
Abstract
Pyroptosis is an active cell death process mediated by gasdermin family proteins including Gasdermin A (GSDMA), Gasdermin B (GSDMB), Gasdermin C (GSDMC), Gasdermin D (GSDMD), Gasdermin E (GSDME, DFNA5), and DFNB59. Emerging evidences have shown that pyroptosis contributes to many pulmonary diseases, especially lung cancer, and pneumonia. The exact roles of pyroptosis and gasdermin family proteins are tremendously intricate. Besides, there are evidences that pyroptosis contributes to these respiratory diseases. However, it often plays a dual role in these diseases which is a cause for concern and makes it difficult for clinical translation. This review will focus on the multifaceted roles of pyroptosis in respiratory diseases.
Similar content being viewed by others

Introduction
The scope of cell death has been greatly expanded in recent years. More molecularly oriented definitions of terms including necroptosis, mitochondrial permeability transition (MPT)-driven necrosis, ferroptosis, parthanatos, entotic cell death, NETotic cell death, lysosome-dependent cell death, autophagy-dependent cell death, immunogenic cell death, cellular senescence, and mitotic catastrophe [1,2,3,4,5]. Pyroptosis is an active programmed cell death process with a strong inflammatory response and manifests as cell membrane pore formation, which eventually leads to chromatin fragmentation, cell swelling, and plasma membrane lysis [6]. At a molecular level, the gasdermin protein family plays an important role in cell membrane pore formation and the activation of pyroptosis [7]. As a large family, gasdermin has six members: Gasdermin A (GSDMA), Gasdermin B (GSDMB), Gasdermin C (GSDMC), Gasdermin D (GSDMD), Gasdermin E (GSDME, DFNA5), and DFNB59 in the human genome [7]. Initially, pyroptosis was discovered to be involved in immune defense against infections [8]. However, the role of pyroptosis soon spread to many other diseases including some respiratory diseases. With further study, more modes of pyroptosis have been explored in pulmonary diseases [9]. In this review, we provide a detailed discussion of the double-edged role of pyroptosis in the regulation of pulmonary diseases and the challenges encountered in clinical translation.
Pyroptosis
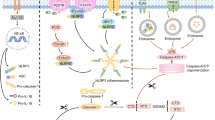
In 1992, an atypical form of cell death was observed in macrophages [10]. A subsequent study revealed that caspase-1 activation was involved in this cell death [11]. In 2001, caspase-1-dependent cell death was identified as a proinflammatory programmed event and named pyroptosis by Brad T. Cookson and Molly A. Brennan [12]. Caspase-1 activation prompts the transition of pro-interleukin (IL)-1β to mature IL-1β and induces the production of IL-1β and IL-18 [13, 14]. IL-1β and IL-18 are finally released outside cells and induce strong inflammatory responses [8]. This caspase-1-mediated pyroptosis is referred to as canonical pyroptosis (Fig. 1). AS a multi-protein complex, the inflammasome plays a central role in the inflammatory response. It assembles in response to pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). The assembly and activation of inflammasomes are an important step in initiating canonical pyroptosis by inducing the self-cleavage and activation of caspase-1 [6]. The context of pyroptosis is expanded when caspase-11 was found to trigger a kind of mouse macrophage death that resembled the cell death induced by caspase-1 [15]. Caspase-4/5 was also found to have a similar function with the caspase-11 which is homologous to caspase-4/5. Unlike caspase-1 activated by ligands of inflammasomes, Caspase-4/5/11 activated by cytosolic lipopolysaccharide (LPS) induces pyroptosis in a noncanonical pathway. The noncanonical pathway is suggested to play an important role in cell immunological responses to intracellular Gram-negative bacteria and some metabolic diseases associated with mitochondrial dysfunction (Fig. 1) [6].

Activation mechanism of pyroptosis in the respiratory system in the canonical pathway and the non-canonical pathway. In canonical pathway, the NLRP3 inflammasome is taken as a representative example. The NLRP3 inflammasome consists of NLRP3, an important member of NLRs (NOD-like receptors), ASC, and pro-caspase-1. In addition to NLRs and pyrin, absent in melanoma 2 (AIM2) can also form inflammasomes to activate pyroptosis
The identification of the gasdermin family yields insight into the mechanism of pyroptosis. The activation mechanism of GSDMD has been preliminarily unmasked, while those of others are largely unknown [7]. GSDMD plays an important role in either the canonical pathway or the non-canonical pathway. Danger signals or microbial infection can activate Caspase1/4/5/11. When activated, caspase1/4/5/11 can specifically cleave their direct substrate GSDMD into two parts: the C-terminal domain of GSDMD (GSDMD-CT) and the N-terminal domain of GSDMD (GSDMD-NT), a conserved domain [16]. GSDMD-NT triggers pyroptosis and GSDMD-CT inhibits the activation of GSDMD-NT by folding back on it. GSDMD-NT has the ability to connect to phosphoinositides and cardiolipin [16]. With oligomerization and membrane binding, GSDMD-NT leads to the lysis of membranes or the leakage of liposomes and forms pores in the membrane [16,17,18]. Subsequent alteration of osmotic pressure eventually causes cell swelling and lysis. The distribution of phosphoinositides is not symmetric on the plasma membrane. Therefore, GSDMD causes only lysis and leakage from within cells. IL-1β, as an important response of inflammasome activation, has also been demonstrated to be affected by GSDMD in some studies. GSDMD impacts the release of mature IL-1β, but not its maturation [16].
