean cohort also had CDH23 mutations (2/13 [15 %]) resulting in autosomal recessive hearing loss with negative GJB2 and SLC26A4 mutations [18]. Most of the patients had progressive, moderate-to-profound, high-tone hearing loss with some residual hearing in the lower frequencies, without vestibular or retinal dysfunction [12, 19].
Rigorous and costly bioinformatic analyses are needed to screen for these mutations, and despite significant reductions in the cost of some NGS techniques, routine clinical screening for CDH23 remains expensive and not cost-effective. Nevertheless, p.P240L, a predominant CDH23 mutation in Japanese and Koreans, is of interest as it accounts for ~43.3 % and >50 % of total CDH23 mutations, respectively [13, 18, 19]. The presence of a predominant allele would facilitate identification of DFNB12 subjects. However, whether p.P240L is a hotspot mutation, a founder mutation, or both, remains to be determined.
In this study, the relative genetic contribution of the CDH23 mutation to nonsyndromic arSNHL was estimated in a Korean pediatric hearing loss cohort, and the genetic load of GJB2 and SLC26A4 was documented. Moreover, whether p.P240L, the most common CDH23 mutation in Koreans, represented a common disease-associated founder mutation was investigated. The clinical characteristics (onset of hearing loss, progression, audiologic evaluation) of the hearing-impaired subjects carrying CDH23 mutations were also documented.
Methods
Ethical considerations
This study was approved by the Institutional Review Boards (IRBs) at the Seoul National University Bundang Hospital (SNUBH) (IRB-B-1007-105-402) and the Seoul National University Hospital (SNUH) (IRBY-H-0905-041-281). Written informed consent was obtained from all participating subjects. In case of the children, written informed consent was obtained from their parents or guardians on their behalf.
Study participants and audiometric evaluation
A total of 438 subjects who visited hereditary hearing loss clinics of tertiary referral centers: (SNUH and SNUBH) for their SNHL from September 2010 to October 2014 were initially enrolled in this study. Clinical evaluations were performed in this cohort, including gender, date of birth, medical history, physical examination, pure tone audiometry and when available, imaging studies (temporal bone computed tomography or magnetic resonance imaging).
All of the enrolled subjects underwent an audiologic evaluation. The pure-tone thresholds were recorded at 0.25, 0.5, 1, 2, 4 and 8 kHz. However, some infants could be recorded at only limited frequencies due to poor cooperation. The hearing threshold was calculated by averaging the thresholds of 0.5, 1, 2 and 4 kHz, and was classified as subtle (16–25 dB), mild (26–40 dB), moderate (41–70 dB), severe (71–95 dB), or profound (>95 dB).
Based on the results of the above clinical and audiologic evaluations, pediatric subjects with prelingual onset, severe-to-profound SNHL segregating either in a sporadic or an autosomal recessive fashion were selected for this study. Individuals with unilateral or phenotypic markers in the inner ear structure, such as an incomplete partition type III, were excluded. However, subjects with an enlarged vestibular aqueduct (EVA) were included to compare the CDH23 mutation frequency with those of mutations in SLC26A4 and other deafness genes in the same cohort (Fig. 1). Consequently, 128 unrelated children with seemingly nonsyndromic severe-to-profound sporadic, or arSNHL without other notable abnormality were finally included in this study. All recruited subjects were Korean.

A diagnostic flow chart of the enrolled subjects. Of a total of 438 subjects with various onset and degree of sensorineural hearing loss (SNHL) diagnosed from 2010 to 2014 at SNUH or SNUBH, 128 with the prelingual onset of a severe-to-profound degree of hearing loss, and with an autosomal recessive (AR) or sporadic hereditary pattern were included in this study. The 310 subjects either with pre-lingual mild to moderate degree SNHL or with post-lingual SNHL subjects are excluded from the present study. SLC26A4 and/or GJB2 were sequenced, and the p.P240L of CDH23 was screened in subjects without SLC26A4 or GJB2 mutations. One of the subjects (SB166-208) with post-lingual SNHL identified to carry p.P240L of CDH23 is included in the STR marker genotyping analysis. Next, targeted resequencing (TRS) of the 80˗204 known deafness genes was performed. Asterisks indicates MYO15A compound heterozygotes; SB77-133, SH10-28, SHJ23, dagger indicates MYO7A compound heterozygotes; SH156-272, SH91-202, SB71-123. Control group I (272 subjects) was assigned for p.P240L mutation screening and control group II (40 subjects) was assigned for STR genotyping. All control groups showed normal hearing.
