
Abstract
Background
Febuxostat is a selective inhibitor of xanthine oxidase (XO). XO is a critical source of reactive oxygen species (ROS) during myocardial ischemia/reperfusion (I/R) injury. Inhibition of XO is therapeutically effective in I/R injury. Evidence suggests that febuxostat exerts antioxidant effects by directly scavenging ROS. The present study was performed to investigate the effects of febuxostat on myocardial I/R injury and its underlying mechanisms.
Methods
We utilized an in vivo mouse model of myocardial I/R injury and an in vitro neonatal rat cardiomyocyte (NRC) model of hypoxia/reoxygenation (H/R) injury. Mice were randomized into five groups: Sham, I/R (I/R + Vehicle), I/R + FEB (I/R + febuxostat), AL + I/R (I/R + allopurinol) and FEB (febuxostat), respectively. The I/R + FEB mice were pretreated with febuxostat (5 mg/kg; i.p.) 24 and 1 h prior to I/R. NRCs received febuxostat (1 and 10 µM) at 24 and 1 h before exposure to hypoxia for 3 h followed by reoxygenation for 3 h. Cardiac function, myocardial infarct size, serum levels of creatine kinase (CK) and lactate dehydrogenase (LDH), and myocardial apoptotic index (AI) were measured in order to ascertain the effects of febuxostat on myocardial I/R injury. Hypoxia/reperfusion (H/R) injury in NRCs was examined using MTT, LDH leakage assay and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. The underlying mechanisms were determined by measuring ROS production, mitochondrial membrane potential (ΔΨm), and expression of cytochrome c, cleaved caspases as well as Bcl-2 protein levels.
Results
Myocardial I/R led to an elevation in the myocardial infarct size, serum levels of CK and LDH, cell death and AI. Furthermore, I/R reduced cardiac function. These changes were significantly attenuated by pretreatment with febuxostat and allopurinol, especially by febuxostat. Febuxostat also protected the mitochondrial structure following myocardial I/R, inhibited H/R-induced ROS generation, stabilized the ΔΨm, alleviated cytosolic translocation of mitochondrial cytochrome C, inhibited activation of caspase-3 and -9, upregulated antiapoptotic proteins and downregulated proapoptotic proteins.
Conclusions
This study revealed that febuxostat pretreatment mediates the cardioprotective effects against I/R and H/R injury by inhibiting mitochondrial-dependent apoptosis.
Similar content being viewed by others

Background
Myocardial ischemia/reperfusion (I/R) injury is characterized by altered metabolic disorders and structural damage during reperfusion following myocardial ischemia [1]. The myocardial injury paradoxically reduces the benefit of procedures, such as thrombolytic therapy, percutaneous coronary intervention, and coronary bypass surgery [2]. Therefore, amelioration of ischemia- and reperfusion-induced myocardial injury is a clinical imperative.
An increase in reactive oxygen species (ROS) production is one of the key events in I/R injury [3]. Excessive ROS generation leads to mitochondrial injury, including loss of mitochondrial membrane potential (ΔΨm) and results in a series of events, particularly apoptosis [4]. Consequently, inhibition of ROS production and protection of mitochondria from oxidative damage are effective strategies to ameliorate myocardial I/R injury. Xanthine oxidase (XO) is involved in the generation of O2− in response to hypoxia. During reperfusion, XO is also a critical source of ROS [5]. Febuxostat, a non-purine selective XO inhibitor, has been shown to have beneficial effects in renal I/R injury [6], suggesting that febuxostat reduced oxidative stress and suppressed apoptosis. However, the effect of febuxostat pretreatment on ischemia- and reperfusion-induced myocardial injury via mitochondrial apoptosis remains unclear.
Therefore, the objectives of the present study were: (1) to determine if febuxostat exerts cardioprotective effects in I/R injury and neonatal rat cardiomyocytes (NRCs) subjected to H/R injury; and (2) to investigate the mechanisms underlying the cardioprotective effects of febuxostat.