Other gasdermins have a similar architecture of two domains with GSDMD, except for DFNB59. The N-terminal domain of these GSDMD-like members can induce pyroptosis [7]. However, they lack caspase1/4/5/11 cleavage sites and their activation mechanisms are still unknown [16, 19]. Gasdermins function in the respiratory system, even with high epithelial expression specific to the skin and gastrointestinal tract [20]. Polymorphisms of GSDMB have been linked to some chronic inflammatory diseases in the respiratory tract, such as asthma [21]. GSDMC has also been implicated in lung cancer progression [22]. A new biological function of GSDMC was also revealed. Under hypoxia and TNFα treatment, GSDMC can be cleaved by caspase-8 mediated by nuclear programmed cell death 1 (PD1), which enhances GSDMC expression and switches apoptosis to pyroptosis [23]. GSDME was originally identified as DFNA5, a deafness gene and now seems to have many penetrated aspects of lung cancer. GSDME works as a mediator of p53 and has a potential role as a tumor suppression gene [24]. Methylation modification of GSDME is common in cancers that silence its expression [25]. Recently, some studies have reported that GSDME can be cleaved by caspase-3 specifically and switch caspase-3-mediated apoptosis to pyroptosis in many cases [26, 27]. Caspase-3 and GSDME work as a “switch” to shift cell death from apoptosis to pyroptosis (Fig. 2).

Pyroptotic pathway. GSDM superfamily members have six members: Gasdermin A (GSDMA), Gasdermin B (GSDMB), Gasdermin C (GSDMC), Gasdermin D (GSDMD), Gasdermin E (GSDME, DFNA5), and DFNB59. Gasdermins except for DFNB59 have similar architecture of two domains: an N-terminal and a C-terminal. N-terminal domains have pore-forming activities and activate pyroptosis. Pyroptosis has been reported to be involved in many pulmonary diseases. (Solid line: promoting effect, dotted line: inhibiting effect)
Association between pyroptosis and pulmonary diseases
Respiratory diseases are one of the most important causes of global population death and mainly include inflammatory diseases, neoplastic diseases, and some autoimmune diseases [28,29,30]. Emerging evidence shows that pyroptosis contributes to these respiratory diseases [6]. However, it often plays a dual role in these diseases. Here, we provided some evidence and views of controversial pyroptosis in different pulmonary diseases.
Lung cancer
The relationship between proinflammatory pyroptosis and lung cancer appears remarkably complicated [9]. Inflammation has been associated with the development and progression of various types of cancer. However, there are complex interactions between inflammatory processes and tumor development or progression [31, 32]. Recently, numerous studies have also found that promoting pyroptosis can inhibit tumor growth and reverse drug resistance [33]. In the adverse reactions related to tumor treatment, pyroptosis plays a role in promoting adverse reactions. Accumulated evidence indicates that pyroptosis exhibits a dual nature in lung cancer and its treatment.
Inflammation is the established factor in carcinogenesis of lung cancer [34]. Inflammasome proteins, including NLRP3 and AIM2, contribute to tumorigenesis by modulating immunity and cross-talk between the microenvironment and lung epithelial cells [35]. Additionally, some pyroptosis can occur in the tumor immune microenvironment through the caspase-1/GSDMD pathway, which will promote numerous malignant phenotypes in tumors including migration, invasion, and metastasis [36]. On the other hand, activated NLRP3 inflammasome aggregation of Ca2+ and the generation of reactive oxygen species (ROS) can promote pyroptosis and inhibit the proliferation of non-small cell lung cancer (NSCLC) [37]. Non-canonical pathway activated by caspase-4 has also been reported to eliminate NSCLC cells in vivo and in vitro [38].
Defects in apoptosis are known to be the overarching reason for the failure of anti-tumor treatment [39, 40]. With deeper research, we have found that pyroptosis is also involved in many anti-tumor treatments and plays a role in enhancing treatment efficacy [41]. Many chemotherapy drugs were reported to induce GSDME-mediated pyroptosis. After receiving chemotherapy, caspase-3 cleaves GSDME to generate a GSDME-N fragment and switches apoptosis to pyroptosis. Apoptotic appearance is overridden by pyroptosis in the case of high GSDME expression [27]. This phenomenon is observed in lung cancer treated with paclitaxel and cisplatin [41]. Pyroptosis induced by cisplatin seems to be higher than that induced by paclitaxel. It has also been reported that lower GSDME expression has an appreciable correlation with worse outcomes for NSCLC patients treated with platinum. GSDME-mediated pyroptosis may increase the sensitivity of platinum by promoting T cell infiltration [42]. The efficacy of platinum could also be enhanced by combination with BI2536, a PLK1 kinase inhibitor, by inducing pyroptosis and affecting DNA damage repair [43]. Some noncoding RNAs can also sensitize chemoresistant cancer cells to cisplatin by activating pyroptosis [44]. Additionally, GSDME executes caspase-3-meditated pyroptosis in EGFR-altered, ALK-rearranged, and KRAS-mutant tumors with genotype-matched regimens. The co-occurrence of pyroptosis and apoptosis contributes to the response to TKIs [45]. However, this pyroptosis appeared to marginally affect the treatment efficacy. In addition to the uncertain therapeutic effects of pyroptosis, it still has other concerning dual aspects. GSDME-mediated pyroptosis can act on normal tissues and exacerbate chemotherapy-induced toxicity. GSDME knockout can attenuate cisplatin-induced acute kidney injury and weight loss [27, 46]. Inhibiting pyroptosis can also reduce drug-induced nausea and vomiting caused by cisplatin and can decrease GSDME-mediated intestinal epithelial cell death through the regulation of ROS/JNK/Bax signaling pathway [47]. Additionally, in therapy-induced liver damage and myocardial injury, reducing pyroptosis was observed to alleviate these treatment-related toxicities [48].
