
Abstract
Toll-like receptors (TLRs) are critical sensors for the detection of potentially harmful microbes. They are instrumental in initiating innate and adaptive immune responses against pathogenic organisms. However, exaggerated activation of TLR receptor signaling can also be responsible for the onset of autoimmune and inflammatory diseases. While positive regulators of TLR signaling, such as protein serine/threonine kinases, have been studied intensively, only little is known about phosphatases, which counterbalance and limit TLR signaling. In this review, we summarize protein phosphorylation events and their roles in the TLR pathway and highlight the involvement of protein phosphatases as negative regulators at specific steps along the TLR-initiated signaling cascade. Then, we focus on individual phosphatase families, specify the function of individual enzymes in TLR signaling in more detail and give perspectives for future research. A better understanding of phosphatase-mediated regulation of TLR signaling could provide novel access points to mitigate excessive immune activation and to modulate innate immune signaling.

Video Abstract
Similar content being viewed by others

Background
Toll like receptors (TLRs) sense pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) by innate immune cells, but also by various other cell types [1]. Upon ligand binding, TLRs trigger downstream signaling cascades, which impinge on the activation of transcription factors such as activation protein-1 (AP-1), interferon regulatory factors (IRFs), and nuclear factor kappa-light chain enhancer of activated B-cells (NF-κB). The latter is a major driver of the production of type I interferon and pro-inflammatory cytokines [2]. Consequently, the initiation of TLR signaling triggers inflammation and activates innate and adaptive immune responses [3,4,5]. As the excessive activation of this pathway can lead to life-threatening conditions, TLR signaling has to be tightly regulated [6]. In fact, an elaborated regulatory network controls TLR signaling: while activation relies on protein phosphorylation, ubiquitination, and selective protein–protein interactions, these processes are counterbalanced by the activity of protein phosphatases and deubiquitinating enzymes. The last decades have witnessed a dramatic increase in our understanding of different protein serine/threonine kinases and their target molecules in TLR signaling [7, 8]. However, we have still limited knowledge of specific protein serine/threonine phosphatases (PSPs) involved in counteracting TLR-initiated kinase signaling [9]. Given the widespread occurrence and important role of protein phosphorylation in TLR signaling, it is apparent that numerous PSPs participate in the regulation of these events. Intriguingly, the identities of most of these enzymes and their particular substrates still await clarification. In this review, we want to summarize the current knowledge about PSPs and their role in TLR signaling. Following an overview of phosphorylation events along the TLR signaling cascade, we will highlight the involved kinases and phosphatases. In the second part, we provide a more detailed description of individual PSP families, to identify gaps in our knowledge and suggest directions for future research in this area.
Part I: the role of protein phosphorylation in TLR signaling
Balancing phosphorylation and dephosphorylation of proteins is pivotal to initiate, drive, and terminate TLR signaling. Right from the initial events at the plasma membrane, cytoplasmic protein kinases with an activity directed against serine or threonine residues in their substrates (Ser/Thr protein kinases) are key drivers of TLR signaling. Representatives of this group of enzymes can be found in complex with the receptor at the plasma membrane, in a large, multi-component signalosome in the cytosol, and as regulators of gene expression in the nucleus. In fact, three main Ser/Thr protein kinase families are conserved elements of TLR signaling: Interleukin-1 receptor-associated kinases (IRAKs), the transforming growth factor-β-activated kinase 1 (TAK-1), and the IκB kinase (IKK) complex. However, additional serine/threonine kinases such as NIK, MST4, TBK-1, TANK, and RIPK also contribute to TLR-initiated processes [6, 7, 10,11,12]
IRAK family members (IRAK1, IRAK2, IRAKM, IRAK4) play a central role in TLR/IL-1 signaling. Upon ligand binding to the TLR and association with the adapter molecule MyD88, IRAKs are promptly recruited to MyD88 through death-domain interactions [13]. IRAK4 is a decisive kinase in the TIR signaling pathway and is the first enzyme to be recruited to the Myddosome complex by MyD88/TIRAP or TRIF/TRAM. Human patients with inherited IRAK-4 deficiency as well as IRAK4 knock-out mice exhibit severe functional defects in TLR pathways [14]. In line with these in vivo findings, cells lacking IRAK4 show impaired TLR pathway responses [15, 16]. Clustering of IRAK4 together with MyD88-bound TLRs results in dimerization of the kinase and trans-phosphorylation of residues within the kinase domain. Therefore, mutations in IRAK4 that abrogate IRAK4 kinase activity (IRAK4 G298D) or disrupt the interaction with MyD88 and IRAK1 (IRAK4 R12C) lead to reduced IL-1-induced signaling and cytokine production [17]. Upon activation, IRAK4 recruits IRAK1 (earlier phase) and IRAK2 (late phase) as well as TRAF6 and Pellino to the Myddosome complex [16, 18,19,20]. Upon binding, IRAK1 is phosphorylated by IRAK4 on a key threonine residue (Thr209) in the kinase domain. This phosphorylation triggers conformational changes in IRAK1, which facilitate autophosphorylation in the kinase activation loop (Thr387) as well as hyperphosphorylation in the so-called proline, serine and threonine (ProST) region of IRAK1. Once activated, IRAK1 is released from the Myddosome, but remains associated with TRAF6, an E3 ubiquitin ligase. IRAK1, but also IRAK2 and IRAK4, can phosphorylate TRAF6 and the E3 ubiquitin ligase Pellino on several residues [21, 22]. For instance, Pellino 1 has been reported to be phosphorylated on seven residues within the forkhead-associated domain and two residues within the RING-like domain [23]. Activation of the E3 ubiquitin ligases Pellino and TRAF6 via phosphorylation and their concomitant K63-linked poly-ubiquitination promotes the recruitment of additional Ser/Thr kinases [24]. At the same time, TRAF6 and Pellino act back on their upstream kinase, IRAK1, to induce the poly-ubiquitination and proteasomal degradation of this enzyme, thereby creating a negative feedback loop [25].
Signal propagation by poly-ubiquitinated TRAF6 occurs through association with the Transforming Growth Factor-β-activated kinase 1 (TAK1) in complex with TAK1 binding protein-1/2 (TAB1/2). The TAK1/TAB complex is activated by binding to K63 poly-ubiquitin chains, such as those attached to TRAF6, and by phosphorylation at Thr184, Thr187 and Ser412 in the TAK1 kinase domain [26,27,28]. Active TAK-1 in turn phosphorylates the IκB kinase β (IKKβ) at key serine residues within the activation loop at Ser176 and Ser180 [8]. IKKβ resides together with a closely related kinase, IKKα, and the scaffold protein IKKγ (also termed NEMO) in a cytosolic complex [8]. Upon stimulation by TAK1, the activity of IKKβ is directed towards the Inhibitor of NF-κB (IκBα), which is phosphorylated by active IKK on serine residues Ser32 and Ser36 [29]. Phosphorylation at these amino-terminal serine residues marks IκBα for poly-ubiquitination via Lys48-connected poly-ubiquitin chains, which initiate the efficient proteasomal degradation of this NF-κB inhibitor. Once the NF-κB heterodimer (p50/p65) is released from its protein inhibitor IκBα, the nuclear localization signal within NFκB is accessible and the transcription factor can translocate to the nucleus. There, NF-κB stimulates the transcription of a large set of genes via binding to characteristic kB sites located in the promoter regions of target genes [30, 31].
Already this short summary of canonical TLR-initiated processes illustrates the multitude of phosphorylation events, which govern this particular signaling pathway inside cells and which are summarized in Fig. 1. It is also evident from this description that Ser/Thr kinases are main actors in TLR signaling. Clearly, these kinase-mediated phosphorylation events have to be counterbalanced by serine-threonine-directed protein phosphatases to control the magnitude and duration of pathway output. However, in contrast to the detailed knowledge we have about the involved kinases, only little is known about protein phosphatases, which are directed against particular substrate proteins and specific phospho-residues in the TLR signaling cascade. This lack of knowledge is even more striking given that the human genome contains only a limited set of serine/threonine-directed protein phosphatases (see Part II of this review), which contrasts the > 400 distinct serine-threonine kinases found in mammals [32, 33]. Some of the known dephosphorylation events concern IRAK1, the kinase embedded within the Myddosome [20]. IRAK1 has been co-immunoprecipitated together with the serine/threonine phosphatase PP2A and treatment with okadaic acid, a potent inhibitor of PP2A activity, or siRNA-mediated depletion of the catalytic subunit of PP2A results in increased ubiquitination and degradation of IRAK1 in response to IL1β stimulation [34]. These findings suggest that the PP2A-regulated phospho-sites on IRAK1 are involved in orchestrating IRAK1 poly-ubiquitination and turnover by the proteasome. This would imply that PP2A activity stabilizes IRAK1 and sustains TLR signaling, while diminishing PP2A activity should negatively impact TLR signaling. On the contrary, PP1 and PP2A have been shown to completely dephosphorylate IRAK1 in vitro suggesting that they can act on several phospho-sites within this enzyme [35, 36]. As the phosphorylation state of particular residues can either have positive effects on IRAK1 activity (e.g. due to phosphorylation of IRAK1 residue Thr-387 within the kinase activation loop) or negative consequences (e.g. due to phosphorylation-dependent poly-ubiquitination leading to proteasomal degradation) it is instrumental to clearly define the particular site(s) of action of a given protein phosphatase to understand the consequences of this enzyme’s activity on TLR signaling output. Interestingly, the individual IRAK1 phospho-residues targeted by either PP2A or PP1 and the kinetics of their activity have not been elucidated so far [37]. This appears to be true for most of the various protein phosphatases, which have been implied in the TLR signaling pathway. Besides PP1 and PP2AC, the phosphatase enzymes known to affect TLR signaling are PP2B, PP4, PP6, PPM1A, PPM1B, PPM1D, PPM1E, PPM1F, PPM1L, and PPM1M. In the second part of this review, we will address the individual phosphatases and detail their known points of action along the TLR signaling cascade, but also highlight current shortcomings in our knowledge.