We recruited two control groups: the control group I included randomly chosen 40 Korean adult subjects with pure tone audiograms indicating normal hearing thresholds and the control group II comprised independent 272 adult subjects with available whole exome sequencing data and with allegedly normal hearing. In this control group II, whole exome sequencing was performed prior to this study to make a molecular diagnosis of diseases irrelevant to SNHL and their hearing status was evaluated by the interview. The control group I and II was recruited for the analysis on the haplotypes around p.P240 of CDH23 and calculation of the normal carrier frequency of p.P240L, respectively.
DNA samples and mutation analysis
Molecular genetic studies were conducted in 128 probands and their family members. Genomic DNA was extracted from peripheral blood using standard protocols (Gentra Puregene Blood Kit, Qiagen, cat. 158389; Venlo, Limburg, Netherlands). The nonsyndromic prelingual arSNHL subjects with an enlarged vestibular aqueduct (EVA) underwent direct PCR-based Sanger sequencing of SLC26A4. Sanger sequencing of GJB2, 3 and 6 was performed on subjects without noticeable phenotypic markers, as described previously [20] (Fig. 1).
Non-EVA samples that were also negative for mutations in connexins GJB2, GJB3, and GJB6 were subject to Sanger sequencing of p.P240L, which was the most frequent mutant allele of CDH23 in Korean cochlear implantees, as reported in our previous study [17] (Fig. 1). The primer sequence for Sanger sequencing to screen p.P240L of CDH23 was CDH23-8F(5′-CCACCATCACTCAACCTAAG-3′) and CDH23-8R (5′-ACTAACAGGGCCCATCCCTA-3). Next, targeted resequencing of 80, 129 (in collaboration with Otogenetics (Norcross, GA, USA) or 200 known deafness genes [17] including CDH23, was performed on the remaining non-EVA samples without a conclusive molecular diagnosis after sequencing of GJB2, GJB3, and GJB6, as described previously [17, 21], with the exception of those samples in which homozygous p.P240L mutations were detected at the screening of p.P240L by Sanger sequencing (Fig. 1). An additional file shows the supplementary lists of the genes included in the three targeted panels (see Additional file 1). This process identified DFNB12 children carrying two probable pathogenic CDH23 variants in a trans configuration.
STR marker genotyping
The haplotype of the p.P240L allele of CDH23 in Koreans was determined to ascertain whether the mutant allele was a common founder. For this purpose, we chose four STR markers flanking the CDH23 gene (Fig. 2a) which included two markers (D10S1650 and D10S1694) previously reported to be hypervariable and informative [5, 11]. Primers for four STR markers flanking the CDH23 gene were generated based on the UCSC Genome Browser as described [21]. The sequences of the microsatellite markers were: D10S584 (5′-TCAATGGGAATGGATACC-3′) (5′-GCAGATCCGAACATGG-3′), D10S1650 (5′-GAAGCCTGTGGTCTAATGAG-3′) (5′-TTCTGGCCTCTGCAGC-3′), D10S606 (5′-TTTGAACCTGGGAGACG-3′) (5′-CATGGACATTCTGCTGC-3′), D10S1694 (5′-CCTGTCTGGCCCAGGTA-3′) (5′-AGTAGGGGTGCTGCTTGA-3′) [5, 11]. Loci were amplified using AmpliTaq DNA polymerase (Invitrogen Life Technologies, Carlsbad, CA, USA) in a Perkin Elmer 9700 thermal cycler (Perkin Elmer, Waltham, MA, USA). The genotypes of these loci on 10 chromosomes were defined using an AmpliTaq gold (Applied Biosystems, Waltham, MA, USA) touchdown protocol under the following conditions: 10 min at 95 °C; 10 cycles of 30 s at 94 °C, 30 s at 65 °C, and 30 s at 72 °C; and a further 20 cycles under the same conditions but with an annealing temperature lowered from 0.5 to 55 °C, 15 cycles of annealing at 55 °C, and a 72 °C final extension for 10 min. STR marker genotyping was performed as described previously [22].