Pyroptosis, as a highly inflammatory cell death, is also involved in radiation-induced tissue damage. Increasing caspase-1 activation is observed in marginal zone cells of radiotherapy [49]. GSDMD and inflammasomes such as AIM2 and NLRP3 are involved in this pyroptosis [49, 50]. Sepsis can also be promoted by caspase-11 mediated pyroptosis [51]. Cyclic GMP-AMP synthase (cGAS) influences caspase-11 in a non-canonical pathway of pyroptosis to aggravate this life-threatening complication in radiotherapy [52]. Other studies have also reported that GSDME expression enhances the sensitivity of cancer cells to radiation treatment by recruiting and activating NK cells to enhance antitumor immunity [53]. In addition, this GSDME-mediated pyroptosis induces radiation-related toxicity. Knocking GSDME protects against tissue damage and weight loss [53].
Pyroptosis makes important contributions to the transformation of cold tumors into hot tumors [54]. Many inflammatory cytokines and DAMPs are secreted during the processes of pyroptosis such as HMGB1, IL-1β, and IL-18. Inflammatory cytokines, including IL-1β and IL-18, are secreted through the GSDM-mediated pore. IL-1β can activate CD8 + T cells and promote the generation of Th1 CD4 + T-cells [55, 56]. IL-18 can also play immunoregulatory roles in inducing Interferon-γ (IFN-γ), polarizing Th1 cells and recruiting and activating natural killer (NK) cells [57]. Unlike cytokines secreted through traversing the GSDM-mediated pore, HMGB1 is regulated indirectly by GSDMD [58]. It could increase related molecules required for CD8 + T-cell priming and migrate tumor-infiltrating to draining lymph nodes to increase anti-tumor T cell responses [59]. These findings explain why pyroptosis elicits alterations in the tumor microenvironment which plays a crucial role in cancer progression and response to therapy. According to a recent study, a new mechanism of GSDMB in immunotherapy is revealed. Specifically, IFNγ can upregulate GSDMB expression and lymphocyte-derived granzyme A (GZMA) can cleave it. This result provides a new insight into enhancing antitumor immunity [60]. Notably, cytokines including IL-18 and IL-1β, which is recognized as effector molecules of pyroptosis, play a various function in different microenvironments. This introduces a lot of uncertainty in the clinical application of pyroptosis.
Increasing evidence suggests that inhibiting pyroptosis can, to some extent, control tumor progression. However, it is still premature to determine that pyroptosis can play a leading role in certain anti-tumor treatments. Conversely, in normal tissues, pyroptosis contributes to treatment-related adverse effects. This further underscores the dual nature of pyroptosis.
Pneumonia
As an open organ, the bronchial and alveolar epithelium of lung is exposed to air pollutants and pathogenic microbes. To defend against the invasion of pathogens, pattern recognition receptors (PRRs), which can be activated by PAMPs and DAMPs, are equipped with bronchial epithelial cells, dendritic cells, and alveolar macrophages [28, 61]. These receptors assemble inflammasomes with apoptosis-associated speck-like protein and procaspase-1. Inflammasomes occupy a key position in maintaining a delicate balance between immune responses and tissue injuries and/or infections. Once PRRs are activated, inflammasomes can induce pyroptosis and produce cytokines [28, 61] (Fig. 2).
Excessive inflammatory responses are always deemed to be an important cause of death following severe pneumonia. As a form of dysregulated hyperinflammation, cytokine release syndrome is the most significant cause of mortality in patients with severe pneumonia including COVID-19. Lactate dehydrogenase (LDH) and cytokines are highly elevated in these patients [62]. Growing evidence favors pyroptosis involvement [63]. Pyroptosis confers host protection to lung epithelial cells in some patients with infection [64,65,66]. Besides, pyroptosis, as an immune response against infections, plays a protective role in host defense in the initial period of pneumonia [67]. Nevertheless, excessive pyroptosis leads to tissue injury and host lethality [68]. In a mouse IAV infection model, persistent NLRP3 inflammasome activation leads to lung injury, which is not associated with viral titer [69]. Angiotensin-converting enzyme 2 (ACE2) is an important receptor of SARS-CoV-2 and a negative regulator of the renin-angiotensin-aldosterone system (RAAS) which is a hormone system that regulates blood pressure and fluid balance in the body. SARS-CoV-2 spike protein and cells expressing ACE2 form syncytia. Syncytia can activate the cascade from caspase-9 to caspase-3/7, resulting in GSDME-mediated pyroptosis [70]. Others have reported that the fusion activates NLRP3 inflammasome, which triggers GSDMD-mediated pyroptosis [71]. Non-Structural Protein 6 (NSP6) of SARS-CoV-2, as a key determinant of pathogenicity, is also reported to trigger GSDMD-mediated pyroptosis by activating NLRP3 inflammasome targeting ATP6AP1 [72, 73]. Some supportive evidences have even shown that angiotensin II elevated by RAAS can activate NLRP3 inflammasome [74,75,76]. The complement cascade can interact with SARS-CoV-2 and then be cleaved into fragments such as C3a and C5a anaphylatoxins. These fragments could also activate NLRP3 inflammasome. Coagulopathy is another life-threatening complication of SARS-CoV-2 and some influenza virus infections [77]. Proinflammatory cytokines, which can promote various procoagulation factors, are insufficient to explain the dramatic coagulation reaction. GSDMD-mediated pyroptosis can trigger blood clotting and cause massive thrombosis in both the canonical pathway and the non-canonical pathway [78]. This systemic fibrin deposition in the model of endotoxemia is reminiscent of coagulopathy seen in COVID-19.