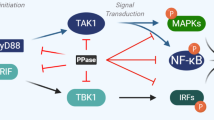
Phosphorylation events in TLR signaling. TLRs recognize their respective ligands at the cell surface or endosomes, leading to recruitment of MyD88. A receptor complex (Myddosome or Triffosome) is formed with IRAK4 and IRAK1 (and IRAK2 in the late phase). IRAK4 phosphorylates IRAK1, and the TRAF6-IRAK1 complex dissociates from MyD88. IRAK1 undergoes autophosphorylation and is degraded by the proteasome upon Pellino-1-mediated polyubiquitination. Pellino-1 and TRAF6 recruit TAK1-TAB2-TAB1 complex and IKKα/β-NEMO Complex. TAK1 is autophosphorylated and phosphorylates IKKα/β-NEMO Complex. Subsequently, IKKα/β phosphorylates IκBα bound to NF-κB p65. The latter is released and translocates to the nucleus. In the TRIF dependent pathway, TRAF6 recruits either RIPK1 and activates the IKK-IκBα-NF-κB p65 axis, or IRF5. On the other hand, TRAF3 recruits NAP1-TANK-TAB1 and NEMO-IKKε-TBK1 complexes, upon which TANK phosphorylates and activates IKKε and TBK1. Both kinases trigger IRF3 or IRF7-mediated type-I interferon production. Phosphorylated tyrosine, serine and threonine residues are represented by white, green, and circles red, respectively. Activation (ubiquitination or phosphorylation) and translocation are indicated by plain or dashed arrows, respectively
Part II: protein serine/threonine phosphatases in TLRs signaling
Negative regulation of TLR signaling is mediated by several mechanisms. Besides inhibition of kinase activity and poly-ubiquitin-initiated protein degradation, the dephosphorylation by protein phosphatases is a rapid and reversible means to limit TLR pathway output. Therefore, there is growing interest in understanding the role of different protein phosphatases in TLR signaling. Protein serine/threonine phosphatases (PSPs) in general are subdivided into three families based on structural homology: phospho-protein phosphatases (PPPs; with 13 PPPs encoded in the human genome), metal-dependent protein phosphatases (PPMs; with 16 PPMs found in humans) and aspartate-based phosphatases (including FCP/SCP- and HAD-type enzymes; with 21 members) [33, 38, 39]. As protein phosphorylation is one of the major posttranslational modifications, protein phosphatases are involved in every physiological process including cell division, cell proliferation and differentiation, programmed cell death as well as the regulation of the immune response. Here, we will focus on those family members with a known direct contribution to TLR signaling.
PPP family members in TLR pathway regulation
Human PPP-family serine/threonine protein phosphatases include PP1, PP2A, PP2B (also known as calcineurin), PP4, PP5, PP6 and PP7. In general, these enzymes contain a highly conserved catalytic core domain, which can combine with a variety of regulatory subunits to determine substrate specificity and subcellular localization [40]. PP1, PP2A, PP4, and PP6 contain three characteristic sequence motifs within the central catalytic domain, differing mainly in their C- and N-terminal regions (Fig. 2). PP2B is distinct in that it contains a Ca2+-calmodulin (CaM) binding motif (CBD) and an autoinhibitory sequence in its C-terminal region and two divergent regions in the catalytic domain. PP5 contains in its N-terminal domain three tetratricopeptide repeats (TPR). Finally, PP7 differs from all the other phosphatases in that it contains EF-hand motifs in the C-terminal domain and a large insert within the catalytic core domain [38, 39].

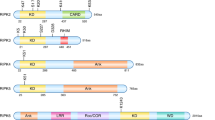
Structural organisation of members of the human PPP family. The major isoforms are depicted schematically. PPPs share high homology in the amino-terminal part of the catalytic core domain, but the carboxy-terminal parts are less similar. Autoinhibitory regions (AI), Ca2+-binding EF-hands, tetratricopeptide repeats (TPR), putative nuclear localisation signals (NLS), Peptidyl-Prolyl-Isomerase like domain (PPI-like), calcineurin B and calmodulin (CaM) binding sites are indicated. The number of regulatory subunits of each phosphatases is given on the right
Protein phosphatase 1 (PP1)
PP1 is a ubiquitously expressed protein serine/threonine phosphatase with three homologous variants of the catalytic subunit (PP1α, PP1β, and PP1γ) encoded in the human genome. A single catalytic PP1 subunit combines with one out of nearly 200 regulatory subunits to determine substrate specificity and/or sub-cellular localization [40, 41]. In TLR-mediated innate immune responses, PP1 has been reported to play both the roles of a negative and a positive regulator.
Focusing on PP1 as a negative regulator of the TLR-mediated inflammatory response, the PP1-GADD34-CUE domain–containing 2 (CUEDC2) complex sequesters IKK-α and IKK-β in the cytoplasm and maintains them in a non-phosphorylated, inactive form. Upon stimulation with a TLR ligand (e.g. LPS), IKK is ‘recruited’ away from the PP1-GADD34-CUEDC2 module by TRAF2/TRAF6 and then is phosphorylated and activated by the TNF receptor signaling complex. Subsequently, when active IKK is released from the TNF receptor signaling complex, IKKs kinase domain binds to the carboxyl terminus of CUEDC2, presumably via TRAF2/6 [42, 43]. Via its regulatory subunit GADD34, PP1 can bind CUEDC2 and dephosphorylate IKK (IKK residues phospho-Ser-180/Ser-181), returning the kinase to its default, inactive state [44]. Interestingly, IKK does not seem to be the only kinase in the TLR-signaling cascade, which is targeted by PP1. PP1 also acts on phospho-Ser-412 of TAK-1 in macrophages and murine embryonic fibroblasts and all three catalytic subunits of PP1 (α-γ) can physically interact with TAK1 [45]. Again, PP1 mediated TAK-1 dephosphorylation depends on the regulatory subunit GADD34, which comprises a TAK-1-binding region between amino acids 242–540 neighbouring the PP1 interaction motif [45]. Phosphorylation of TAK1 residues Thr-178, Thr-184, Thr-187, and Ser-192 in the kinase activation loop is involved in maximal TAK1 activity [26], while Ser-412 is located outside of the kinase domain between the TAB1 and TAB2/3 regions of TAK1. Nevertheless, dephosphorylation of phospho-Ser-412 by PP1 appears as a key regulatory step to modulate TLR-mediated pro-inflammatory cytokine induction [45, 46]. In the context of viral infections, PP1 also negatively regulates TLR-induced IFN-α and IFN-β production. Wang and colleagues [47] identified two conserved sequences (RVLF408, RVFF422) located at the N-terminal and C-terminal ends of the DNA-binding domain of human IRF7, which match canonical PP1-binding motifs. Indeed, PP1 and IRF7 physically interact and PP1 targets four IRF7 key phosphorylation sites (S471, S472, S477, S479) [47]. Dephosphorylation of IRF7 by PP1 impairs its transcriptional activity and reduces IFN-α production upon viral infection [47]. Moreover, TLR (RLR) ligands could downregulate the kinetics of PP1 activity in macrophages. In fact, authors suggested that by means of its phosphatase activity, PP1 inhibits the full activation of IRF3 (S385, S396) leading to the decrease in TLR-mediated IFN-β expression [48].
Surprisingly, PP1γ has also been described as a positive regulator of NF-κB activation. Upon TLR-initiated MyD88 signaling, PP1γ associates with TRAF6 and promotes oligomerization of this ubiquitin E3 ligase [49]. It is suggested that PP1γ association and/or activity induce a conformational change in TRAF6 to increase its auto-ubiquitination as well as ubiquitination of the TRAF6 substrate IKKγ (NEMO), which would in the end lead to enhanced NF-κB signaling [49]. The positive contribution to NF-kB-mediated gene expression requires PP1γ phosphatase activity, but TRAF6 phospho-sites targeted by PP1γ have not been delineated [49]. Furthermore, it is unclear if a regulatory subunit of PP1γ is involved in this process. Through these events, PP1γ might contribute to physiological stimulation of NF-kB, but PP1-mediated activation of TRAF6 seems to be of particular importance in certain tumor types such as glioma cells [50] and liver cancer [51].
Protein phosphatase 2A (PP2A)
Similar to PP1, two variants of the catalytic subunit of the serine/threonine phosphatase PP2A, termed PP2Acα and PP2Acβ, are encoded in the human genome. Either of the catalytic subunits forms the central enzyme, which combines with the scaffold subunit (PP2Aa⍺ or PP2Aaβ) and one out of 26 different regulatory subunits to build the holoenzyme complex. Again, the regulatory subunits determine the spatio-temporal specificity of PP2A enzyme activity [52,53,54]. A negative role of PP2A in TLR-mediated signaling was initially postulated, when application of the protein serine/threonine phosphatase inhibitor ocadaic acid resulted in increased levels of LPS-induced cytokines and chemokines [55, 56]. Additionally, reduction of PP2A expression by specific siRNAs resulted in a gain of function with regard to LPS-induced TNFα secretion further suggesting that PP2A limits TLR-induced responses [56]. Subsequently, biochemical studies have detected an interaction between PP2A and IRAK1 upon IL1β stimulation, with direct consequences for IRAK1 phosphorylation levels [34]. Potentially, there are additional targets of PP2A in the TLR signaling cascade, such as IKKα/β/γ [57], IκB-α [58], NFκB p65 [59] and MAPKs [56, 60], which have also been reported to associate with PP2A. Clear evidence for a physiological role of PP2A in TLR signaling comes from gene disruption of one of the two catalytic subunits (PP2Acα) in myeloid cells [61]. Though knock-out macrophages derived from these mice still express PP2Acβ, the total PP2A phosphatase activity was reduced by ~ 60%. This reduction was coupled to an increase in LPS-stimulated phosphorylation of proteins in the MAPK pathway (p38, JNK/c-Jun, and ERK [p44/42]) and the NF-κB pathway (IKKα/β, NF-κB p65, and IκBα) [61]. In vivo, the lack of PP2Ac⍺ in myeloid cells translated into an exaggerated inflammatory response and higher sensitivity towards LPS, with detrimental consequences for these animals upon bacterial infection [61].