a Location of p.P240L of CDH23 and 4 STR markers linked to the p.P240L mutation on chromosome 10q22.1. Four STR makers were located in the flanking regions of the CDH23 gene. The D10S1650 and D10S1694 were previously reported STR markers to be hypervariable and informative. b Pedigrees showing the segregation of the p.P240L mutation and the four STR marker haplotypes. Haplotypes for STR markers linked to p.P240L from five probands (black arrow) are indicated in bold in either open or shaded boxes). A single haplotype for four STR markers (D10S584, D10S1650, D10S606, and D10S1694) is definitely shared by four alleles linked to p.P240L (shaded box) including one allele from a postlingual-hearing-loss adult (SB116-208), indicative of a very strong founder allele in Koreans. The single common haplotype can be potentially assigned to two of three alleles (open box) carrying p.P240L from two subjects (SB56-103 and SH97-211. Definite haplotypes could be assigned based on information from parental samples or affected siblings in SH164-359, SH59-133, and SB116-208. Despite a lack of parental or sibling samples, a most-likely probable haplotype could be assigned based on the haplotypes of other subjects carrying p.P240L in SB56-103 and SH97-211.
One multiplex family (SB116) with co-segregating post-lingual onset of SNHL and a heterozygous p.P240L mutant allele of CDH23 was also included in this analysis. STR marker analyses were conducted in 11 subjects from five SNHL families (four prelingual and one post-lingual) in total, segregating p.P240L of CDH23, and also in 40 adult Korean subjects with documented normal hearing thresholds.
Statistical analysis
The allele frequency of p.P240L was compared between prelingually onset, severe-to-profound sporadic/autosomal recessive SNHL in a pediatric population and 272 normal hearing controls (control group II). Differences in STR genotypes and the distribution of haplotypes between mutant chromosomes carrying p.P240L and 80 wild-type Korean chromosomes from 40 normal-hearing subjects (control group I) were analyzed by Fisher’s exact test. One postlingual adult DFNB12 subject (SB116-208) was additionally included for the STR genotyping, while the subject was excluded for calculation of the allele frequency of p.P240L among pediatric prelingual severe to profound SNHL. Contributions of some genotypes were compared among subjects, but not between chromosomes, due to the lack of parental samples, as described previously [17, 23]. Especially, haplotypes of normal controls were constructed in a conservative way that was toward minimizing the statistical difference of haplotypes between p.P240L carriers with SNHL and normal controls.
Statistical analyses were performed using SPSS 18.0 (SPSS Inc., Chicago, USA). The level of statistical significance was defined as a p value of <0.05.
Results
The contribution of mutations in CDH23 to profound nonsyndromic arSNHL in Korean children
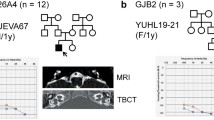
Among 128 children with prelingual onset, nonsyndromic, sporadic or autosomal recessive, severe-to-profound SNHL, 22 (17.2 %) had two mutant alleles of GJB2 (DFNB1) and 23 (18.0 %) carried two mutant alleles of SLC26A4 (DFNB4). The detection frequency of CDH23 mutations (SH 59-133 [17], SH 164-359 [17], SB56-103 and SH 97-211) in our pediatric cohort was 4/128 (3.1 %), which was slightly higher than the 3/128 (2.3 %) and 3/128 (2.3 %) of MYO15A and MYO7A, respectively, in the same cohort. Considering the fact that we did not screen CDH23 from 22 subjects with two mutant alleles of GJB2, the actual frequency of CDH23 might be even slightly high than this figure. Our result showed that CDH23 was one of the leading causes of prelingual-onset profound nonsyndromic arSNHL in children and was a component of the suite of genes that includes SLC26A4 and GJB2, in the Korean pediatric cohort (Fig. 1). The mean age at detection of hearing loss was 2.25 years, with a range of 1–4 years. All four subjects with CDH23 mutations showed a similar phenotype and prelingual profound hearing loss with minimal residual hearing (Fig. 3). None of the four CDH23 carriers exhibited symptoms of nyctalopia, and ophthalmologic examinations revealed no definite abnormalities.