In pneumonia caused by other pathogens, pyroptosis still plays an important role. Streptococcus pneumoniae can cause cell death by activating both an apoptotic pathway and a pyroptotic pathway. In the pyroptotic pathway, pneumoniae activates NLRP3 inflammasome to mediate IL-1β production and release through hydrogen peroxide produced by pneumococci as a product of the pyruvate oxidase SpxB [79]. In another study of H7N9 influenza, GSDME-mediated pyroptosis was revealed to contribute to the pulmonary cytokine storm of virus infection [80].
Asthma
Asthma is a common chronic lung disease, that affects more than 300 million people worldwide [81]. Many studies indicate some correlation between asthma and pyroptosis. GSDMA and GSDMB on chromosome 17q are linked to asthma [82, 83]. The chromosome 17q region is the most consistently associated and powerful region with asthma susceptibility. In recent years, many studies have identified that GSDMB seems to play an important role in asthma susceptibility and severity. Single nucleotide polymorphisms in GSDMB display a strong correlation with GSDMB expression levels and the severity of asthma. Higher expression of GSDMB is correlated with antiviral pathways and exacerbations of asthma [84]. GSDMB has been demonstrated to be highly expressed in human asthmatic lungs, specifically in bronchial epithelial cells, but not to be significantly expressed in alveolar epithelial cells, fibroblasts, and smooth muscle [85, 86]. The overexpression of GSDMB can upregulate genes correlated with airway remodeling and airway hyperresponsiveness including 5-LO, TGF-β1, and MMP-9 [87]. A splice variant causing the deletion of exon 6, which encodes 13 amino acids in the N-terminal domain, has been reported to decrease the risk of asthma, suggesting that abolishing GSDMB-mediated pyroptotic activity may play a role in asthma [88]. However, at the present stage, direct proof of how pyroptosis affects asthma is still lacking.
Other pulmonary diseases
Pyroptosis is reported to promote lung injury in acute respiratory distress syndrome (ARDS) [89]. Pyroptosis is also involved in other acute lung injuries (ALIs) such as brain injury-induced acute lung injury and pancreatitis-induced acute lung injury [90, 91]. Some natural products from various medicinal plants such as Emodin, Syringaresinol (SYR) and Honokiol (HKL), and 5-Androstenediol (5-AED), a natural steroid hormone can suppress pyroptosis to resist lung injury [92,93,94,95]. The pyroptosis of pulmonary artery smooth muscle cells can promote pulmonary hypertension [96]. Chronic obstructive pulmonary disease (COPD) is also considered to have a certain correlation with pyroptosis [97]. Additionally, nicotine has been found to influence the progression of COPD by GSDMD-induced pyroptosis [98].
Therapeutic implications
Cell death facilitates maintaining physiological homeostasis and healthy development by removing cells suffering from damage or infection. Excessive cell death can also contribute to human pathologies [99]. The dual nature of pyroptosis has been a long-standing and intriguing topic. Pyroptosis is involved in the protection of the host, especially in the initial stage of some inflammatory respiratory diseases through immune defense. An excessive inflammatory response could cause ultimately tissue damage [61]. Uncontrolled pyroptosis is responsible for system-wide inflammation in a large number of lung inflammatory diseases. For lung cancer, from the perspective of inflammation, pyroptosis carries a potential risk of carcinogenesis. On the other hand, from the cell death perspective, pyroptosis can promote tumor cell death. At the same time, pyroptosis is also a significant factor causing adverse reactions in tumor therapy. Thus, how to harness this double-edged sword to yield more positive effects is worth our contemplation.
Currently, many basic trials have demonstrated that the specific compounds can directly or indirectly target pyroptosis. These compounds mainly include some common chemotherapy drugs, such as cisplatin, doxorubicin, and 5-FU [100]. These chemotherapy drugs have been applied in clinical treatment for decades. The pyroptosis recently discovered that the occurring alongside apoptosis in these drugs lacks sufficient specificity, but it is still meaningful. Knowledge of the pyroptosis pathway activated by these drugs can help optimize treatment strategies, overcome resistance, and reduce the side effects of chemotherapy. Other pyroptosis inducers are largely in the experimental stage including some natural products. From the perspective of drug development targeting pyroptosis, we are currently at the first step from bench to bedside.Precise drug delivery is another viable solution. A delivery platform based on macrophage was developed to achieve targeted tumor drug delivery and controlled release [101]. Some burgeoning nanoplatforms can also stably deliver drugs to activate pyroptosis [102]. Precisely activating pyroptosis in the tumor site can effectively prevent damage to normal tissues caused by pyroptosis.
A deep understanding of the pyroptosis pathway is also crucial. In specific diseases, only by understanding which mode of pyroptosis can exert a greater effect can better utilize pyroptosis for therapeutic benefits. For inflammatory diseases, the timing of pyroptosis inhibition is particularly crucial. Inhibiting pyroptosis too early might impact the early activation of the immune response. Conversely, inhibiting pyroptosis too late might not effectively curb the cytokine storm. For the clinical translation and application of pyroptosis, the deepening of theoretical knowledge about the pyroptosis pathway and the development of targeted drugs are both particularly important.