Methods
Materials and reagents
Male C57BL/6 mice (20–25 g) were obtained from the Laboratory Animal Center of Guangdong Province. One hundred eight mice were included in the study. Febuxostat was purchased from Teijin (Tokyo, Japan), and diluted in 0.5% methylcellulose (Sigma, St. Louis, MO, USA). Dulbecco Modified Eagle’s Medium (DEME), 5-bromo-2-deoxyuridine (BrdU), fetal bovine serum (FBS), 2,3,5-triphenyltetrazolium chloride (TTC) and Evan’s Blue dye were purchased from Sigma-Aldrich (St. Louis, MO, USA). Serum creatine kinase (CK) and lactate dehydrogenase (LDH) commercial kits were obtained from Biovision (Mountain View, CA, USA). Caspase-3 and caspase-9 activity were measured using commercial kits (Biovision, Mountain View, CA, USA). The Hoechst staining kit was obtained from Promega (Beijing, China). ROS and Mitochondrial Membrane Potential Assay Kits were obtained from Beyotime (Jiangsu, China). Antibodies against Bcl-2 (1:1,000), Bcl-XL (1:1,000), Bax (1:1,000), Bak (1:1,000), Cytochrome C (1:1,000), Caspase-9 (1:1,000), Caspase-3 (1:1,000), and GAPDH were purchased from Cell Signaling Technology (Beverly, MA, USA). COX IV (1:1,000) was from Bioworld Corporation (Dublin, USA).
Experimental animals and myocardial I/R model
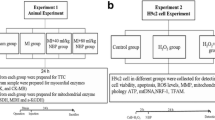
All animals received humane care in accordance with the Guide for the Care and Use of Laboratory Animals published by the United States National Institute of Health (NIH Publication No. 85-23, revised 1996). All investigations were approved by the Bioethics Committee of Southern Medical University, Guangzhou, China. The I/R model was developed as described previously [7, 8]. Briefly, mice were anesthetized by an intraperitoneal injection of ketamine and pentobarbital sodium, and connected to a rodent ventilator. The left anterior descending (LAD) coronary artery was surgically ligated by passing a 7–0 silk suture under the LAD. Regional ischemia was confirmed by visual inspection of pale color of the myocardium and ST segment elevation on electrocardiogram. Animals in the Sham and FEB group were also anesthetized and a suture was passed under the LAD, without occlusion.
Experimental protocols in vivo
Mice were randomized into three groups: (1) Sham, which received sham operation without coronary artery ligation and used as normal control; (2) I/R (I/R + Vehicle), which were pretreated with 0.5 ml of vehicle (0.5% methylcellulose); (3) I/R + FEB (I/R + febuxostat), the mice were pretreated with febuxostat (5 mg/kg) [9] in 0.5 ml methylcellulose; (4) ALL + I/R (I/R + allopurinol), allopurinol (30 mg/kg) in 0.5 ml methylcellulose; and (5) FEB (febuxostat), which received sham operation without coronary artery ligation. All pretreatments were administered through intraperitoneal injection 24 and 1 h before ischemia induction. Groups 2, 3, 4 were subjected to myocardial ischemia for 45 min, followed by reperfusion for 2 h. Following reperfusion, a portion of the blood was collected by cardiopuncture, and the heart was harvested and washed with ice-cold normal saline.
In vitro studies
Neonatal rat cardiomyocytes (NRCs) were prepared from newborn (1- to 2-day old) Sprague–Dawley rats as previously described [10]. Briefly, ventricles of the newborn rats were isolated aseptically, and then digested using trypsin and collagenase, and purified by differential pre-plating. The resuspended cells were maintained in DMEM with 10% (v/v) fetal bovine serum, BrdU (100 μmol/L), followed by transfer to a culture dish under conditions of 95% atmosphere and 5% CO2 at a temperature of 37°C. Hypoxia/reoxygenation (H/R) was performed in vitro as described previously [11, 12]. The in vitro model of H/R included hypoxia for 3 h, and oxygenation for 3 h. The following groups were tested. (1) Control group: cells were cultured in a standard incubator (95% O2, 5% CO2, 37°C); (2) H/R: cells were incubated in the hypoxic chamber for 3 h (95% N2, 5% CO2, 37°C), and then reoxygenated in a standard incubator for 3 h (95% O2, 5% CO2, 37°C); (3) Vehicle (H/R + Veh): cells were cultured with methylcellulose 24 and 1 h prior to H/R; (4, 5) H/R + Feb: cells were exposed to febuxostat (1 and 10 µM) [9] at 24 and 1 h respectively prior to H/R; and (6) H/R + All: cells were administered allopurinol (10 µM) before hypoxia induction. (7) Feb: cells were exposed to febuxostat 10 µM at 24 and 1 h in a standard incubator (95% O2, 5% CO2, 37°C).
Assessment of myocardial infarct size
Evan’s Blue-triphenyltetrazolium chloride (TTC) double staining methods were used to determine myocardial infarct size as described previously [13]. Following reperfusion for 2 h, the LAD was re-occluded and 0.2–0.3 ml of 2% solution of Evan’s Blue dye was injected into the right jugular vein to identify the area prone to ischemic damage, termed area at risk (AAR). When the right side of the heart turned blue, the heart was rapidly excised and rinsed in normal saline. The left ventricle (LV) was isolated and frozen at −20°C for 30 min. The LV was then cut into five 1-mm thick slices, which were incubated in 1% TTC for 15 min at 37.0°C. The infarct area (INF; white) and the area at risk (AAR; red and white) from each segment were measured using an image analyzer. Ratios of area at risk vs. left ventricle (AAR/LV), infarct area vs. area at risk (INF/AAR) and infarct area vs. left ventricle (INF/LV) were calculated.