One negative feed-back-loop that involves PP2A and that dampens TLR-initiated signaling is based on the lipid ceramide, which can be produced from membrane sphingolipids during inflammation [62, 63]. Ceramide either binds to PP2A directly or associates with and thereby displaces an inhibitor of PP2A termed SET [64, 65]. In both cases, PP2A phosphatase activity is unleashed by ceramide diminishing phosphorylation of multiple signaling proteins. For example during LPS-induced lung inflammation, PP2Acα is responsible for dampening cytokine release and ceramide treatment of macrophages activates PP2A to alleviate some of the acute responses and damage in the lung tissue [66]. However, despite the cumulative evidence for an important regulatory role of this protein phosphatase, the direct target(s) of PP2Ac⍺ activity and the identity of PP2Acα-associated regulatory subunits involved in the TLR signaling cascade are currently unclear.
Interestingly, PP2Ac⍺ can also associate with and de-phosphorylate serine residues in the TIR domain of MyD88 [67]. Phosphorylation of MyD88 serine residues Ser-242 and Ser-244 seems to stabilize the TLR-MyD88 complex, while PP2A-mediated dephosphorylation of these residues dampens TLR-initiated signals. This activity of PP2A is only detected during a process termed endotoxin tolerance, when immune cells have been first exposed to a low dose of LPS (0.1 µg/ml), which results in increased PP2A activity after 24 h. When these LPS-experienced cells are re-stimulated with higher doses of LPS (1 µg/ml for 15 min) the increased PP2A activity and the PP2A-mediated de-phosphorylation of MyD88 suppress TLR downstream signaling and inflammatory cytokine release [67]. Presumably, distinct PP2A holoenzymes are responsible for the different effects of this phosphatase on the initial TLR signal as described before versus the effect of PP2A on signal outcome upon TLR re-stimulation during LPS tolerance. However, it is also conceivable that differential posttranslational modification of a single PP2A holoenzyme might direct its activity towards distinct kinases in initial TLR stimulation versus the de-phosphorylation of the adapter protein MyD88 upon re-stumulation with TLR ligands.
Protein phosphatase 2B (PP2B)/calcineurin
PP2B is a Ca2+/calmodulin-dependent serine/threonine phosphatase, which is also known as Calcineurin. In vertebrates, three genes (PPP3CA, PPP3CB, and PPP3CC) encode three related catalytic subunits termed Calcineurin A or PP2BC. The genes PPP3CA and PPP3CB are ubiquitously expressed, while PPP3CC transcripts are only found in testis and brain [68]. In addition, two distinct forms of the regulatory subunit, Calcineurin B, are available: Calcineurin B1, encoded by the PPP3R1 gene, is found in all cell types, while the product of the PPP3R2 gene, the protein phosphatase 3 regulatory subunit-like protein, is only found in testis [69]. The regulatory subunit Calcineurin B contains four Ca2+-binding EF-hand domains, while the catalytic subunit Calcineurin A contains two Calmodulin-binding sites, which are involved in regulating PP2B phosphatase activity in a Ca2+/Calmodulin-dependent manner [70]. Interestingly, while knock-out mice lacking either Calcineurin catalytic subunit are viable, the deletion of the regulatory subunit Calcineurin B1 leads to embryonic lethality [71]. Nevertheless, lack of either PPP3A or PPP3B impairs T cell function [72,73,74]. In T cells, PP2B dephosphorylates the transcription factor NFAT to promote its translocation to the nucleus, where it induces expression of cytokines such as IL-2 [75]. In addition, PP2B-mediated dephosphorylation of Bcl-10 contributes to NF-kB activity in response to TCR stimulation [76, 77]. The positive regulatory role of PP2B for T cell activation is in line with the use of PP2B inhibitors such as cyclosporin A or FK506 as immunosuppressants [68]. PP2B also has a role in the TLR-mediated innate immune response, with both negative and positive contribution to the TLR signaling pathway depending on cell type and stimulus.
For instance, PP2B acts as a negative regulator of the TLR signaling pathway in human and murine macrophages, astrocytes, and fibroblasts [78]. In macrophages, inhibition or knock-down of PP2B enhances the expression of inflammatory cytokines in response to LPS, poly(I:C), peptidoglycan, or CpG DNA, while overexpression of constitutively active Calcineurin blocks NF-κB activation by TLR ligands [79]. Protein–protein interaction and functional analysis in peritoneal macrophages revealed that PP2B interacts with TLR2, TLR4, MyD88 and TRIF, but not with TLR3 and TLR9 [79]. Accordingly, PP2B negatively regulates both MyD88-dependent and MyD88-independent TLR signaling pathways. Similar to macrophages, treatment of endothelial cells with PP2B inhibitors leads to elevated TLR4-dependent gene expression [80]. Furthermore, the treatment of kidney tubular cells with cyclosporin A or FK506 (Tacrolimus) resulted in activation of NF-κB-mediated inflammatory responses accompanied by increased TLR4/MyD88/IRAK signaling suggesting that PP2B negatively regulates this pathway [81]. In a similar manner, the application of calcineurin inhibitors led to elevated coronary arteritis in a mouse model of Kawasaki disease and this effect depended on the presence of MyD88 [82]. Though the direct target of PP2B activity in the TLR4 signaling cascade is currently unknown, overexpression of the constitutively active phosphatase reduces the phosphorylation of IRAK-1 in a dose-dependent manner [79] suggesting that PP2B acts at a position close to the activated receptor to diminish NF-kB activation. Furthermore, increased phosphorylation and degradation of IκBɑ was observed upon PP2B inhibition, suggesting a further mode of action of how this phosphatase could suppress NF-κB activity [83].
In contrast to these negative regulatory effects of PP2B on TLR signaling and NF-κB mediated gene expression events, several studies have indicated a positive contribution of PP2B to this pathway, which would be analogous to the situation in T-cell receptor signaling. For example, primary macrophages lacking a negative regulator of calcineurin-1 termed RCAN-1 show elevated inflammation in response to bacterial LPS due to enhanced TLR4-MyD88-NF-kB signaling [84]. Furthermore, PP2B inhibition by cyclosporin A selectively diminishes TLR-9-mediated IL10 expression in human B-cells [85] and treatment of mice with the PP2B inhibitor FK506 protects from LPS-induced toxicity and leads to a LPS tolerant phenotype of isolated macrophages [86]. Along the same line, peripheral blood mononuclear cells from patients receiving PP2B inhibitors show impaired inflammatory cytokine production in response to TLR2, TLR4, and TLR7/8 stimulation [87].
Together, these investigations do not draw a clear picture of PP2B’s involvement in TLR signaling. One should bear in mind that several of these studies to a large extend rely on prolonged use of Calcineurin inhibitors such as Tacrolimus or cyclophilin A. Importantly, knock-out macrophages with genetic ablation of the regulatory subunit Calcineurin B1 indicate that such pharmacological inhibitors can have effects, even when the primary target molecule is not present [82]. Therefore, contrasting reports on the role of PP2B in TLR signaling should be interpreted with caution and future emphasis should be placed on clearly delineating bone-fide protein phosphorylation sites affected by the action of PP2B/calcineurin in TLR signaling.
Protein phosphatase 4 (PP4)
PP4 is a conserved protein serine/threonine phosphatase, which is expressed in all cell types and which is found throughout the cytoplasm and the nucleus, showing enrichment around the centrosome and microtubule nucleation points [88,89,90]. PP4 is composed of the PP4 catalytic subunit (PP4c), whose association with different PP4R regulatory subunits determines substrate specificity. With regard to innate immunity, PP4 has been initially reported as an activator of NF-kB-dependent transcriptional responses by dephosphorylating the c-Rel subunit of the NF-kB transcription factor [91]. Similarly, PP4 seems to act on phospho-Thr-435 of the NF-kB subunit p65 and this event correlated with increased NF-kB transcriptional activity [92]. This supposedly positive effect of PP4 on NF-kB signaling contrasts the situation with PP2A, which by dephosphorylating the RelA subunit of the NF-kB transcription factor negatively affects NF-kB transcriptional responses [59].
However, the role of PP4 in TLR signaling appears to be complex, as numerous examples of PP4′s negative regulatory function in the TLR signaling pathway became apparent. For example, the regulatory subunit R1 of PP4 (PP4R1) physically interacts with TRAF2 and TRAF6 upon LPS stimulation in murine macrophages [93, 94]. PP4 dephosphorylates specific sites in TRAF2 (Ser-11) and presumably in TRAF6, thereby blocking TRAF6 auto-ubiquitination [94]. Reduced ubiquitination of TRAF6 interferes with recruitment of TAK1 and the IKK complex ultimately reducing the activation of NF-κB [93, 94]. In line with this model, the holoenzyme PP4C-PP4R1 is proposed to regulate IKK activity and to suppress the oncogenic NF-kB signaling in T cell lymphomas. In fact, T cell activation and proliferation trigger PP4R1 expression, which is lost in cutaneous T cell lymphoma (CTCL) [95]. PP4R1 recruits PP4C to IKK and mediates the dephosphorylation of the IKK complex at residues Ser-176/Ser-180. Small interfering RNA-mediated PP4R1 depletion causes sustained and increased IKK activity and T cell hyperactivation. Furthermore, deficiency for PP4R1 in CTCL results in constitutive IKK-NF-kB signaling and thus forms an important molecular event maintaining the malignant phenotype of a subset of CTCL cells [95]. Also in the context of antiviral immunity, PP4C displays a negative regulatory function and suppresses TLR mediated production of type I IFN, as siRNA-mediated silencing of PP4C results in elevated responses towards LPS or poly(I:C) [96]. In particular, PP4C targets and dephosphorylates TBK1 at residue Ser-172. This dephosphorylation inhibits TBK1 kinase activity and thereby restrains IRF3 activation and type I IFN production [96]. Similarly, other viral agents, including polyomaviruses, exploit the PP4R1/PP4C module to interfere with NF-kB activation, thereby evading antiviral immune responses [97,98,99]. In these cases, the viral small T antigen connects PP4R1 and PP4C with NEMO, the scaffold of the IKK complex, and this interaction is instrumental to inhibit NF-kB activation [100]. This is reminiscent of the situation in T-cells, where PP4R1/PP4C are shown to act on IKKα and IKKβ [95] a scenario that is compatible with the findings from polyomavirus-infected cells. Together, several lines of evidence support a negative regulatory role of PP4 together with the regulatory subunit PP4R1 for NF-kB activation, while regulatory subunits of PP4 involved in promoting NF-kB signaling are currently not clearly established.