Conclusion
Pyroptosis, as a potential target for the treatment of pulmonary diseases, has gradually become clear. However, what needs to be emphasized is the dual nature of pyroptosis. For lung cancer, on the one hand, pyroptosis can promote the death of cancer cells and enhance antitumor immunity. On the other hand, pyroptosis also increases adverse drug reactions. For other pulmonary inflammatory diseases, pyroptosis can play both protective roles and negative roles in different periods of various diseases. Besides, pyroptosis has a potential effect on asthma (Fig. 3). In conclusion, pyroptosis is involved in a number of pulmonary diseases in different pathways. However, the understanding of pyroptosis in respiratory diseases is still limited. The future challenge lies in defining the details of the molecular mechanism of pyroptosis and how the details can improve the outcomes of diseases. The exploration of a balance point of pyroptosis for treatment should be a long-standing and open research area.

Role of pyroptosis in the lung. This schematic representation shows the important role of pyroptosis in the regulation of pulmonary diseases including pneumonia, asthma, lung cancer COPD, and ALI/ARDS
Availability of data and materials
No datasets were generated or analysed during the current study.
References
Lee SY, Ju MK, Jeon HM, Jeong EK, Lee YJ, Kim CH, Park HG, Han SI, Kang HS. Regulation of tumor progression by programmed necrosis. Oxid Med Cell Longev. 2018;2018:3537471.
Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14:48.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, et al. Ferroptosis: a regulated cell death Nexus linking metabolism, Redox Biology, and Disease. Cell. 2017;171:273–85.
Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 2019;26:99–114.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. Molecular mechanisms of cell death: recommendations of the nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin D in pyroptosis. Acta Pharm Sin B. 2021;11:2768–82.
Shi J, Gao W, Shao F, Pyroptosis. Gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42:245–54.
Lacey CA, Mitchell WJ, Dadelahi AS, Skyberg JA. Caspase-1 and caspase-11 mediate pyroptosis, inflammation, and control of brucella joint infection. Infect Immun. 2018;86:e00361–18.
Xia X, Wang X, Cheng Z, Qin W, Lei L, Jiang J, Hu J. The role of pyroptosis in cancer: pro-cancer or pro-host? Cell Death Dis. 2019;10:650.
Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358:167–69.
Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci USA. 1999;96:2396–401.
Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–4.
Thornberry NA, Herbert GB, JR C. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–74.
Cordoba-Rodriguez R, Fang H, Lankford CS, Frucht DM. Anthrax lethal toxin rapidly activates caspase-1/ICE and induces extracellular release of interleukin (IL)-1beta and IL-18. J Biol Chem. 2004;279:20563–66.
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8.
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71.
Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–6.
Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K, Sumiyama K, Sagai T, Shiroishi T. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics. 2007;89:618–29.
Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, von Mutius E, Farrall M, Lathrop M, Cookson W, Consortium G. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–21.
Wei J, Xu Z, Chen X, Wang X, Zeng S, Qian L, Yang X, Ou C, Lin W, Gong Z, Yan Y. Overexpression of GSDMC is a prognostic factor for predicting a poor outcome in lung adenocarcinoma. Mol Med Rep. 2020;21:360–70.
Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, Nie L, Chen Y, Wang YC, Liu C, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22:1264–75.
Masuda Y, Futamura M, Kamino H, Nakamura Y, Kitamura N, Ohnishi S, Miyamoto Y, Ichikawa H, Ohta T, Ohki M, et al. The potential role of DFNA5, a hearing impairment gene, in p53-mediated cellular response to DNA damage. J Hum Genet. 2006;51:652–64.
Yokomizo K, Harada Y, Kijima K, Shinmura K, Sakata M, Sakuraba K, Kitamura Y, Shirahata A, Goto T, Mizukami H, et al. Methylation of the DFNA5 gene is frequently detected in colorectal cancer. Anticancer Res. 2012;32:1319–22.
Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103.
Opitz B, van Laak V, Eitel J, Suttorp N. Innate immune recognition in infectious and noninfectious diseases of the lung. Am J Respir Crit Care Med. 2010;181:1294–309.
Chung F, Barnes N, Allen M, Angus R, Corris P, Knox A, Miles J, Morice A, O’Reilly J, Richardson M. Assessing the burden of respiratory disease in the UK. Respir Med. 2002;96:963–75.
Wang C, Xiao F, Qiao R, Shen YH. Respiratory medicine in China: progress, challenges, and opportunities. Chest. 2013;143:1766–73.
Diakos CI, Charles KA, McMillan DC, Clarke SJ. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014;15:e493–503.
Hou J, Karin M, Sun B. Targeting cancer-promoting inflammation - have anti-inflammatory therapies come of age? Nat Rev Clin Oncol. 2021;18:261–79.
Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, Yu T, Wu X, Shi Y, Ma P, Shu Y. Pyroptosis: a new frontier in cancer. Biomed Pharmacother. 2020;121:109595.
Gomes M, Teixeira AL, Coelho A, Araújo A, Medeiros R. The role of inflammation in lung cancer. Adv Exp Med Biol. 2014;816:1–23.
Pretre V, Papadopoulos D, Regard J, Pelletier M, Woo J. Interleukin-1 (IL-1) and the inflammasome in cancer. Cytokine. 2022;153:155850.
Yang J, Liu S, Li Y, Fan Z, Meng Y, Zhou B, Zhang G, Zhan H. FABP4 in macrophages facilitates obesity-associated pancreatic cancer progression via the NLRP3/IL-1β axis. Cancer Lett. 2023;575:216403.