Echocardiography
Echocardiography was performed 1 week before and after I/R induction. An echocardiography system with a Sonos 4500 and a 15–16 MHz transducer (Philips Corporation) was used. Transthoracic echocardiography was performed to obtain both 2-dimensional and M-mode images. To determine cardiac structure and function, left ventricular end diastolic dimension (LVEDD), left ventricular end systolic dimension (LVESD), left ventricular ejection fraction (LVEF), and fractional shortening (FS%) were analyzed from images as previously described [8, 14]. FS% was calculated as (LVEDD − LVESD)/LVEDD × 100%.
Serum creatine kinase (CK) and lactate dehydrogenase (LDH) levels
In order to determine the degree of myocardial injury, the serum myocardial enzymes LDH and CK were measured using commercial kit reagents according to the manufacturers’ instructions.
TUNEL assay and assessment of caspases activity
Myocardial apoptosis was assessed using DeadEndTM Fluorometric terminal deoxynucleotidyl-transferase dUTP nick-end labeling (TUNEL) assay. The total cardiomyocyte nuclei were identified by Hoechst 33258, and apoptotic nuclei were labeled with green fluorescein dye. In order to examine caspase-3 and caspase-9 activity, we used commercial kit reagents with procedures outlined by the manufacturer (Biovision, Mountain View, CA). Samples of whole left ventricular homogenate were prepared and tissues were homogenized in the cell lysis buffer followed by centrifugation. The supernatant was collected and used for the assay. Protein concentration in the lysate was measured and 100 µg lysate protein was used in 50 µl Cell Lysis Buffer for each assay. Caspase activity was monitored using a Microplate Reader at 405 nm.
Cell viability
Cell viability was determined by the MTT assay, with 1 × 104 cells/well seeded into 96-well plates. Following the experimental interventions, MTT solution was added into each well (5 mg/ml) and the plates were incubated for 4 h at 37°C. The viability was then measured by evaluating the absorbance at 570 nm. An LDH kit (Sigma) was used to measure the extent of cellular injury as previously described [15].
Determination of NRCs apoptosis by TUNEL
NRCs apoptosis was assessed using the DeadEnd™ Fluorometric TUNEL System according to the manufacturer’s protocol. Cells were fixed with 4.0% formaldehyde in PBS for 25 min at 4°C, and incubated with 10 μM Hoechst 33258 for 15 min. Cells were observed under fluorescence microscopy.
Measurement of ROS and Mitochondrial membrane potential (ΔΨm)
ROS and Mitochondrial Membrane Potential Assay Kits (S0033 and C2006) were used to measure ROS and ΔΨm of NRCs according to the manufacturer’s instructions. Techniques to measure ROS were performed as previously described [16]. Briefly, cells were incubated with the 1:1000 ROS-sensitive dye 2′,7′-dichloruorescein-diacetate (DCFH-DA) dilution, and then incubated for 20 min at 37°C. To measure ΔΨm, cells were seeded into laser confocal petri dishes. After treatment, the dishes were incubated with JC-1 staining solution (5 μg/ml) in an incubator for 20 min at 37°C. The cells were then washed twice with JC-1 staining buffer and confocal laser scanning microscopy (OLYMPUS FV1000) was used for detection.
Electron transmission microscopy
After reperfusion for 2 h, the LVs were harvested, cut into 1-mm3 sections on ice and fixed with 2.5% glutaraldehyde. Osmium tetroxide (1% in 0.1 mol/l cacodylate) was used for post-fixation. Tissues were dehydrated with a series of ethanol rinses. Samples were embedded and sliced. The slices were stained and observed using electron transmission microscopy (PHILIPS CM10, Holland). Mitochondrial area was measured using the Scanning Probe Imaging Processor. In each specimen, the shape parameters of 30 mitochondria were measured.
Western blot
Protein extracts from NRCs were subjected to Western blot. Protein concentrations were measured with a BCA Protein Assay Kit. Equal amounts of protein were loaded into lanes and were separated using SDS-PAGE, followed by transfer to a polyvinylidene fluoride membrane. The membranes were then blocked with 5% skim milk solution, followed by overnight incubation at 4°C with the appropriate primary antibody. The membranes were probed the following day with secondary antibodies for 1 h at room temperature, and then washed with Tris-buffered saline/0.1% Tween-20. The signals were detected using an electrochemiluminesence (ECL) system and scanned. The relative intensity of the bands was quantified using the Image J 3.0 system.