Protein phosphatase 6 (PP6)
PP6, closely related to PP2A and PP4, is a Ser/Thr phosphatase with a bimetallic catalytic center and is expressed in most human cells and tissues [38, 101]. PP6 comprises a catalytic core domain and three regulatory subunits, namely PP6R1, PP6R2 and PP6R3, and is involved NF-kB signaling regulation. The PP6R1 is highly expressed in hematopoietic and lymphoid cells [102].
Proteomic and gene depletion analyses revealed that PP6 specifically down-regulates TAK1 activity by dephosphorylating the phospho-Thr-187 residue in the activation loop of the kinase [103]. As phosphorylation at Thr-187 is connected to increased TAK-1-induced NF-κB activity [104], it is reasonable to assume that PP6 is a negative regulator of this pathway. However, it is currently unknown, which PP6 regulatory protein is involved in this process and how the overall outcome of TLR-initited NF-κB activation is affected by PP6 activity.
PPM family members in TLR pathway regulation
The PPM serine/threonine phosphatase family comprises 16 members (PPM1A-N) and most of them are widely expressed in human tissues [105, 106]. Structurally, these phosphatases are monomeric Mg2+/Mn2+ dependent phosphatases with a highly conserved core catalytic domain. Importantly, they generally differ from the PPP phosphatases by not forming holoenzymes. Instead, all PPM members possess N-terminal or C-terminal non-enzymatic domains, which contribute to substrate specificity and/or structural stability (Fig. 3) [38, 106,107,108]. Some PPM members contain, besides their phosphatase domain, typical structural folds such as a pleckstrin homology (PH) domain and a leucine-rich repeat (LRR) region in PHLPP [109, 110]. Moreover, a few PPMs contain distinct structural insertions within the catalytic core and a recent study involving PPM1G suggests that this enzyme can form a PPP-type holoenzyme using the regulatory domain B56δ of PP2A to operate in the context of adherens junction integrity [111].

Structural organisation of members of the human PPM family. Representative members of the PPM family are depicted schematically. The PP2C-catalytic core domains of each protein are represented by the green boxes and the delimiting amino acid residues are given. The amino acids involved in chelating metal-ions are highlighted with red, blue and brown circles. N-myristoylation sites (yellow circle), sequence motifs, and protein- or lipid-binding domains are indicated
Generally, PPM phosphatases are involved in diverse cellular processes such as cell proliferation, growth, survival, apoptosis, regulation of metabolism, and stress signaling [105, 106, 112,113,114,115,116]. However, only few studies have investigated the role of PPM phosphatases in the immune response and in particular in TLR-initiated signaling cascades.
Protein phosphatase Mg2+/Mn2+ dependent 1A (PPM1A/α)
PPM1A is ubiquitously expressed in human tissues and, together with PPM1B, is the PPM family member, which has been studied most intensively with respect to innate immunity. [117] In the context of antiviral immunity, PPM1A was assigned an important role in governing antiviral defense and balancing anti-viral signal transduction. This phosphatase antagonizes TANK-binding kinase (TBK1)-mediated phosphorylation and aggregation of STING, which is orchestrating innate immune responses upon detection of cytosolic DNA as a sign of viral infection or mitochondrial damage [118, 119]. In addition, PPM1A antagonizes RIG-I-like receptors (RLRs), which respond to RNA viruses. Cytosolic double-stranded RNA-sensing by RLRs activates TBK1/IKKε kinases via mitochondria-associated anti-viral signaling proteins (MAVS). Analogous to counteracting TBK1 in response to DNA, PPM1A dephosphorylates the TBK1/IKKε complex and MAVS, thereby limiting the immune response against RNA viruses. As a result, PPM1A knock-out mice are resistant against RNA viruses, whereas zebrafish overexpressing PPM1A are more susceptible to RNA virus infection [120].
Likewise, PPM1A also limits TLR-initiated responses, as cytokine release upon LPS stimulation or upon Mycobacterium tuberculosis infection are reduced in PPM1A-overexpressing THP-1 monocytic cells [121]. In contrast, knock-down of PPM1A results in elevated levels of TNFα-release and PPM1A expression is upregulated in response to TLR stimulation by the agonists imiquimod and Pam3CSK4 [122]. These results could indicate that PPM1A operates in a negative feedback-loop to prevent an exaggerated and prolonged TLR response and to restore pre-inflammatory conditions [122].
The ability of PPM1A to dampen TLR signaling appears to hinge on the dephosphorylation of IKKβ at residues p-Ser-177 and p-Ser-181 [123]. PPM1A activity towards IKKβ terminates TNFα-induced NF-κB signaling and the same situation has been reported for PPM1B [123]. Therefore, PPM1A-mediated control of TBK1 and IKKβ phosphorylation can explain the negative regulatory role of this enzyme in TLR signaling.
Protein phosphatase Mg2+/Mn2+ dependent 1B (PPM1B/β)
PPM1B is structurally similar to PPM1A and its genetic deletion abrogates embryonic development [124]. PPM1B plays a prominent role in inflammation by restoring the balance between apoptotic and anti-apoptotic signaling in response to TNFα in various cell lines. Together with PPM1A, PPM1B functions by specifically associating with and dephosphorylating the IKKβ complex at p-Ser-177/p-Ser-181 [125] and the upstream kinase TAK1 to terminate TNFα-induced NF-κB activity [123, 125, 126]. The adapter protein 14–3-3ε seems to be involved in the spatial and temporal co-ordination of the PPM1B—TAK1 interaction [127]. Accordingly, PPM1B is part of a negative feedback loop, which helps to limit and resolve NF-κB pathway activity at later time points following cytokine stimulation [125]. The negative feedback excerted by PPM1B on the NFκB pathway can be counter-regulated itself via PKA-mediated phosphorylation of PPM1B at Ser-195, which results in proteasomal degradation of PPM1B [128]. With regard to TLR signaling, there are striking parallels between PPM1B and PPM1A: Upon viral infection, PPM1B blocks the antiviral response by increased association with and dephosphorylation of TBK1 at Ser-172, allowing enhanced virus replication [129]. This negative regulatory function of PPM1B is exploited by certain viral proteins, which direct this phosphatase to suppress interferon production [130]. Though PPM1B has mainly been studied in the context of antiviral responses, it is highly plausible that this enzyme also controls TLR-initiated signaling during encounters with pathogenic bacteria, an area clearly demanding further exploration.
Protein phosphatase Mg2+/Mn2+ dependent 1D (PPM1D/Wip1)
The wildtype p53-induced phosphatase 1 (Wip1, encoded by PPM1D) is a well-studied oncogenic member of the PPM family. It is rapidly and transiently expressed upon DNA-damaging agents and ionizing or UV irradiation in a p53-dependent manner [131,132,133]. As its name implies, Wip1 has a key role in restoring pre-stress cell homeostasis by controlling critical cellular functions such as proliferation, cell cycle arrest and programmed cell death after p53-dependent stress stimuli [134,135,136,137,138].
Wip1 is essential for immune cell development and differentiation including T and B cells, neutrophils and macrophages by regulating p53-dependent and p53-independent p38 MAPK-STAT1 pathways [138,139,140,141]. PPM1D knock-out mice exhibit both neutrophilia and an abnormal lymphoid histopathology in thymus and spleen, accompanied by severe defects in immune cell functions [140,141,142]. Not surprisingly, PPM1D-depleted mice also show increased susceptibility to pathogens and viral infections [143].
For example, PPM1D negatively regulates pro-inflammatory cytokine production of neutrophils after bacterial infection [144]. Accordingly, neutrophils from Wip1-deficient mice release increased amounts of pro-inflammatory cytokines and facors such as elastase, lactoferrin, and myeloperoxidase upon LPS stimulation. Apparently due to this gain in bactericidal potency, Wip1-KO mice can better contain Staphylococcus aureus infection in a skin abscess model [144]. On the other hand, the enhanced neutrophil functions result in pronounced LPS-induced lung damage accompanied by increased neutrophil infiltration and inflammation in the Wip1-KO mice [144]. Ppm1d-/-mice display a pro-inflammatory phenotype in skin and intestine with elevated levels of the cytokines TNFα, IL-6, IL-12 and IL-17 [143]. Finally, Wip1 appears to be involved in LPS-induced neuro-inflammation in a blood–brain-barrier (BBB) model, where Wip1 expression levels decreased upon LPS stimulation and the decrease in Wip1 is accompanied by augmented levels of the inflammatory cytokines TNFα, IL-1β, IL-12 and IL-6 [145].
While Wip1 expression is positively regulated by NF-κB [146], the p65 subunit of the transcription factor itself seems to be the main target of Wip1 activity [147]. In particular, Wip1 dephosphorylates p-Ser-536 of the NF-κB p65 subunit and this phospho-residue is essential for the transactivation function of p65 in recruiting the co-activator p300 [147]. Thus, this p65-directed activity of Wip1/ PPM1D can nicely explain the negative effect of this phosphatase on the expression of NF-κB-dependent inflammatory factors such as IL-1, IL-6 and IL-8 [147]. Together, the phosphatase Wip1 appears as a further PPM family member involved in negative feedback regulation of TLR-induced NF-κB signaling.