Yuan R, Zhao W, Wang QQ, He J, Han S, Gao H, Feng Y, Yang S. Cucurbitacin B inhibits non-small cell lung cancer in vivo and in vitro by triggering TLR4/NLRP3/GSDMD-dependent pyroptosis. Pharmacol Res. 2021;170:105748.
Yokoyama S, Nakayama S, Xu L, Pilon AL, Kimura S. Secretoglobin 3A2 eliminates human cancer cells through pyroptosis. Cell Death Discov. 2021;7:12.
Hata AN, Yeo A, Faber AC, Lifshits E, Chen Z, Cheng KA, Walton Z, Sarosiek KA, Letai A, Heist RS, et al. Failure to induce apoptosis via BCL-2 family proteins underlies lack of efficacy of combined MEK and PI3K inhibitors for KRAS-mutant lung cancers. Cancer Res. 2014;74:3146–56.
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26.
Zhang CC, Li CG, Wang YF, Xu LH, He XH, Zeng QZ, Zeng CY, Mai FY, Hu B, Ouyang DY. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis. 2019;24:312–25.
Peng Z, Wang P, Song W, Yao Q, Li Y, Liu L, Li Y, Zhou S. GSDME enhances cisplatin sensitivity to regress non-small cell lung carcinoma by mediating pyroptosis to trigger antitumor immunocyte infiltration. Signal Transduct Target Ther. 2020;5:159.
Wu M, Wang Y, Yang D, Gong Y, Rao F, Liu R, Danna Y, Li J, Fan J, Chen J, et al. A PLK1 kinase inhibitor enhances the chemosensitivity of cisplatin by inducing pyroptosis in oesophageal squamous cell carcinoma. EBioMedicine. 2019;41:244–55.
Ren N, Jiang T, Wang C, Xie S, Xing Y, Piao D, Zhang T, Zhu Y. LncRNA ADAMTS9-AS2 inhibits gastric cancer (GC) development and sensitizes chemoresistant GC cells to cisplatin by regulating miR-223-3p/NLRP3 axis. Aging. 2020;12:11025–41.
Lu H, Zhang S, Wu J, Chen M, Cai MC, Fu Y, Li W, Wang J, Zhao X, Yu Z, et al. Molecular targeted therapies elicit concurrent apoptotic and GSDME-Dependent pyroptotic tumor cell death. Clin Cancer Res. 2018;24:6066–77.
Shen X, Wang H, Weng C, Jiang H, Chen J. Caspase 3/GSDME-dependent pyroptosis contributes to chemotherapy drug-induced nephrotoxicity. Cell Death Dis. 2021;12:186.
Liao X, Ye B, Hu W, Han J, Zhao Y, Dai Y, Wu X, Mo Z, Wei L, Nie K. Xiaobanxia decoction alleviates chemotherapy-induced nausea and vomiting by inhibiting GSDME-mediated pyroptosis. J Ethnopharmacol. 2024;318:116970.
Zhong Z, Gao Y, Zhou J, Wang F, Zhang P, Hu S, Wu H, Lou H, Chi J, Lin H, Guo H. Inhibiting mir-34a-5p regulates doxorubicin-induced autophagy disorder and alleviates myocardial pyroptosis by targeting Sirt3-AMPK pathway. Biomed Pharmacother. 2023;168:115654.
Liu YG, Chen JK, Zhang ZT, Ma XJ, Chen YC, Du XM, Liu H, Zong Y, Lu GC. NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis. 2017;8:e2579.
Xiao J, Wang C, Yao JC, Alippe Y, Yang T, Kress D, Sun K, Kostecki KL, Monahan JB, Veis DJ, et al. Radiation causes tissue damage by dysregulating inflammasome-gasdermin D signaling in both host and transplanted cells. PLoS Biol. 2020;18:e3000807.
Chen R, Zeng L, Zhu S, Liu J, Zeh HJ, Kroemer G, Wang H, Billiar TR, Jiang J, Tang D, Kang R. cAMP metabolism controls caspase-11 inflammasome activation and pyroptosis in sepsis. Sci Adv. 2019;5:eaav5562.
Wu M, Shi J, He S, Wang D, Zhang N, Wang Z, Yang F, He J, Hu D, Yang X, Yuan C. cGAS promotes sepsis in radiotherapy of cancer by up-regulating caspase-11 signaling. Biochem Biophys Res Commun. 2021;551:86–92.
Tan G, Lin C, Huang C, Chen B, Chen J, Shi Y, Zhi F. Radiosensitivity of colorectal cancer and radiation-induced gut damages are regulated by gasdermin E. Cancer Lett. 2022;529:1–10.
Lei Q, Wang D, Sun K, Wang L, Zhang Y. Resistance mechanisms of Anti-PD1/PDL1 therapy in solid tumors. Front Cell Dev Biol. 2020;8:672.
Jain A, Song R, Wakeland EK, Pasare C. T cell-intrinsic IL-1R signaling licenses effector cytokine production by memory CD4 T cells. Nat Commun. 2018;9:3185.
Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, Caucheteux S, Ratner-Hurevich M, Berzofsky JA, Nir-Paz R, Paul WE. IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells. J Exp Med. 2013;210:491–502.
Rosenbaum SR, Wilski NA, Aplin AE. Fueling the fire: inflammatory forms of cell death and implications for Cancer Immunotherapy. Cancer Discov. 2021;11:266–81.
Volchuk A, Ye A, Chi L, Steinberg BE, Goldenberg NM. Indirect regulation of HMGB1 release by gasdermin D. Nat Commun. 2020;11:4561.