Statistical analyses
Data were expressed as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) followed by either a Bonferroni post hoc test or Student’s t test was used for statistical significance of multiple treatments as appropriate. A value of P < 0.05 was considered statistically significant.
Results
Effect of febuxostat on myocardial injury in vivo
Figure 1 illustrates the effect of febuxostat treatment on infarct size and necrosis in mice. Representative myocardial infarct size images are shown in Figure 1a. Compared with Sham, I/R induced a significant increase in INF/AAR (INF/AAR: 38.3 ± 3.8 vs. 0; INF/LV: 20.5 ± 2.4 vs. 0; P < 0.01, respectively). However, infarct size in the I/R + FEB and I/R + ALL groups was markedly reduced compared with I/R (P < 0.01). There was no difference in AAR/LV between groups. The difference between the sham and FEB groups was not significant (P > 0.05).

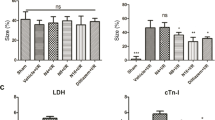
Effects of febuxostat on infarct size and necrosis in mice. a Representative images of myocardial infarct size and the ratio of AAR/LV, INF/AAR, and INF/LV. b Serum CK levels. c Serum LDH levels. The results were expressed as mean ± SD. (n = 7). *P < 0.01 vs. Sham, # P < 0.01, vs. I/R, ¶ P < 0.01 vs. I/R + ALL.
Figure 1b, c shows the effect of febuxostat on serum CK and LDH activities. In comparison with Sham group, I/R treatment increased serum CK and LDH activities (P < 0.01). Febuxostat treatment effectively blocked the increase of CK and LDH activities compared with I/R mice (CK: 121.9 ± 16.6 vs. 196.3 ± 15.3 mU/ml; LDH: 765.5 ± 166.1 vs. 1549.8 ± 365.8 mU/ml; P < 0.01, respectively), similar to allopurinol (CK: 138.2 ± 7.18 vs. 196.3 ± 15.3 mU/ml; LDH: 1005.9 ± 53.1 vs. 1549.8 ± 365.8 mU/ml; P < 0.01, respectively). Compared with I/R + ALL mice, I/R + FEB markedly decreased CK and LDH activities (P < 0.05). There was no significant difference between the Sham group and FEB group (P > 0.05).
Apoptosis is the major mechanism of cell death following I/R injury [17]. TUNEL and caspases activities were used to assess the level of apoptosis. Figure 2 illustrates the effect of febuxostat treatment on apoptosis in mice. In comparison with Sham group, I/R treatment increased the apoptotic index (34.9 ± 2.6 vs. 4.5 ± 0.9%, P < 0.01). Pretreatment with either febuxostat or allopurinol effectively attenuated the increase in apoptotic index (Febuxostat: 26.9 ± 3.7 vs. 34.9 ± 2.6%; Allopurinol: 29.4 ± 1.9 vs. 34.9 ± 2.6%, P < 0.01, respectively). Compared with I/R + ALL mice, I/R +FEB markedly decreased in apoptotic index (P < 0.01). Caspase-3 and -9 activities were increased in the myocardium following I/R compared to Sham (Caspase-3: 2.9 ± 0.2 vs. 1; Caspase-9: 3.5 ± 0.4 vs. 1, P < 0.01, respectively). The Caspase-3 and -9 activities in the febuxostat and allopurinol-pretreated groups was significantly lower than that in I/R group (P < 0.01). In comparison with I/R + ALL, I/R + FEB markedly decreased the Caspase-3 and -9 activities (Caspase-3: 2.1 ± 0.2 vs. 2.5 ± 0.2; Caspase-9: 2.2 ± 0.1 vs. 2.9 ± 0.2, P < 0.01, respectively). There was no significant difference between the Sham and FEB groups in apoptotic index and Caspases activities (P > 0.05).

Effects of febuxostat on apoptosis in mice. a Representative photomicrographs (×200) of TUNEL staining in myocardial I/R tissue. Green fluorescence indicates TUNEL-positive apoptotic nuclei; blue fluorescence indicates total cardiomyocyte nuclei. b Apoptotic index. c Myocardial caspase-3 activity. d Myocardial caspase-9 activity. The results were expressed as mean ± SD (n = 7). The results were expressed as mean ± SD (n = 7). *P < 0.01 vs. Sham, # P < 0.01, vs. I/R, ¶ P < 0.01 vs. I/R + ALL.