Protein phosphatase Mg2+/Mn2+ dependent 1E (PPM1E)
The relatively large, 755 amino acids containing PPM family member PPM1E localizes to the nucleus and is predominantly expressed in brain and testis [148]. Only one prominent study on inflammatory pathway control by PPM1E has been published so far. PPM1E seems to indirectly affect TLR signaling, and its action has opposing consequences compared to other PPM family members [149]. In particular, expression of miR-135-5b in human cultured monocytes downregulates PPM1E expression and thereby activates AMPKα signaling via increased phosphorylation of AMPKa residue Thr-172. This yields in a marked attenuation of LPS-induced TNFα expression by suppression of ROS production and NF-κB activation [149]. Therefore, PPM1E positively contributes to LPS-induced responses by keeping the negative player—AMPK—under control.
Protein phosphatase, Mg2+/Mn2+ dependent 1F (PPM1F)
The ubiquitously expressed cytoplasmic PPM1F is implicated in determining integrin-mediated cell adhesion, migration and survival and plays a key role in controlling neuronal functions by regulating Ca2+/Calmodulin-dependent kinase cascades [150,151,152,153,154,155]. Accordingly, ppm1f-/-mice show severe developmental defects and die around day E10.5 in utero [155]. PPM1F function in innate immunity has not been intensively studied, but recently PPM1F has been recognized as negative regulator of the IKK-NF-κB pathway in response to DNA-damage by dephosphorylating p-Thr-187 of TAK1 [156]. Accordingly, cells with low expression levels of PPM1F exhibit higher TAK1 activity and, in turn, show increased nuclear translocation of NF-κB and upregulation of anti-apoptotic proteins to promote cell survival and thus chemo-resistance [156]. Consequently, PPM1F might also play a role in the TLR-TAK1-NF-κB axis. Supporting this idea, Zhang et al. reported that the production of inflammatory cytokines and chemokines (IL-6, TNFα and CXCL10) was significantly augmented in THP1 PPM1F knock-down macrophages upon LPS-TLR4 stimulation [157]. Hence, it is conceivable that PPM1F suppresses the TLR4-TAK1-NF-κB signaling pathway similar to other PPM family members. Nevertheless, the molecular substrate(s) and the target residue(s) of PPM1F upon LPS stimulation still remain to be clarified.
Protein phosphatase Mg2+/Mn2+ dependent 1L (PPM1L/ε)
PPM1L is a phosphatase localized at the endoplasmic reticulum (ER) [158]. Similar to other family members, PPM1L appears to be capable to suppress the TAK1-NF-κB pathway. Li and colleagues analyzed IL-1 / IL-1-receptor signaling and found that the activities of JNK and p38 were constantly counterregulated by PPM1L [159]. PPM1L associates with and dephosphorylates TAK1 and thereby inhibits MKK4/MKK6 binding to TAK1. On the other hand, the activity of PPM1L is diminished upon IL-1 treatment of cells resulting in transient activation of transcription factors such as AP-1 [159]. Accordingly, PPM1L appears as a further anti-inflammatory regulator [105, 159]. In line with that, a recent study assigned PPM1L a function overlapping with the activities of PPM1A and PPM1B: inhibiting NF-κB signaling via dephosphorylation of IKKβ. Clearly, PPM1L directly binds IKKβ and dephosphorylates p-Ser-177/p-Ser-181 in IKKβ, which are phosphorylated in macrophages by TAK1 upon TLR signaling [160]. Accordingly, PPM1L activity diminishes the release of inflammatory cytokines IL-1β, IL-6, TNF-α, and IL-12 [160], an activity, which could help to prevent excessive inflammatory responses.
Protein phosphatase, Mg2+/Mn2+ dependent 1 M (PPM1M/η)
PPM1M accumulates in the nucleus and its function correlates with other members of the PPM family such as PPM1A and B in selectively acting on IKKβ downstream of TAK1 to suppress IL-1-induced NF-κB activation [161]. Accordingly, this phosphatase appears to suppress the TLR-initiated TAK1-NF-κB axis in a way similar to other PPM family members.
Protein phosphatase Mg2+/Mn2+ dependent 1 N (PPM1N)
PPM1N is one of the least studied members of the PPM family. However, genome-wide investigations revealed that PPM1N expression in bone marrow macrophages was up-regulated upon B. abortus infection [162], suggesting a function of PPM1N either in the immune response towards bacteria or in inflammation.
Conclusion
Identifying the involved protein phosphatases and their molecular targets in TLR signaling provides a rational basis to expand existing therapeutic strategies in acute and chronic inflammation as well as autoimmune diseases [163]. A detailed knowledge of the protein phosphatases, which counteract the various kinases downstream of TLR stimulation, could also open up novel opportunities to cope with viral or bacterial pathogens that evade host immune responses. The serine/threonine phosphatases involved in TLR signaling and their point of interception as presented in this review is summarized in Fig. 4. It has become clear that numerous pathogenic bacteria interfere with innate immunity by exploiting host protein phosphatases. In this regard, the protein tyrosine phosphatase SHP-1 is activated by Bordetella pertussis to block iNOS expression and NO production in TLR-activated macrophages and also exploited by Neisseria gonorrhoeae or Moraxella catarrhalis to block IL-1β production via regulation of the SYK-TLR4-CEACAM1 complex [164, 165]. Similarly, Listeria monocytogenes directly hijacks PPM1A and PPM1B to dephosphorylate and translocate SIRT2 into the nucleus to promote effective infection [166]. Since these two Ser/Thr phosphatases are also an integral part of a negative regulatory feedback loop in TLR signaling, it is plausible that Listeria—triggered PPM1A and PPM1B activities also interfere with TLR-initiated innate immune signaling.

TLR-initiated signaling and the involved protein Ser/Thr phosphatases (Figs 3, 4). Summary of serine and threonine phosphorylation and de-phosphorylation events in TLR-signaling. IRAK1 phosphorylation by IRAK4 is reverted by PP2A. TRAF6 activity is inhibited by PP1 and PP4. TAK-1 has multiple phosphorylation sites, which are targeted by PP1 (Ser-432), PP6 (Thr-187), PPM1B (Ser-192), PPM1F (Thr-187), PPM1L (Thr-187), and PPM1M (unknown residues). PP1-mediated inactivation of TAK1 occurs in four sequential steps (Top frame on left). GADD34 binds to phosphorylated TAK1 (1), and recruits PP1C (2). The TAK1-GADD34-PP1C complex is formed (3) and PP1C catalyses TAK1 dephosphorylation and inactivation (4). Subsequently, the NEMO-IKK⍺/β complex phosphorylation by TAK1 is reverted by PPM1A, PPM1B, PPM1L and PPM1M on residues Ser-177 and Ser-181 and by PP1 on Ser-180/Ser-181. In the ground state, NEMO-IKKα/β complex is in a non-phosphorylated inactive form associated with GADD34/CUEDC2/PP1C (Lower frame on left). Upon stimulation, the NEMO-IKKα/β complex is released from PP1C by TRAF6 recruiment (1), IKKα and IKKβ are phosphorylated by TAK1 and subsequently trigger degradation of IκBα (2). CUEDC2 is recruited to phosphorylated IKKα/β (3), recruits GADD34 (4) and GADD34 in turn recruits PP1C (5). Finally, PP1C dephosphorylates IKKα/β and the whole complex retunrs to the ground state (6). The phosphorylation of IκBα is countered by PP6. NF-κB is de-phosphorylated by PP1, PP4 (Thr-465) and PPM1D (Ser-536). Tyrosine, serine and threonine residues are represented by white, green and red circles, respectively. Dephosphorylation is indicated by a red stop line, activation (ubiquitination or phosphorylation) and translocation are indicated by plain and dashed arrows, respectively
In general, pinpointing the contribution of a particular protein phosphatase to specific cellular events remains a challenge for several reasons: (i) in contrast to protein kinases there is a lack of potent and selective phosphatase inhibitors; (ii) the action of a phosphatase can in many cases only be revealed upon a positive stimulus, hampering the direct evaluation of phosphatase action; (iii) the low complexity and essential function of catalytic subunits, especially in the case of PPPs, often leads to lethal phenotypes in knock-out approaches; and (iv) redundancy between closely related enzymes, as seen for PPMs, often results in similar phenotypes upon overexpression.
The absence of highly specific enzyme inhibitors is a major obstacle for the phosphatase field in general. In the past, inhibitors such as ocadaic acid or microcystins have been widely used, but the explanatory power of such findings is often limited as PP1, PP2A, PP4, and PP6, with their extensive homology, are all sensitive to the same class of inhibitors. Thus, it is desirable, but also more demanding, to complement such pharmacologial approaches with genetic manipulation of individual phosphatases. However, one has to be aware that genetic approaches requiring prolonged selection, e.g. generation of clonal knock-out or knock-down cell lines, can trigger compensatory mechanisms that entail altered expression of related enzymes. Such compensatory activity by other, related phosphatases with overlapping substrate spectrum can easily mask a phenotype. Therefore, novel small molecule inhibitors or activators of specific phosphatases that allosterically interfere with enzyme activity of the catalytic subunit are welcome additions to the investigators toolbox [167, 168]. Furthermore, innovative strategies triggering the rapid and selective degradation of a particular phosphatase allow instant interrogation of protein function in the model system of choice [169]. Together, these approaches are poised to overcome current limitations in phosphatase research.
Given the prominent role of protein phosphorylation as a regulatory principle in TLR signaling, it is obvious, that manipulation of phosphatase activity represents a promising strategy to control innate immune responses. As we have discussed, there are numerous examples of protein phosphatases exerting negative regulatory roles in TLR signaling, but the opposite situation, a positive contribution to NF-κB activation, is also known for some phosphatase enzymes. Accordingly, a suite of small molecule stimulators as well as inhibitors of defined protein phosphatases would be ideally suited to allow a transient tuning of the TLR pathway in either direction: towards increased signaling output, when the ultimate goal is to limit infectious agents, or towards dampened responses in a situation of exaggerated or chronic inflammation. Though such approaches are still in their infancy, it is about time to unleash the translational potential of protein phosphatases in innate immunity.