Moriya T, Kitagawa K, Hayakawa Y, Hemmi H, Kaisho T, Ueha S, Ikebuchi R, Yasuda I, Nakanishi Y, Honda T, et al. Immunogenic tumor cell death promotes dendritic cell migration and inhibits tumor growth via enhanced T cell immunity. iScience. 2021;24:102424.
Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, Wang Y, Li D, Liu W, Zhang Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368:eaaz7548.
Brusselle GG, Provoost S, Bracke KR, Kuchmiy A, Lamkanfi M. Inflammasomes in respiratory disease: from bench to bedside. Chest. 2014;145:1121–33.
Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, Wang T, Zhang X, Chen H, Yu H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130:2620–9.
Yap JKY, Moriyama M, Iwasaki A. Inflammasomes and pyroptosis as therapeutic targets for COVID-19. J Immunol. 2020;205:307–12.
Wang J, Sahoo M, Lantier L, Warawa J, Cordero H, Deobald K, Re F. Caspase-11-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLoS Pathog. 2018;14:e1007105.
Breitbach K, Sun GW, Kohler J, Eske K, Wongprompitak P, Tan G, Liu Y, Gan YH, Steinmetz I. Caspase-1 mediates resistance in murine melioidosis. Infect Immun. 2009;77:1589–95.
Wang J, Shao Y, Wang W, Li S, Xin N, Xie F, Zhao C. Caspase-11 deficiency impairs neutrophil recruitment and bacterial clearance in the early stage of pulmonary Klebsiella pneumoniae infection. Int J Med Microbiol. 2017;307:490–6.
Chen YJ, Wang SF, Weng IC, Hong MH, Lo TH, Jan JT, Hsu LC, Chen HY, Liu FT. Galectin-3 enhances avian H5N1 Influenza A Virus-Induced Pulmonary inflammation by promoting NLRP3 inflammasome activation. Am J Pathol. 2018;188:1031–42.
Damjanovic D, Small CL, Jeyanathan M, McCormick S, Xing Z. Immunopathology in influenza virus infection: uncoupling the friend from foe. Clin Immunol. 2012;144:57–69.
Zhao C, Zhao W. NLRP3 Inflammasome-A key player in antiviral responses. Front Immunol. 2020;11:211.
Ma H, Zhu Z, Lin H, Wang S, Zhang P, Li Y, Li L, Wang J, Zhao Y, Han J. Pyroptosis of syncytia formed by fusion of SARS-CoV-2 spike and ACE2-expressing cells. Cell Discov. 2021;7:73.
Ratajczak MZ, Bujko K, Ciechanowicz A, Sielatycka K, Cymer M, Marlicz W, Kucia M. SARS-CoV-2 entry receptor ACE2 is expressed on very small CD45(-) precursors of hematopoietic and endothelial cells and in response to Virus spike protein activates the Nlrp3 inflammasome. Stem Cell Rev Rep. 2021;17:266–77.
Sun X, Liu Y, Huang Z, Xu W, Hu W, Yi L, Liu Z, Chan H, Zeng J, Liu X, et al. SARS-CoV-2 non-structural protein 6 triggers NLRP3-dependent pyroptosis by targeting ATP6AP1. Cell Death Differ. 2022;29:1240–54.
Sun X, Yu J, Wong SH, Chan MTV, Zhang L, Wu WKK. SARS-CoV-2 targets the lysosome to mediate airway inflammatory cell death. Autophagy. 2022;18:2246–8.
Zhao M, Bai M, Ding G, Zhang Y, Huang S, Jia Z, Zhang A. Angiotensin II stimulates the NLRP3 inflammasome to Induce Podocyte Injury and mitochondrial dysfunction. Kidney Dis. 2018;4:83–94.
Pinar AA, Scott TE, Huuskes BM, Tapia Caceres FE, Kemp-Harper BK, Samuel CS. Targeting the NLRP3 inflammasome to treat cardiovascular fibrosis. Pharmacol Ther. 2020;209:107511.
Wang J, Feng Y, Huo H, Zhang X, Yue J, Zhang W, Yan Z, Jiao X. NLRP3 inflammasome mediates angiotensin II-induced islet beta cell apoptosis. Acta Biochim Biophys Sin. 2019;51:501–8.
Deshpande C. Thromboembolic findings in COVID-19 autopsies. Pulmonary thrombosis or embolism? Ann Intern Med. 2020;173:394–5.
Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, Grover SP, Zhang X, Li L, Xiang B, et al. Inflammasome activation triggers blood clotting and host death through Pyroptosis. Immunity. 2019;50:1401–11.
Surabhi S, Jachmann LH, Shumba P, Burchhardt G, Hammerschmidt S, Siemens N. Hydrogen peroxide is crucial for NLRP3 inflammasome-mediated IL-1beta production and cell death in pneumococcal infections of bronchial epithelial cells. J Innate Immun. 2021;14:192–206.
Wan X, Li J, Wang Y, Yu X, He X, Shi J, Deng G, Zeng X, Tian G, Li Y, et al. H7N9 virus infection triggers lethal cytokine storm by activating gasdermin E-mediated pyroptosis of lung alveolar epithelial cells. Natl Sci Rev. 2022;30:nwab137.
Diseases GBD, Injuries C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the global burden of Disease Study 2019. Lancet. 2020;396:1204–22.
Das S, Miller M, Broide DH. Chromosome 17q21 genes ORMDL3 and GSDMB in asthma and immune diseases. Adv Immunol. 2017;135:1–52.