Effect of febuxostat on cardiac function
Echocardiograms were obtained after 1 week in order to determine the effect of febuxostat on cardiac function following I/R. Seven mice were enrolled in each group. I/R-induced reduction in cardiac function was significantly restored by pretreatment with febuxostat and allopurinol (P < 0.01, respectively) (Table 1).
Effect of febuxostat on H/R injury in NRCs
Figure 3a shows viability of NRCs following H/R as detected by an MTT assay Compared with Control group, The viability of H/R and H/R + Veh cells was significantly reduced (65.5 ± 1.3 vs. 100%; 66.6 ± 2.2 vs. 100%, P < 0.01, respectively). Pretreatment with febuxostat and allopurinol significantly alleviated this effect (Febuxostat: 80.0 ± 2.7 vs. 65.5 ± 1.3%; Allopurinol: 77.6 ± 2.3 vs. 65.5 ± 1.3%, P < 0.01, respectively), especially 10 µM febuxostat restoring cell survival to 96.8 ± 1.4%.

Effects of febuxostat on H/R injury in NRCs. a MTT assay following H/R. b Leakage of LDH in culture medium. c Apoptotic index in NRCs. d Typical morphological changes in apoptosis under fluorescence microscope (×200). All values indicate mean ± SD. *P < 0.01 vs. Sham, # P < 0.01, vs. I/R, ¶ P < 0.01 vs. H/R + All. These experiments were performed in quintuplicate with similar results.
LDH activity was measured to assess cardiomyocyte injury at the end of reoxygenation. LDH activity of H/R cells was markedly increased compared with controls (36.2 ± 0.3 vs. 13.3 ± 1.0, P < 0.01). However, febuxostat and allopurinol treatment significantly inhibited the increase in LDH activity (Febuxostat: 28.5 ± 1.0 vs. 36.2 ± 0.3; Allopurinol: 31.8 ± 1.3 vs. 36.2 ± 0.3, P < 0.01, respectively). Compared with H/R + All mice, H/R + Feb markedly decreased LDH activity (P < 0.01). There was no significant difference between the Control and Feb groups (P > 0.05) (Figure 3b).
The anti-apoptotic effects of febuxostat were detected using the TUNEL assay. Compared with the control, significant increases in the percentage of apoptotic cells were observed in H/R (54.6 ± 3.8 vs. 9.9 ± 0.3, P < 0.01). However, febuxostat and allopurinol administration suppressed H/R-induced damage of NRCs (Febuxostat: 38.2 ± 2.3 vs. 54.6 ± 3.8; Allopurinol: 45.7 ± 5.4, P < 0.01, respectively). In comparison with H/R + All, 10 µM febuxostat treatment significantly decreased AI (P < 0.01) (Figure 3c).
Effect of febuxostat on mitochondrial integrity
The mitochondria of Sham mice were regular with unbroken cristae. I/R injury induced mitochondrial swelling and loss of cristae as shown in Figure 3a. However, in the febuxostat- treated group, the outer mitochondrial membranes were intact with regular organization. Febuxostat pretreatment also reduced the degree of mitochondrial swelling. Compared with Sham group, the mitochondrial structure of the FEB group was not significantly altered (Figure 4a). The mean area of mitochondria of I/R group was markedly increased compared with Sham (0.40 ± 0.02 vs. 0.27 ± 0.03, P < 0.01). However, febuxostat pretreatment significantly inhibited the increase (0.32 ± 0.02 vs. 0.40 ± 0.02, P < 0.01). There was no significant difference between the Sham and FEB groups (P > 0.05) (Figure 4b).

Effect of febuxostat on mitochondrial structure in mice. a Representative electron micrographs (×50,000) of mitochondrial structures. Sham the structure of mitochondria was generally very good. I/R I/R-induced mitochondrial structural disruption (swelling, rupture and loss of cristae). I/R + FEB febuxostat treatment ameliorated I/R-induced mitochondrial impairment. FEB the structure of mitochondria was basically intact. b Mean area of mitochondria. All values represent mean ± SD. *P < 0.01 vs. Sham, # P < 0.01 vs. I/R, n = 4/group, these experiments were performed with similar results.
Effect of febuxostat on ROS production and ΔΨm
We examined the overall ROS generation using the dye DCFH-DA. H/R led to enhanced staining of DCFH-DA, whereas febuxostat treatment decreased staining of DCFH-DA (2.6 ± 0.5 vs. 6.2 ± 0.4, P < 0.01) (Figure 5a).