Availability of data and materials
Not applicable.
Abbreviations
- AMPKα:
-
5′ AMP-activated protein kinase
- AP-1:
-
Activation protein-1
- CUEDC2:
-
Coupling of ubiquitin conjugation to ER degradation (CUE) domain-containing protein 2
- DAMPs:
-
Damage-associated molecular patterns (DAMPs)
- FCP/SCP:
-
TFIIF-associating component of RNA polymerase II C-terminal domain IL-
- GADD34:
-
Growth arrest and DNA damage-inducible protein
- HAD:
-
Halo acid dehydrogenase
- 1R:
-
Interleukin 1 receptor
- IL-1R:
-
Interleukin-1 receptor
- IRFs:
-
Interferon regulatory factors
- IRAKs:
-
Interleukin-1 receptor-associated kinases
- IκB:
-
Inhibitory κappa B
- IKK:
-
IκB kinase
- LPS:
-
Lipopolysaccharides
- MAPK:
-
Mitogen-activated protein kinase
- MAVS:
-
Mitochondrial antiviral-signaling protein
- MKK:
-
Mitogen-activated protein kinase kinase
- MST4:
-
Mammalian STE20-like protein kinase 4
- MyD88:
-
Myeloid differentiation primary response 88
- NEMO:
-
NF-Kappa-B essential modulator
- NFAT:
-
Nuclear factor of activated T-cells
- NF-κB:
-
Nuclear factor kappa-light chain enhancer of activated B-cells
- NIK:
-
NF-kappa-B-inducing kinase
- PAM3CSK4:
-
Tripalmitoylated lipopeptide
- PAMPs:
-
Pathogen-associated molecular patterns
- PPM:
-
Metal-dependent protein phosphatase
- PPP:
-
Phosphoprotein phosphatases
- PSPs:
-
Protein serine/threonine phosphatases
- RCAN-1:
-
Regulator of calcineurin 1
- RIPK:
-
Receptor-interacting serine/threonine-protein kinase
- RLR:
-
Retinoic acid-inducible Gene-I-like receptor
- STAT1:
-
Signal transducer and activator of transcription 1
- STING:
-
Stimulator of interferon genes protein
- TAK-1:
-
Transforming growth factor-β-activated kinase 1
- TANK:
-
TRAF family member-associated NF-Kappa-B activator
- TBK-1:
-
TANK-binding kinase 1
- TCR:
-
T cell receptor
- TIR:
-
Toll/interleukin-1 receptor
- TLR:
-
Toll-like receptor
- TRAF:
-
Tumor necrosis factor receptor-associated factor
- TRIF:
-
TIR-domain-containing adapter-inducing interferon-β
- TRAM:
-
TRIF-related adaptor molecule
- TAB1/2:
-
TAK1-binding protein 1/2
- TNF-α:
-
Tumor necrosis factor alpha
- WIP1:
-
Wild-type p53-induced phosphatase 1
References
O’Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors—redefining innate immunity. Nat Rev Immunol. 2013;13:453–60.
Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–51.
Pelka K, De Nardo D. Emerging Concepts in Innate Immunity. Methods Mol Biol. 2018;1714:1–18.
Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell. 2020;180:1044–66.
Kawai T, Akira S. Signaling to NF-kappaB by toll-like receptors. Trends Mol Med. 2007;13:460–9.
Brown J, Wang H, Hajishengallis GN, Martin M. TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dental Res. 2011;90:417–27.
Chau TL, Gioia R, Gatot JS, Patrascu F, Carpentier I, Chapelle JP, O’Neill L, Beyaert R, Piette J, Chariot A. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem Sci. 2008;33:171–80.
Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158.
Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012;33:449–58.
Jiao S, Zhang Z, Li C, Huang M, Shi Z, Wang Y, Song X, Liu H, Li C, Chen M, et al. The kinase MST4 limits inflammatory responses through direct phosphorylation of the adaptor TRAF6. Nat Immunol. 2015;16:246–57.
Park HH, Morgan MJ, Kang HC, Kim YS. Regulation of RIP3 protein stability by PELI1-mediated proteasome-dependent degradation. BMB Rep. 2018;51:484–5.
Gu M, Liu Z, Lai R, Liu S, Lin W, Ouyang C, Ye S, Huang H, Wang X. RKIP and TBK1 form a positive feedback loop to promote type I interferon production in innate immunity. EMBO J. 2016;35:2553–65.
Rhyasen GW, Starczynowski DT. IRAK signalling in cancer. Br J Cancer. 2015;112:232–7.
Rekhter M, Staschke K, Estridge T, Rutherford P, Jackson N, Gifford-Moore D, Foxworthy P, Reidy C, Huang XD, Kalbfleisch M, et al. Genetic ablation of IRAK4 kinase activity inhibits vascular lesion formation. Biochem Biophys Res Commun. 2008;367:642–8.
Suzuki N, Saito T. IRAK-4-a shared NF-kappaB activator in innate and acquired immunity. Trends Immunol. 2006;27:566–72.
De Nardo D, Balka KR, Cardona Gloria Y, Rao VR, Latz E, Masters SL. Interleukin-1 receptor-associated kinase 4 (IRAK4) plays a dual role in myddosome formation and Toll-like receptor signaling. J Biol Chem. 2018;293:15195–207.
De S, Karim F, Kiessu E, Cushing L, Lin LL, Ghandil P, Hoarau C, Casanova JL, Puel A, Rao VR. Mechanism of dysfunction of human variants of the IRAK4 kinase and a role for its kinase activity in interleukin-1 receptor signaling. J Biol Chem. 2018;293:15208–20.
Kawagoe T, Sato S, Matsushita K, Kato H, Matsui K, Kumagai Y, Saitoh T, Kawai T, Takeuchi O, Akira S. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat Immunol. 2008;9:684–91.
Pauls E, Nanda SK, Smith H, Toth R, Arthur JSC, Cohen P. Two phases of inflammatory mediator production defined by the study of IRAK2 and IRAK1 knock-in mice. J Immunol. 2013;191:2717–30.
Balka KR, De Nardo D. Understanding early TLR signaling through the Myddosome. J Leukocyte Biol. 2019;105:339–51.
Moynagh PN. The Pellino family: IRAK E3 ligases with emerging roles in innate immune signalling. Trends Immunol. 2009;30:33–42.
Moynagh PN. The roles of Pellino E3 ubiquitin ligases in immunity. Nat Rev Immunol. 2014;14:122–31.
Smith H, Peggie M, Campbell DG, Vandermoere F, Carrick E, Cohen P. Identification of the phosphorylation sites on the E3 ubiquitin ligase Pellino that are critical for activation by IRAK1 and IRAK4. Proc Natl Acad Sci USA. 2009;106:4584–90.
Ordureau A, Smith H, Windheim M, Peggie M, Carrick E, Morrice N, Cohen P. The IRAK-catalysed activation of the E3 ligase function of Pellino isoforms induces the Lys63-linked polyubiquitination of IRAK1. Biochem J. 2008;409:43–52.
Gottipati S, Rao NL, Fung-Leung WP. IRAK1: a critical signaling mediator of innate immunity. Cell Signal. 2008;20:269–76.
Scholz R, Sidler CL, Thali RF, Winssinger N, Cheung PC, Neumann D. Autoactivation of transforming growth factor beta-activated kinase 1 is a sequential bimolecular process. J Biol Chem. 2010;285:25753–66.
Ouyang C, Nie L, Gu M, Wu A, Han X, Wang X, Shao J, Xia Z. Transforming growth factor (TGF)-beta-activated kinase 1 (TAK1) activation requires phosphorylation of serine 412 by protein kinase A catalytic subunit alpha (PKACalpha) and X-linked protein kinase (PRKX). J Biol Chem. 2014;289:24226–37.
Dai L, Aye Thu C, Liu XY, Xi J, Cheung PC. TAK1, more than just innate immunity. IUBMB Life. 2012;64:825–34.
Ferreiro DU, Komives EA. Molecular mechanisms of system control of NF-kappaB signaling by IkappaBalpha. Biochemistry. 2010;49:1560–7.
Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733.
Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–34.
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34.
Sacco F, Perfetto L, Castagnoli L, Cesareni G. The human phosphatase interactome: an intricate family portrait. FEBS Lett. 2012;586:2732–9.
Dobierzewska A, Giltiay NV, Sabapathi S, Karakashian AA, Nikolova-Karakashian MN. Protein phosphatase 2A and neutral sphingomyelinase 2 regulate IRAK-1 protein ubiquitination and degradation in response to interleukin-1beta. J Biol Chem. 2011;286:32064–73.
Kollewe C, Mackensen AC, Neumann D, Knop J, Cao P, Li S, Wesche H, Martin MU. Sequential autophosphorylation steps in the interleukin-1 receptor-associated kinase-1 regulate its availability as an adapter in interleukin-1 signaling. J Biol Chem. 2004;279:5227–36.
Yamin TT, Miller DK. The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphorylation. J Biol Chem. 1997;272:21540–7.
Dunne A, Carpenter S, Brikos C, Gray P, Strelow A, Wesche H, Morrice N, O’Neill LA. IRAK1 and IRAK4 promote phosphorylation, ubiquitination, and degradation of MyD88 adaptor-like (Mal). J Biol Chem. 2010;285:18276–82.
Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–84.
Cohen PTW. Phosphatase families dephosphorylating serine and threonine residues in proteins. In: Handbook of cell signaling (Second Edition). 2010: 659–675.
Moorhead GB, De Wever V, Templeton G, Kerk D. Evolution of protein phosphatases in plants and animals. Biochem J. 2009;417:401–9.
Heroes E, Lesage B, Gornemann J, Beullens M, Van Meervelt L, Bollen M. The PP1 binding code: a molecular-lego strategy that governs specificity. FEBS J. 2013;280:584–95.