Pedigrees and audiograms of the subjects carrying the CDH23 mutation. All four carriers of p.P240L in CDH23 showed severe-to-profound autosomal recessive prelingual SNHL (arSNHL) with minimal residual hearing.
The p.P240L mutant allele of CDH23 predominated in the pediatric cohort
Interestingly, all four subjects carried p.P240L as either a homozygote or compound heterozygotes, suggesting that this mutant allele was either a strong common founder or a mutational hotspot in this population (Table 1) (Fig. 3).
To ascertain whether p.P240L was a causative variant in these subjects, the carrier frequency of the p.P240L allele was calculated among 272 adult controls with allegedly normal hearing thresholds (control group II). None of the normal control subjects carried the mutant allele, which further confirmed its pathogenic potential (6/256 vs 0/544, p = .001, by Fischer’s exact test) (Table 2).
The rate of concordance in STR markers linked to the p.P240L mutation
p.P240L was assumed to act as a common founder mutation or a mutational hot spot. To ascertain whether p.P240L was a common founder, p.P240L-linked haplotypes of four STR markers (D10S584, D10S1650, D10S606, and D10S1694) were constructed and analyzed in five unrelated probands from five families (four prelingual [SH 59, SH164, SB56, and SH97] and one postlingual [SB116]), and in 40 adult Korean normal-hearing subjects (control group I) (Table 3; Fig. 2b). For two pediatric probands (SH164-359 and SH59-133) and one adult proband (SB116-208), haplotypes could definitely be assigned, based on information from the parents and affected siblings (Fig. 2b). For the remaining two pediatric probands (SH56-103 and SH97-211), precise haplotypes could not be assigned due to a lack of parental or sibling samples. Instead, their most likely probable haplotype was inferred from the other p.P240L-carrying subjects in our cohort (Fig. 2b). In detail, our three p.P240L-carrying probands (SH164-359, SH59-133, and SB116-208) with definitive STR haplotypes shared a common single haplotype (shaded box in Fig. 2b) in their all four alleles linked to p.P240L. Based on this, the other two subjects (SB56-103 and SH97-211) were also assumed to have this common haplotype (Fig. 2b). However, these two subjects (SB56-103 and SH97-211) might have a different haplotype linked to p.P240L against our assumption. Nonetheless, at least four (57.1 %) (shaded box in Fig. 2b) (two alleles from SH164-359, and each allele from SH59-133 and SB116-208) of the seven p.P240L-linked alleles (shaded or open box in Fig. 2b) showed identical haplotypes (Fig. 2b). In contrast, the normal controls showed diverse haplotypes for these STR markers, and the identical haplotype present in the p.P240L carriers could not be assigned to any of the normal controls (0/80) (Table 3). Consequently, p.P240L was significantly associated with a single haplotype in our cohort even under a conservative setting where SH56-103 and SH97-211 were assumed to have a different haplotype (57.1 % [4/7] vs. 0 % [0/80], p < .0001 by Fisher’s exact test). However, the identical haplotype could be inferred also in two subjects (SH56-103 and SH97-211) without any additional samples from their families by assuming the most likely haplotype, making the proportion of shared identical haplotypes 85.7 % (6/7). Conversely, when only three STR markers were considered (D10S584, D10S1650, and D10S606), seven alleles from the normal controls (8.75 %, 7/80) could potentially be matched to an identical haplotype that was strongly associated with p.P240L. Even under this assumption, a highly significant statistical association was detected between p.P240L and a single haplotype (p < .001 by Fisher’s exact test) (Table 3).