Effect of febuxostat treatment on ROS production and mitochondrial membrane potential (ΔΨm). a ROS staining was performed using DCFH-DA. Left: representative images of ROS staining; right: ROS fluorescence intensity. b Changes in mitochondrial membrane potential (ΔΨm) are indicated by JC-1 staining; left: Red fluorescence represents the mitochondrial aggregate form of JC-1, indicating intact mitochondrial membrane potential. Green fluorescence represents the monomeric form of JC-1, indicating dissipation of ΔΨm. Right: ratio of red/green fluorescence intensity. Both ROS and ΔΨm staining were observed with a laser confocal microscope (×400) (Bar 50 µm). All values indicate mean ± SD. **P < 0.01 vs. Con, ## P < 0.01 vs. H/R.
The mitochondrial potential (ΔΨm) was measured in order to investigate mitochondrial function. It was dramatically reduced in H/R and H/R + Veh treated cells. However, febuxostat treatment restored the ΔΨm compared with H/R cells 0.7 ± 0.1 vs. 0.2 ± 0.02, P < 0.01) (Figure 5b).
Effect of febuxostat on cytochrome C, cleaved caspase-3 and -9 expression
Western blot analyses indicated that H/R induced a statistically significant increase in cytochrome C released into the cytoplasm compared with the control cells. Febuxostat treatment reduced the release of mitochondrial cytochrome C following H/R injury of NRCs (P < 0.01). We also observed the effects of febuxostat treatment on the expression of caspase-3 and -9. There was no significant difference in proprotein expression levels between groups. Cleaved protein expression was significantly increased in H/R cells, while febuxostat treatment reduced the expression of cleaved caspase-3 and -9 (Figure 6a–d).

Effect of febuxostat treatment on the expression of proteins regulating mitochondria-mediated apoptosis. a Representative Western blots from each group. b The levels of Cyto-C (cyto/mito).c The levels of cleaved caspase-3. d The levels of cleaved caspase-9. e The ratio of Bcl-2/Bax. f The levels of Bcl-2 and Bcl-XL. g The levels of Bax and Bak. Levels of proteins were quantified using densitometry and normalized against COX IV or GADPH. Data are presented as mean ± SD. **P < 0.01 vs. Con, ## P < 0.01 vs. H/R. These experiments were performed in quintuplicate with similar results.
Effect of febuxostat on the expression levels of apoptosis-related proteins
In order to investigate whether febuxostat pretreatment protected against H/R-induced apoptosis, we examined the expression of anti-apoptotic (Bcl-2 and Bcl-XL) and pro-apoptotic proteins (Bax and Bak). H/R treatment dramatically reduced Bcl-2 and Bcl-XL, increased Bax and Bak expression and decreased the ratio of Bcl-2/Bax. Following febuxostat treatment, the expression of anti-apoptotic proteins was upregulated, while the level of pro-apoptotic proteins was downregulated. As shown in Figure 6e, the ratio of Bcl-2/Bax was also increased following febuxostat treatment.
Discussion
The present study yielded several important findings. First, febuxostat pretreatment ameliorated myocardial injury in a mouse I/R model and alleviated H/R injury in cultured NRCs. Second, febuxostat treatment decreased ROS production and inhibited subsequent apoptosis. The underlying protective mechanisms may be attributed to deactivation of a mitochondrial-dependent apoptotic pathway. The role of febuxostat alleviating myocardial ischemia reperfusion injury was superior to allopurinol. This could be due to by higher bioavailability and more potent XO inhibitory effect of febuxostat. Beyond that, febuxostat has fewer side effects than allopurinol.
ROS production is a key mechanism in the injury associated with I/R [18]. During reperfusion, XO is one of the main sources of ROS. XO inhibition with allopurinol modulates ROS production and intracellular Ca2+ overload in H/R-injured neonatal rat cardiomyocytes [19]. Treatment with allopurinol has been shown to decrease the infarct areas of myocardial I/R injury in the dog, with XO proposed as the source of free radicals in the myocardium [20]. Febuxostat, a new XO inhibitor, attenuates the pressure overload in LV [21] and protects the kidneys from I/R injury [6] via reduction in ROS production. These observations were consistent with our results that inhibition of XO by febuxostat reduced ROS production.
Together with the reduction of ROS, TUNEL-positive apoptotic cells were also suppressed in febuxostat-treated groups. The possible protective mechanisms of XO inhibitor-induction are mediated by reduced ROS production and mitochondrial protection [22]. This finding is supported by our observation that fubuxostat pretreatment inhibited mitochondrial apoptotic pathway. Hypoxia alters the mitochondrial structure and triggers changes in permeability [23], resulting in functional impairment of the mitochondria, including dissipation of the ΔΨm, release of cytochrome C into the cytoplasm [24] and mitochondrial swelling [25]. Cytochrome C activates caspases, resulting in apoptosis [26, 27]. Our results suggested that the ΔΨm was decreased in H/R cells, but febuxostat restored the Δψm to normal levels. In addition, cytoplasmic cytochrome C and its downstream cleaved caspases were increased in H/R cells, while pretreatment with febuxostat ameliorated these increases. These data demonstrated that cardioprotection by febuxostat was partly accomplished via inhibition of mitochondrial apoptosis.