Yao F, Long LY, Deng YZ, Feng YY, Ying GY, Bao WD, Li G, Guan DX, Zhu YQ, Li JJ, et al. RACK1 modulates NF-kappaB activation by interfering with the interaction between TRAF2 and the IKK complex. Cell Res. 2014;24:359–71.
Devin A, Lin Y, Yamaoka S, Li Z, Karin M, Liu Z. The alpha and beta subunits of IkappaB kinase (IKK) mediate TRAF2-dependent IKK recruitment to tumor necrosis factor (TNF) receptor 1 in response to TNF. Mol Cell Biol. 2001;21:3986–94.
Li HY, Liu H, Wang CH, Zhang JY, Man JH, Gao YF, Zhang PJ, Li WH, Zhao J, Pan X, et al. Deactivation of the kinase IKK by CUEDC2 through recruitment of the phosphatase PP1. Nat Immunol. 2008;9:533–41.
Gu M, Ouyang C, Lin W, Zhang T, Cao X, Xia Z, Wang X. Phosphatase holoenzyme PP1/GADD34 negatively regulates TLR response by inhibiting TAK1 serine 412 phosphorylation. J Immunol. 2014;192:2846–56.
Gallardo F, Bertran J, Lopez-Arribillaga E, Gonzalez J, Menendez S, Sanchez I, Colomo L, Iglesias M, Garrido M, Santamaria-Babi LF, et al. Novel phosphorylated TAK1 species with functional impact on NF-kappaB and beta-catenin signaling in human Cutaneous T-cell lymphoma. Leukemia. 2018;32:2211–23.
Wang L, Zhao J, Ren J, Hall KH, Moorman JP, Yao ZQ, Ning S. Protein phosphatase 1 abrogates IRF7-mediated type I IFN response in antiviral immunity. Eur J Immunol. 2016;46:2409–19.
Gu M, Zhang T. lin W, Liu Z, Lai R, Xia D, Huang H, Wang X: Protein phosphatase PP1 negatively regulates the Toll-like receptor- and RIG-I-like receptor-triggered production of type I interferon by inhibiting IRF3 phosphorylation at serines 396 and 385 in macrophage. Cell Signal. 2014;26:2930–9.
Opaluch AM, Schneider M, Chiang CY, Nguyen QT, Maestre AM, Mulder LC, Secundino I, De Jesus PD, Konig R, Simon V, et al. Positive regulation of TRAF6-dependent innate immune responses by protein phosphatase PP1-gamma. PLoS ONE. 2014;9:e89284.
Bao Z, Duan C, Gong C, Wang L, Shen C, Wang C, Cui G. Protein phosphatase 1gamma regulates the proliferation of human glioma via the NF-kappaB pathway. Oncol Rep. 2016;35:2916–26.
Wan C, Wu M, Zhang S, Chen Y, Lu C. alpha7nAChR-mediated recruitment of PP1gamma promotes TRAF6/NF-kappaB cascade to facilitate the progression of Hepatocellular Carcinoma. Mol Carcinog. 2018;57:1626–39.
Nematullah M, Hoda MN, Khan F. Protein phosphatase 2A: a double-faced phosphatase of cellular system and its role in neurodegenerative disorders. Mol Neurobiol. 2018;55:1750–61.
Clark AR, Ohlmeyer M. Protein phosphatase 2A as a therapeutic target in inflammation and neurodegeneration. Pharmacol Ther. 2019;201:181–201.
Zhou J, Pham HT, Walter G. The formation and activity of PP2A holoenzymes do not depend on the isoform of the catalytic subunit. J Biol Chem. 2003;278:8617–22.
Shanley TP, Vasi N, Denenberg A, Wong HR. The serine/threonine phosphatase, PP2A: endogenous regulator of inflammatory cell signaling. J Immunol. 2001;166:966–72.
Sun L, Stoecklin G, Van Way S, Hinkovska-Galcheva V, Guo RF, Anderson P, Shanley TP. Tristetraprolin (TTP)-14-3-3 complex formation protects TTP from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor-alpha mRNA. J Biol Chem. 2007;282:3766–77.
Li S, Wang L, Berman MA, Zhang Y, Dorf ME. RNAi screen in mouse astrocytes identifies phosphatases that regulate NF-kappaB signaling. Mol Cell. 2006;24:497–509.
Kray AE, Carter RS, Pennington KN, Gomez RJ, Sanders LE, Llanes JM, Khan WN, Ballard DW, Wadzinski BE. Positive regulation of IkappaB kinase signaling by protein serine/threonine phosphatase 2A. J Biol Chem. 2005;280:35974–82.
Yang J, Fan GH, Wadzinski BE, Sakurai H, Richmond A. Protein phosphatase 2A interacts with and directly dephosphorylates RelA. J Biol Chem. 2001;276:47828–33.
Zhao B, Sun L, Haas M, Denenberg AG, Wong HR, Shanley TP. PP2A regulates upstream members of the c-jun N-terminal kinase mitogen-activated protein kinase signaling pathway. Shock. 2008;29:181–8.
Sun L, Pham TT, Cornell TT, McDonough KL, McHugh WM, Blatt NB, Dahmer MK, Shanley TP. Myeloid-specific gene deletion of protein phosphatase 2A magnifies MyD88- and TRIF-dependent inflammation following endotoxin challenge. J Immunol. 2017;198:404–16.
Oaks J, Ogretmen B. Regulation of PP2A by sphingolipid metabolism and signaling. Front Oncol. 2014;4:388.
Li C, Wang A, Wu Y, Gulbins E, Grassme H, Zhao Z. Acid sphingomyelinase-ceramide system in bacterial infections. Cell Physiol Biochem. 2019;52:280–301.
Chalfant CE, Szulc Z, Roddy P, Bielawska A, Hannun YA. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J Lipid Res. 2004;45:496–506.
Mukhopadhyay A, Saddoughi SA, Song P, Sultan I, Ponnusamy S, Senkal CE, Snook CF, Arnold HK, Sears RC, Hannun YA, et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J. 2009;23:751–63.
He Z, Du L, Ke Y, Wen C, Zhang Y. PP2ACalpha of alveolar macrophages is a novel protective factor for LPS-induced acute respiratory distress syndrome. Inflammation. 2019;42:1004–14.
Xie L, Liu C, Wang L, Gunawardena HP, Yu Y, Du R, Taxman DJ, Dai P, Yan Z, Yu J, et al. Protein phosphatase 2A catalytic subunit alpha plays a MyD88-dependent, central role in the gene-specific regulation of endotoxin tolerance. Cell Rep. 2013;3:678–88.
Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80:1483–521.
Liu L, Zhang J, Yuan J, Dang Y, Yang C, Chen X, Xu J, Yu L. Characterization of a human regulatory subunit of protein phosphatase 3 gene (PPP3RL) expressed specifically in testis. Mol Biol Rep. 2005;32:41–5.
Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem. 1998;273:13367–70.
Gooch JL, Toro JJ, Guler RL, Barnes JL. Calcineurin A-alpha but not A-beta is required for normal kidney development and function. Am J Pathol. 2004;165:1755–65.
Bueno OF, Brandt EB, Rothenberg ME, Molkentin JD. Defective T cell development and function in calcineurin A beta -deficient mice. Proc Natl Acad Sci USA. 2002;99:9398–403.
Zhang BW, Zimmer G, Chen J, Ladd D, Li E, Alt FW, Wiederrecht G, Cryan J, O’Neill EA, Seidman CE, et al. T cell responses in calcineurin A alpha-deficient mice. J Exp Med. 1996;183:413–20.
Chan VS, Wong C, Ohashi PS. Calcineurin Aalpha plays an exclusive role in TCR signaling in mature but not in immature T cells. Eur J Immunol. 2002;32:1223–9.
Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–47.
Frischbutter S, Gabriel C, Bendfeldt H, Radbruch A, Baumgrass R. Dephosphorylation of Bcl-10 by calcineurin is essential for canonical NF-kappaB activation in Th cells. Eur J Immunol. 2011;41:2349–57.
Palkowitsch L, Marienfeld U, Brunner C, Eitelhuber A, Krappmann D, Marienfeld RB. The Ca2+-dependent phosphatase calcineurin controls the formation of the Carma1-Bcl10-Malt1 complex during T cell receptor-induced NF-kappaB activation. J Biol Chem. 2011;286:7522–34.
Conboy IM, Manoli D, Mhaiskar V, Jones PP. Calcineurin and vacuolar-type H+-ATPase modulate macrophage effector functions. Proc Natl Acad Sci USA. 1999;96:6324–9.
Kang YJ, Kusler B, Otsuka M, Hughes M, Suzuki N, Suzuki S, Yeh WC, Akira S, Han J, Jones PP. Calcineurin negatively regulates TLR-mediated activation pathways. J Immunol. 2007;179:4598–607.
Rodrigues-Diez R, Gonzalez-Guerrero C, Ocana-Salceda C, Rodrigues-Diez RR, Egido J, Ortiz A, Ruiz-Ortega M, Ramos AM. Calcineurin inhibitors cyclosporine A and tacrolimus induce vascular inflammation and endothelial activation through TLR4 signaling. Sci Rep. 2016;6:27915.
Gonzalez-Guerrero C, Ocana-Salceda C, Berzal S, Carrasco S, Fernandez-Fernandez B, Cannata-Ortiz P, Egido J, Ortiz A, Ramos AM. Calcineurin inhibitors recruit protein kinases JAK2 and JNK, TLR signaling and the UPR to activate NF-kappaB-mediated inflammatory responses in kidney tubular cells. Toxicol Appl Pharmacol. 2013;272:825–41.
Murata K, Motomura Y, Tanaka T, Kanno S, Yano T, Onimaru M, Shimoyama A, Nishio H, Sakai Y, Oh-Hora M, et al. Calcineurin inhibitors exacerbate coronary arteritis via the MyD88 signalling pathway in a murine model of Kawasaki disease. Clin Exp Immunol. 2017;190:54–67.