Discussion
The prevalence of recessive CDH23 mutations in our pediatric cohort with severe-to-profound nonsyndromic arSNHL was 3.1 %. This was in agreement with findings for CDH23 variants in other studies on East Asian populations, the prevalences of which ranged from 1.6 to 15.4 %, using cohorts with nonsyndromic arSNHL not necessarily limited to prelingual pediatric deaf subjects (Table 2) [18, 19, 24, 25]. The wide range in the frequency from 1.6 to 15.4 % may be attributed to ethnicity differences among East Asians. Alternatively, an intrinsic difference in the study cohort, such as cohort size or onset age of SNHL, could also account for these differences. In this study, the cohort consisted exclusively of prelingual nonsyndromic arSNHL pediatric subjects. Therefore, significantly higher frequency of CDH23 mutations (15.4 vs 3.1 %) obtained from a different Korean cohort with nonsyndromic arSNHL not limited to prelingual SNHL cases [18] (Table 2) may suggest CDH23 mutations also significantly contribute to adult-onset postlingual arSNHL in Koreans. Indeed, in contrast to the p.P240L mutation, which presented mostly as prelingual severe-to-profound hearing loss, certain CDH23 mutations, such as p.R2029W and p.T1368M, were reported to be associated with postlingual onset of moderate hearing loss, and most CDH23-affected subjects showed progressive hearing loss [13]. These results warrant further investigation in a separate study.
CDH23 mutations were the third-most prevalent molecular etiology in the pediatric prelingual arSNHL cohort, after SLC26A4 and GJB2, which accounted for 18.0 and 17.2 %, respectively. A mutation of p.P240L in CDH23 was detected in all four DFNB12 subjects, as either a compound heterozygote or a homozygote, in the Korean cohort. Screening of p.P240L alone significantly facilitated detection of DFNB12 or DFNB12 candidates in Korean subjects, despite the large size of this gene. Given the prevalence of p.P240L, incorporation of a test for p.P240L in CDH23 as a next step in GJB2 sequencing in non-EVA subjects would be cost-effective (Fig. 1).
Indeed, several studies have reported the predominance of the p.P240L mutation in Japanese and Korean populations affected by the prelingual or postlingual arSNHL [18, 19, 24]. In line with these studies, our results showed that 85.7 % (6/8) of the alleles of the CDH23 mutation carriers in the Korean pediatric population had p.P240L (Table 2). Based on the frequency of this variant, we hypothesized that p.P240L of CDH23 had a founder effect in East Asians. Based on our haplotype analyses, p.P240L was mainly carried on a single common haplotype and the common haplotype linked to p.P240L was not detected at all in normal controls, indicating that the p.P240L mutation arose from a common founder in Koreans.
This is the first documented evidence for the founder mutation hypothesis of p.P240L of CDH23. Previous studies on CDH23 mutations in Caucasian populations did not show clear founder mutations due to the heterogeneous ethnicities of these populations [5, 6, 11, 14]. Instead, most of the CDH23 variants from different ethnic backgrounds were assumed to be private [26, 27]. Several reports identified founder effects of frequently encountered mutations in specific ethnic populations. Regarding mutations in GJB2, several studies reported the frequency of 235delC among East Asians [22], 35delG in the Caucasians [28], and 167delT in Ashkenazi Jews [29], suggesting that these were the result of a founder effect, rather than a mutational hot spot. The most common deafness gene in our cohort, SLC26A4, contained the H723R and IVS7-2A>G founder mutations, which are unique to East Asians [30]. The p.A306 mutation of TMPRSS3 was also recently reported to possess a founder role in Koreans [23].