Febuxostat suppressed apoptosis by another possible mechanism modulated by Bcl-2 family of proteins. Several mitochondrial events are modulated by Bcl-2, especially those linking mitochondrial physiology and apoptosis [28, 29]. Bcl-XL and Bcl-2 (anti-apoptotic proteins) maintain the integrity of the external mitochondrial membrane, preventing the release of cytochrome C from the mitochondria. Conversely, several proapoptotic proteins such as Bax and Bak cause mitochondrial injury, resulting in cell death [30]. In the present study, Western blot revealed that Bcl-XL and Bcl-2 (anti-apoptotic proteins) expression levels were decreased significantly and proapoptotic protein (Bax and Bak) expression was induced by H/R. Febuxostat treatment was also shown to enhance the expression of anti-apoptotic proteins and decrease the expression of proapoptotic proteins, reducing the ratio of Bcl-2/Bax. These results further confirm that febuxostat treatment modulated H/R-induced apoptosis via a mitochondrial-dependent pathway and provide evidence supporting its anti-apoptotic role.
Conclusion
In conclusion, our studies demonstrated for the first time that febuxostat provides cardioprotection following ischemia- and reperfusion-induced myocardial injury by reducing ROS generation and mitochondrial apoptosis (Figure 7). Thus, febuxostat may be a clinically useful agent in myocardial injury.

Schematic depicting protective signaling of febuxostat in I/R-induced apoptosis. Febuxostat inhibits the mitochondrial apoptotic pathway by reducing ROS production and modulating Bcl-2 family proteins.
Abbreviations
- XO:
-
xanthine oxidase
- ROS:
-
reactive oxygen species
- I/R:
-
ischemia/reperfusion
- NRC:
-
neonatal rat cardiomyocyte
- H/R:
-
hypoxia/reoxygenation
- CK:
-
creatine kinase
- LDH:
-
lactate dehydrogenase
- AI:
-
apoptotic index
- FBS:
-
fetal bovine serum
- TTC:
-
2,3,5-triphenyltetrazolium chloride
- DMEM:
-
dulbecco modified eagle’s medium
- LAD:
-
left anterior descending
- INF:
-
infarct area
- AAR:
-
area at risk
- LV:
-
left ventricle
- LVEDD:
-
left ventricular end diastolic dimension
- LVESD:
-
left ventricular end systolic dimension
- LVEF:
-
left ventricular ejection fraction
- FS:
-
fractional shortening
References
Kalogeris T, Baines CP, Krenz M, Korthuis RJ (2012) Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 298:229–317
Gottlieb RA (2011) Cell death pathways in acute ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther 16:233–238
Windecker S, Bax JJ, Myat A, Stone GW, Marber MS (2013) Future treatment strategies in ST-segment elevation myocardial infarction. Lancet 382:644–657
Sinha K, Das J, Pal PB, Sil PC (2013) Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol 87:1157–1180
Li C, Jackson RM (2002) Reactive species mechanisms of cellular hypoxia-reoxygenation injury. AJP Cell Physiol 282:C227–C241
Tsuda H, Kawada N, Kaimori J, Kitamura H, Moriyama T, Rakugi H et al (2012) Febuxostat suppressed renal ischemia-reperfusion injury via reduced oxidative stress. Biochem Biophys Res Commun 427:266–272
Ha T, Hua F, Liu X, Ma J, McMullen JR, Shioi T et al (2008) Lipopolysaccharide-induced myocardial protection against ischaemia/reperfusion injury is mediated through a PI3K/Akt-dependent mechanism. Cardiovasc Res 78:546–553
Aragón JP, Condit ME, Bhushan S, Predmore BL, Patel SS, Grinsfelder DB et al (2011) Beta3-adrenoreceptor stimulation ameliorates myocardial ischemia-reperfusion injury via endothelial nitric oxide synthase and neuronal nitric oxide synthase activation. J Am Coll Cardiol 58:2683–2691
Nomura J, Busso N, Ives A, Tsujimoto S, Tamura M, So A et al (2013) Febuxostat, an inhibitor of xanthine oxidase, suppresses lipopolysaccharide-induced MCP-1 production via MAPK phosphatase-1-mediated inactivation of JNK. PLoS One 8:e75527
Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z et al (2014) Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 129:1139–1151
Wang Y, Li X, Wang X, Lau W, Wang Y, Xing Y et al (2013) Ginsenoside Rd attenuates myocardial ischemia/reperfusion injury via Akt/GSK-3β signaling and inhibition of the mitochondria-dependent apoptotic pathway. PLoS One 8:e70956