Stein MM, Thompson EE, Schoettler N, Helling BA, Magnaye KM, Stanhope C, Igartua C, Morin A, Washington C 3rd, Nicolae D, et al. A decade of research on the 17q12-21 asthma locus: piecing together the puzzle. J Allergy Clin Immunol. 2018;142:749–64.
Li X, Christenson SA, Modena B, Li H, Busse WW, Castro M, Denlinger LC, Erzurum SC, Fahy JV, Gaston B, et al. Genetic analyses identify GSDMB associated with asthma severity, exacerbations, and antiviral pathways. J Allergy Clin Immunol. 2021;147:894–909.
Miller M, Broide DH. Why is ORMDL3 on chromosome 17q21 highly linked to asthma? Am J Respir Crit Care Med. 2019;199:404–6.
Schmiedel BJ, Seumois G, Samaniego-Castruita D, Cayford J, Schulten V, Chavez L, Ay F, Sette A, Peters B, Vijayanand P. 17q21 asthma-risk variants switch CTCF binding and regulate IL-2 production by T cells. Nat Commun. 2016;7:13426.
Broide DH. Immunologic and inflammatory mechanisms that drive asthma progression to remodeling. J Allergy Clin Immunol. 2008;121:560–70.
Panganiban RA, Sun M, Dahlin A, Park HR, Kan M, Himes BE, Mitchel JA, Iribarren C, Jorgenson E, Randell SH, et al. A functional splice variant associated with decreased asthma risk abolishes the ability of gasdermin B to induce epithelial cell pyroptosis. J Allergy Clin Immunol. 2018;142:1469–78.
Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, Gao X, An S, Mittal M, Vogel SM, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. 2017;127:4124–35.
Shao XF, Li B, Shen J, Wang QF, Chen SS, Jiang XC, Qiang D. Ghrelin alleviates traumatic brain injury-induced acute lung injury through pyroptosis/NF-kappaB pathway. Int Immunopharmacol. 2020;79:106175.
Wu XB, Sun HY, Luo ZL, Cheng L, Duan XM, Ren JD. Plasma-derived exosomes contribute to pancreatitis-associated lung injury by triggering NLRP3-dependent pyroptosis in alveolar macrophages. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165685.
Zhuo Y, Yang L, Li D, Zhang L, Zhang Q, Zhang S, Li C, Cui L, Hao J, Li J, Wang X. Syringaresinol resisted sepsis-induced acute lung injury by suppressing pyroptosis via the oestrogen receptor-beta signalling pathway. Inflammation. 2021;45:824–37.
Liu Y, Zhou J, Luo Y, Li J, Shang L, Zhou F, Yang S. Honokiol alleviates LPS-induced acute lung injury by inhibiting NLRP3 inflammasome-mediated pyroptosis via Nrf2 activation in vitro and in vivo. Chin Med. 2021;16:127.
Wu T, Liu W, Fan T, Zhong H, Zhou H, Guo W, Zhu X. 5-Androstenediol prevents radiation injury in mice by promoting NF-kappaB signaling and inhibiting AIM2 inflammasome activation. Biomed Pharmacother. 2020;121:109597.
Liu Y, Shang L, Zhou J, Pan G, Zhou F, Yang S. Emodin attenuates LPS-induced acute lung injury by inhibiting NLRP3 inflammasome-dependent pyroptosis signaling pathway in vitro and in vivo. Inflammation. 2021;45:753–67.
He S, Ma C, Zhang L, Bai J, Wang X, Zheng X, Zhang J, Xin W, Li Y, Jiang Y, et al. GLI1-mediated pulmonary artery smooth muscle cell pyroptosis contributes to hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2020;318:L472–82.
Wang L, Chen Q, Yu Q, Xiao J, Zhao H. TREM-1 aggravates chronic obstructive pulmonary disease development via activation NLRP3 inflammasome-mediated pyroptosis. Inflamm Res. 2021;70:971–80.
Mo R, Zhang J, Chen Y, Ding Y. Nicotine promotes chronic obstructive pulmonary disease via inducing pyroptosis activation in bronchial epithelial cells. Mol Med Rep. 2022;25:92.
Crowley LC, Marfell BJ, Scott AP, Boughaba JA, Chojnowski G, Christensen ME. Waterhouse NJ.Dead cert: measuring cell death. Cold Spring Harb Protoc. 2016;2016:1064–72.
Liu Y, Zhang X, Zhang P, He T, Zhang W, Ma D, Li P, Chen J. A high-throughput Gaussia luciferase reporter assay for screening potential gasdermin E activators against pancreatic cancer. Acta Pharm Sin B. 2023;13:4253–72.
Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL, Yu EA, Schenk EL, Tan W, Zee A, et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell. 2020;182:1232–51.
Hu R, Chen X, Li Z, Zhao G, Ding L, Chen L, Dai C, Chen Y, Zhang B. Liquid nanoparticles for nanocatalytic cancer therapy. Adv Mater. 2023;35:e2306469.
Acknowledgements
The authors express their gratitude to the Natural Science Foundation of Jiangsu Province (Grant No. BK20220314), the Program of Wuxi Health Commission (Q202353), and the Program of Jiangyin Health Commission (M202204).
Funding
No funding.
Author information
Authors and Affiliations
Contributions
X.L. and Y.Q. conceived, designed, and drafted the manuscript. D.W. assisted in the preparation of the figures. Q.W. and H.W. edited and revised the manuscript; all authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liang, X., Qin, Y., Wu, D. et al. Pyroptosis: a double-edged sword in lung cancer and other respiratory diseases. Cell Commun Signal 22, 40 (2024). https://doi.org/10.1186/s12964-023-01458-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01458-w