Most CDH23 missense mutations localized in the calcium-binding sequences were associated with nonsyndromic hearing loss, which was presumed to be attributable to the increased sensitivity of the cochlear function to calcium-dependent cell adhesion, compared to that of the retinal function [15]. p.P240L is a missense mutation that affects a residue localized in the EC3 domain, with the highly conserved calcium-binding motifs, and therefore does not interfere with calcium binding. Instead, the change from the rigid cyclic side chain of proline to a longer and more flexible leucine residue, results in instability in the structure and function of the protein [18]. To date, the functional defects caused by the p.P240L mutation have not been determined in vitro or in vivo. However, this mutation has been identified only in subjects with nonsyndromic hearing loss (DFNB12) [13, 18, 19]. Furthermore, based solely on a previous audiologic phenotypic study, p.P240L carriers usually show congenital and more severe SNHL than do carriers of other missense mutations in CDH23 [13]. In accordance with this finding, the four pediatric subjects in this study with p.P240L showed prelingual and severe-to-profound nonsyndromic SNHL with minimal residual hearing at all frequencies.
The p.P240L allele can be classified as a DFNB12 allele, rather than an USH1D allele, based on a previous hypothesis [11] and the phenotype of p.P240L carriers in East Asian populations [13, 18, 19]. The higher prevalence of this DFNB12 allele of p.P240L compared to other CDH23 mutant alleles implies that in Koreans the nonsyndromic hearing loss (DFNB12) phenotype is more commonly associated with CDH23 mutations than is the syndromic hearing loss phenotype (USH1D).
The geographic or ethnic spectra of founder mutations vary, and whether the most frequent mutation allele represents a founder mutation and/or a mutational hot spot is often unclear. Some founder mutations have a narrow distribution; for example, due to the practice of consanguineous marriage, the high prevalence of GJB2 mutations in some Western cultures can be attributed to a single ancestor mutation [31]. On the other hand, the p.A306T mutation of TMPRSS3 affects a wide ethnic spectrum, encompassing East Asians and Europeans, and hence is predicted to be a mutational hot spot [23, 32, 33]. However, the distribution of several founder alleles, such as the 235delC mutation of GJB2 and the H723R and IVS7-2A>G mutations of SLC26A4, is restricted to East Asians [22, 30]. Only a single cohort of Koreans was investigated in this study, a high prevalence of the p.P240L mutation in East Asians has been reported by others [13, 18, 19, 34], whereas this mutation was rarely reported in other ethnic groups [5, 6, 14]. Therefore, p.P240L is likely a founder mutation exclusive to East Asians. Information on the distinct distribution pattern of the deafness mutations may facilitate determination of the phylogenetic evolution of the deafness genes. Although its value would be limited due to a lack of data on Mongolian or other Asian populations, the different frequency of the p.P240L mutation between East Asian populations and other ethnicities suggests that the p.P240L mutation occurred sometime after the divergence of several ethnic groups from Central or East Asia ~40,000 years ago [35, 36]. Further studies using other genetic polymorphisms, such as single-nucleotide polymorphisms (SNPs) and STR markers adjacent to the p.P240L mutation, would enable a more accurate estimate of the age of this mutation. Further data on the prevalence of CDH23 mutations and the haplotypes in other ethnic groups would enable tracing of the common ancestor or route of divergence of the CDH23 mutation. This information would have phylogenetic applications.
Conclusion
The genetic load of CDH23 mutations in Korean children with prelingual onset, nonsyndromic, sporadic or autosomal recessive, severe-to-profound SNHL was 3.1 %. p.P240L was the most common CDH23 mutation. Genotyping of STR markers linked to p.P240L showed that the p.P240L mutation had a strong founder effect in Koreans. This novel finding may facilitate genetic diagnosis of, and research into, CDH23 mutations, as well as provide information for further phylogenetic studies.