Huang GQ, Wang JN, Tang JM, Zhang L, Zheng F, Yang JY et al (2011) The combined transduction of copper, zinc-superoxide dismutase and catalase mediated by cell-penetrating peptide, PEP-1, to protect myocardium from ischemia-reperfusion injury. J Transl Med 9:73
Wang X, Cao Y, Shen M, Wang B, Zhang W, Liu Y et al (2015) DIDS reduces ischemia/reperfusion-induced myocardial injury in rats. Cell Physiol Biochem 35:676–688
Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M et al (2010) A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res 107:1445–1453
Uchiyama T (2004) Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation 109:3042–3049
Li R, Yan G, Li Q, Sun H, Hu Y, Sun J et al (2012) MicroRNA-145 protects cardiomyocytes against hydrogen peroxide (H2O2)-induced apoptosis through targeting the mitochondria apoptotic pathway. PLoS One 7:e44907
Webster KA (2012) Mitochondrial membrane permeabilization and cell death during myocardial infarction: roles of calcium and reactive oxygen species. Futur Cardiol 8:863–884
Ma S, Zhang Z, Yi F, Wang Y, Zhang X, Li X et al (2013) Protective effects of low-frequency magnetic fields on cardiomyocytes from ischemia reperfusion injury via ROS and NO/ONOO–. Oxidative Med Cell Longev 2013:1–9
Kang S, Lim S, Song H, Chang W, Lee S, Bae S et al (2006) Allopurinol modulates reactive oxygen species generation and Ca2+ overload in ischemia-reperfused heart and hypoxia-reoxygenated cardiomyocytes. Eur J Pharmacol 535:212–219
David E (1985) Chambers DAPG: xanthine oxidase as a source of free radical damage in myocardial ischemia. J Mol Cell Cardiol 17:145–152
Xu X, Hu X, Lu Z, Zhang P, Zhao L, Wessale JL et al (2008) Xanthine oxidase inhibition with febuxostat attenuates systolic overload-induced left ventricular hypertrophy and dysfunction in mice. J Cardiac Fail 14:746–753
Lee W, Lee S (2006) Synergistic protective effect of ischemic preconditioning and allopurinol on ischemia/reperfusion injury in rat liver. Biochem Biophys Res Commun 349:1087–1093
Ong S, Samangouei P, Kalkhoran SB, Hausenloy DJ (2015) The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J Mol Cell Cardiol 78:23–34
Yu W, Sheng M, Xu R, Yu J, Cui K, Tong J et al (2013) Berberine protects human renal proximal tubular cells from hypoxia/reoxygenation injury via inhibiting endoplasmic reticulum and mitochondrial stress pathways. J Transl Med 11:24
Foster KA, Galeffi F, Gerich FJ, Turner DA, Müller M (2006) Optical and pharmacological tools to investigate the role of mitochondria during oxidative stress and neurodegeneration. Prog Neurobiol 79:136–171
Galluzzi L, Morselli E, Kepp O, Kroemer G (2009) Targeting post-mitochondrial effectors of apoptosis for neuroprotection. Biochim Biophys Acta (BBA) Bioenerg 1787:402–413
Chiong M, Wang ZV, Pedrozo Z, Cao DJ, Troncoso R, Ibacache M et al (2011) Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis 2:e244
Kawakami M, Inagawa R, Hosokawa T, Saito T, Kurasaki M (2008) Mechanism of apoptosis induced by copper in PC12 cells. Food Chem Toxicol 46:2157–2164
Anilkumar U, Prehn JHM (2014) Anti-apoptotic BCL-2 family proteins in acute neural injury. Front Cell Neurosci 8:281
Chien CY, Chien CT, Wang SS (2014) Progressive thermopreconditioning attenuates rat cardiac ischemia/reperfusion injury by mitochondria-mediated antioxidant and antiapoptotic mechanisms. J Thorac Cardiovasc Surg 148:705–713
Authors’ contributions
SW drafted the manuscript, designed experiments. AC and PY contributed to the design of the study. SW and YL carried out the experiments and performed statistical analysis. XS analyzed and interpreted data. XW and CZ discussed the data and helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81270218, to Dr. Chen) and National Natural Science Foundation of China (81400190, to Dr. Wang).
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, S., Li, Y., Song, X. et al. Febuxostat pretreatment attenuates myocardial ischemia/reperfusion injury via mitochondrial apoptosis. J Transl Med 13, 209 (2015). https://doi.org/10.1186/s12967-015-0578-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-015-0578-x