References
del Castillo FJ, Rodriguez-Ballesteros M, Alvarez A, Hutchin T, Leonardi E, de Oliveira CA et al (2005) A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J Med Genet 42:588–594
Diaz-Horta O, Duman D, Foster J 2nd, Sirmaci A, Gonzalez M, Mahdieh N et al (2012) Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PLoS One 7:e50628
Lin X, Tang W, Ahmad S, Lu J, Colby CC, Zhu J et al (2012) Applications of targeted gene capture and next-generation sequencing technologies in studies of human deafness and other genetic disabilities. Hear Res 288:67–76
Chaib H, Place C, Salem N, Dode C, Chardenoux S, Weissenbach J et al (1996) Mapping of DFNB12, a gene for a non-syndromal autosomal recessive deafness, to chromosome 10q21-22. Hum Mol Genet 5:1061–1064
Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL et al (2001) Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet 68:26–37
Bolz H, von Brederlow B, Ramirez A, Bryda EC, Kutsche K, Nothwang HG et al (2001) Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet 27:108–112
Wayne S, Der Kaloustian VM, Schloss M, Polomeno R, Scott DA, Hejtmancik JF et al (1996) Localization of the Usher syndrome type ID gene (Ush1D) to chromosome 10. Hum Mol Genet 5:1689–1692
Alagramam KN, Goodyear RJ, Geng R, Furness DN, van Aken AF, Marcotti W et al (2011) Mutations in protocadherin 15 and cadherin 23 affect tip links and mechanotransduction in mammalian sensory hair cells. PLoS One 6:e19183
Kazmierczak P, Sakaguchi H, Tokita J, Wilson-Kubalek EM, Milligan RA, Muller U et al (2007) Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature 449:87–91
Astuto LM, Bork JM, Weston MD, Askew JW, Fields RR, Orten DJ et al (2002) CDH23 mutation and phenotype heterogeneity: a profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am J Hum Genet 71:262–275
Schultz JM, Bhatti R, Madeo AC, Turriff A, Muskett JA, Zalewski CK et al (2011) Allelic hierarchy of CDH23 mutations causing non-syndromic deafness DFNB12 or Usher syndrome USH1D in compound heterozygotes. J Med Genet 48:767–775
Pennings RJ, Topsakal V, Astuto L, de Brouwer AP, Wagenaar M, Huygen PL et al (2004) Variable clinical features in patients with CDH23 mutations (USH1D-DFNB12). Otol Neurotol 25:699–706
Miyagawa M, Nishio SY, Usami S (2012) Prevalence and clinical features of hearing loss patients with CDH23 mutations: a large cohort study. PLoS One 7:e40366
Baux D, Faugere V, Larrieu L, Le Guedard-Mereuze S, Hamroun D, Beroud C et al (2008) UMD-USHbases: a comprehensive set of databases to record and analyse pathogenic mutations and unclassified variants in seven Usher syndrome causing genes. Hum Mutat 29:E76–E87
Oshima A, Jaijo T, Aller E, Millan JM, Carney C, Usami S et al (2008) Mutation profile of the CDH23 gene in 56 probands with Usher syndrome type I. Hum Mutat 29:E37–E46
Ouyang XM, Yan D, Du LL, Hejtmancik JF, Jacobson SG, Nance WE et al (2005) Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population. Hum Genet 116:292–299
Park JH, Kim NK, Kim AR, Rhee J, Oh SH, Koo JW et al (2014) Exploration of molecular genetic etiology for Korean cochlear implantees with severe to profound hearing loss and its implication. Orphanet J Rare Dis 9:167
Woo HM, Park HJ, Park MH, Kim BY, Shin JW, Yoo WG et al (2014) Identification of CDH23 mutations in Korean families with hearing loss by whole-exome sequencing. BMC Med Genet 15:46
Wagatsuma M, Kitoh R, Suzuki H, Fukuoka H, Takumi Y, Usami S (2007) Distribution and frequencies of CDH23 mutations in Japanese patients with non-syndromic hearing loss. Clin Genet 72:339–344
Kim SY, Park G, Han KH, Kim A, Koo JW, Chang SO et al (2013) Prevalence of p.V37I variant of GJB2 in mild or moderate hearing loss in a pediatric population and the interpretation of its pathogenicity. PLoS One 8:e61592
Choi BY, Park G, Gim J, Kim AR, Kim BJ, Kim HS et al (2013) Diagnostic application of targeted resequencing for familial nonsyndromic hearing loss. PLoS One 8:e68692
Yan D, Park HJ, Ouyang XM, Pandya A, Doi K, Erdenetungalag R et al (2003) Evidence of a founder effect for the 235delC mutation of GJB2 (connexin 26) in east Asians. Hum Genet 114:44–50
Chung J, Park SM, Chang SO, Chung T, Lee KY, Kim AR et al (2014) A novel mutation of TMPRSS3 related to milder auditory phenotype in Korean postlingual deafness: a possible future implication for a personalized auditory rehabilitation. J Mol Med (Berl) 92:651–663
Usami S, Nishio SY, Nagano M, Abe S, Yamaguchi T (2012) Deafness Gene Study C: Simultaneous screening of multiple mutations by invader assay improves molecular diagnosis of hereditary hearing loss: a multicenter study. PLoS One 7:e31276
Mizutari K, Mutai H, Namba K, Miyanaga Y, Nakano A, Arimoto Y et al (2015) High prevalence of CDH23 mutations in patients with congenital high-frequency sporadic or recessively inherited hearing loss. Orphanet J Rare Dis 10:60
Liu XZ, Blanton SH, Bitner-Glindzicz M, Pandya A, Landa B, MacArdle B et al (2001) Haplotype analysis of the USH1D locus and genotype-phenotype correlations. Clin Genet 60:58–62
Ebermann I, Lopez I, Bitner-Glindzicz M, Brown C, Koenekoop RK, Bolz HJ (2007) Deafblindness in French Canadians from Quebec: a predominant founder mutation in the USH1C gene provides the first genetic link with the Acadian population. Genome Biol 8:R47
Van Laer L, Coucke P, Mueller RF, Caethoven G, Flothmann K, Prasad SD et al (2001) A common founder for the 35delG GJB2 gene mutation in connexin 26 hearing impairment. J Med Genet 38:515–518
Morell RJ, Kim HJ, Hood LJ, Goforth L, Friderici K, Fisher R et al (1998) Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med 339:1500–1505
Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M et al (2003) Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet 40:242–248
Nance WE, Liu XZ, Pandya A (2000) Relation between choice of partner and high frequency of connexin-26 deafness. Lancet 356:500–501
Weegerink NJ, Schraders M, Oostrik J, Huygen PL, Strom TM, Granneman S et al (2011) Genotype-phenotype correlation in DFNB8/10 families with TMPRSS3 mutations. J Assoc Res Otolaryngol 12:753–766
Elbracht M, Senderek J, Eggermann T, Thurmer C, Park J, Westhofen M et al (2007) Autosomal recessive postlingual hearing loss (DFNB8): compound heterozygosity for two novel TMPRSS3 mutations in German siblings. J Med Genet 44:e81
Tsukada K, Nishio S, Usami S (2010) Deafness Gene Study C: A large cohort study of GJB2 mutations in Japanese hearing loss patients. Clin Genet 78:464–470
Cavalli-Sforza LL, Feldman MW (2003) The application of molecular genetic approaches to the study of human evolution. Nat Genet 33(Suppl):266–275
Tateno Y, Komiyama T, Katoh T, Munkhbat B, Oka A, Haida Y et al (2014) Divergence of East Asians and Europeans estimated using male- and female-specific genetic markers. Genome Biol Evol 6:466–473
Authors’ contributions
SYK and ARK wrote the manuscript. BYC, ARK, NKDK, MYK, EHJ, YEH, MYC and WYP carried out the genetic tests, and BYC, SYK, ARK, and BJK analyzed the data. BYC helped to prepare and revise the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank all the subjects of this study for their participation in this research. This study was supported by the grants of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI14C1867 to B.Y. Choi) and the Research Coordination center for rare diseases (HI12C0014[A120017] to B.Y. Choi).
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
So Young Kim and Ah Reum Kim contributed equally to this work
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kim, S.Y., Kim, A.R., Kim, N.K.D. et al. Strong founder effect of p.P240L in CDH23 in Koreans and its significant contribution to severe-to-profound nonsyndromic hearing loss in a Korean pediatric population. J Transl Med 13, 263 (2015). https://doi.org/10.1186/s12967-015-0624-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-015-0624-8