Zhang Y, Sun X, Muraoka K, Ikeda A, Miyamoto S, Shimizu H, Yoshioka K, Yamamoto K. Immunosuppressant FK506 activates NF-kappaB through the proteasome-mediated degradation of IkappaBalpha. Requirement for Ikappabalpha n-terminal phosphorylation but not ubiquitination sites. J Biol Chem. 1999;274:34657–62.
Pang Z, Junkins RD, Raudonis R, MacNeil AJ, McCormick C, Cheng Z, Lin TJ. Regulator of calcineurin 1 differentially regulates TLR-dependent MyD88 and TRIF signaling pathways. PLoS ONE. 2018;13:e0197491.
Ziegler S, Gartner K, Scheuermann U, Zoeller T, Hantzschmann J, Over B, Foermer S, Heeg K, Bekeredjian-Ding I. Ca(2+)-related signaling events influence TLR9-induced IL-10 secretion in human B cells. Eur J Immunol. 2014;44:1285–98.
Jennings C, Kusler B, Jones PP. Calcineurin inactivation leads to decreased responsiveness to LPS in macrophages and dendritic cells and protects against LPS-induced toxicity in vivo. Innate Immun. 2009;15:109–20.
Howell J, Sawhney R, Testro A, Skinner N, Gow P, Angus P, Ratnam D, Visvanathan K. Cyclosporine and tacrolimus have inhibitory effects on toll-like receptor signaling after liver transplantation. Liver Transpl. 2013;19:1099–107.
Brewis ND, Street AJ, Prescott AR, Cohen PT. PPX, a novel protein serine/threonine phosphatase localized to centrosomes. EMBO J. 1993;12:987–96.
Kloeker S, Wadzinski BE. Purification and identification of a novel subunit of protein serine/threonine phosphatase 4. J Biol Chem. 1999;274:5339–47.
Cohen PT, Philp A, Vazquez-Martin C. Protein phosphatase 4-from obscurity to vital functions. FEBS Lett. 2005;579:3278–86.
Hu MC, Tang-Oxley Q, Qiu WR, Wang YP, Mihindukulasuriya KA, Afshar R, Tan TH. Protein phosphatase X interacts with c-Rel and stimulates c-Rel/nuclear factor kappaB activity. J Biol Chem. 1998;273:33561–5.
Yeh PY, Yeh KH, Chuang SE, Song YC, Cheng AL. Suppression of MEK/ERK signaling pathway enhances cisplatin-induced NF-kappaB activation by protein phosphatase 4-mediated NF-kappaB p65 Thr dephosphorylation. J Biol Chem. 2004;279:26143–8.
Chen L, Dong W, Zou T, Ouyang L, He G, Liu Y, Qi Y. Protein phosphatase 4 negatively regulates LPS cascade by inhibiting ubiquitination of TRAF6. FEBS Lett. 2008;582:2843–9.
Hadweh P, Habelhah H, Kieff E, Mosialos G, Hatzivassiliou E. The PP4R1 subunit of protein phosphatase PP4 targets TRAF2 and TRAF6 to mediate inhibition of NF-kappaB activation. Cell Signal. 2014;26:2730–7.
Brechmann M, Mock T, Nickles D, Kiessling M, Weit N, Breuer R, Muller W, Wabnitz G, Frey F, Nicolay JP, et al. A PP4 holoenzyme balances physiological and oncogenic nuclear factor-kappa B signaling in T lymphocytes. Immunity. 2012;37:697–708.
Zhan Z, Cao H, Xie X, Yang L, Zhang P, Chen Y, Fan H, Liu Z, Liu X. Phosphatase PP4 negatively regulates type I IFN production and antiviral innate immunity by dephosphorylating and deactivating TBK1. J Immunol. 2015;195:3849–57.
Kwun HJ, Shuda M, Camacho CJ, Gamper AM, Thant M, Chang Y, Moore PS. Restricted protein phosphatase 2A targeting by Merkel cell polyomavirus small T antigen. J Virol. 2015;89:4191–200.
DeCaprio JA, Garcea RL. A cornucopia of human polyomaviruses. Nature Rev Microbiol. 2013;11:264–76.
Griffiths DA, Abdul-Sada H, Knight LM, Jackson BR, Richards K, Prescott EL, Peach AH, Blair GE, Macdonald A, Whitehouse A. Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J Virol. 2013;87:13853–67.
Abdul-Sada H, Muller M, Mehta R, Toth R, Arthur JSC, Whitehouse A, Macdonald A. The PP4R1 sub-unit of protein phosphatase PP4 is essential for inhibition of NF-kappaB by merkel polyomavirus small tumour antigen. Oncotarget. 2017;8:25418–32.
Brautigan DL. Protein Ser/Thr phosphatases–the ugly ducklings of cell signalling. FEBS J. 2013;280:324–45.
Ziembik MA, Bender TP, Larner JM, Brautigan DL. Functions of protein phosphatase-6 in NF-kappaB signaling and in lymphocytes. Biochem Soc Trans. 2017;45:693–701.
Kajino T, Ren H, Iemura S, Natsume T, Stefansson B, Brautigan DL, Matsumoto K, Ninomiya-Tsuji J. Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J Biol Chem. 2006;281:39891–6.
Singhirunnusorn P, Suzuki S, Kawasaki N, Saiki I, Sakurai H. Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-beta-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J Biol Chem. 2005;280:7359–68.
Lammers T, Lavi S. Role of type 2C protein phosphatases in growth regulation and in cellular stress signaling. Crit Rev Biochem Mol Biol. 2007;42:437–61.
Kamada R, Kudoh F, Ito S, Tani I, Janairo JIB, Omichinski JG, Sakaguchi K. Metal-dependent Ser/Thr protein phosphatase PPM family: evolution, structures, diseases and inhibitors. Pharmacol Ther. 2020;215:107622.
Das AK, Helps NR, Cohen PT, Barford D. Crystal structure of the protein serine/threonine phosphatase 2C at 20 A resolution. EMBO J. 1996;15:6798–809.
Zhang M, Yogesha SD, Mayfield JE, Gill GN, Zhang Y. Viewing serine/threonine protein phosphatases through the eyes of drug designers. FEBS J. 2013;280:4739–60.
Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;25:917–31.
Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24.
Kumar P, Tathe P, Chaudhary N, Maddika S. PPM1G forms a PPP-type phosphatase holoenzyme with B56delta that maintains adherens junction integrity. EMBO Rep. 2019;20:e46965.
Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh F, Tsukuda H, Taya Y, Imai K. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 2000;19:6517–26.
Ofek P, Ben-Meir D, Kariv-Inbal Z, Oren M, Lavi S. Cell cycle regulation and p53 activation by protein phosphatase 2C alpha. J Biol Chem. 2003;278:14299–305.
Tamura S, Toriumi S, Saito J, Awano K, Kudo TA, Kobayashi T. PP2C family members play key roles in regulation of cell survival and apoptosis. Cancer Sci. 2006;97:563–7.
Stern A, Privman E, Rasis M, Lavi S, Pupko T. Evolution of the metazoan protein phosphatase 2C superfamily. J Mol Evol. 2007;64:61–70.
Lu G, Wang Y. Functional diversity of mammalian type 2C protein phosphatase isoforms: new tales from an old family. Clin Exp Pharmacol Physiol. 2008;35:107–12.
Hanada M, Kobayashi T, Ohnishi M, Ikeda S, Wang H, Katsura K, Yanagawa Y, Hiraga A, Kanamaru R, Tamura S. Selective suppression of stress-activated protein kinase pathway by protein phosphatase 2C in mammalian cells. FEBS Lett. 1998;437:172–6.
Li Z, Liu G, Sun L, Teng Y, Guo X, Jia J, Sha J, Yang X, Chen D, Sun Q. PPM1A regulates antiviral signaling by antagonizing TBK1-mediated STING phosphorylation and aggregation. PLoS Pathog. 2015;11:e1004783.
Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nature Rev Mol Cell Biol. 2020;21:501–21.
Xiang W, Zhang Q, Lin X, Wu S, Zhou Y, Meng F, Fan Y, Shen T, Xiao M, Xia Z, et al. PPM1A silences cytosolic RNA sensing and antiviral defense through direct dephosphorylation of MAVS and TBK1. Sci Adv. 2016;2:e1501889.
Sun J, Schaaf K, Duverger A, Wolschendorf F, Speer A, Wagner F, Niederweis M, Kutsch O. Protein phosphatase, Mg2+/Mn2+-dependent 1A controls the innate antiviral and antibacterial response of macrophages during HIV-1 and Mycobacterium tuberculosis infection. Oncotarget. 2016;7:15394–409.
Smith SR, Schaaf K, Rajabalee N, Wagner F, Duverger A, Kutsch O, Sun J. The phosphatase PPM1A controls monocyte-to-macrophage differentiation. Sci Rep. 2018;8:902.
Sun W, Yu Y, Dotti G, Shen T, Tan X, Savoldo B, Pass AK, Chu M, Zhang D, Lu X, et al. PPM1A and PPM1B act as IKKbeta phosphatases to terminate TNFalpha-induced IKKbeta-NF-kappaB activation. Cell Signal. 2009;21:95–102.
Sasaki M, Ohnishi M, Tashiro F, Niwa H, Suzuki A, Miyazaki J, Kobayashi T, Tamura S. Disruption of the mouse protein Ser/Thr phosphatase 2Cbeta gene leads to early pre-implantation lethality. Mech Dev. 2007;124:489–99.
Prajapati S, Verma U, Yamamoto Y, Kwak YT, Gaynor RB. Protein phosphatase 2Cbeta association with the IkappaB kinase complex is involved in regulating NF-kappaB activity. J Biol Chem. 2004;279:1739–46.
Hanada M, Ninomiya-Tsuji J, Komaki K, Ohnishi M, Katsura K, Kanamaru R, Matsumoto K, Tamura S. Regulation of the TAK1 signaling pathway by protein phosphatase 2C. J Biol Chem. 2001;276:5753–9.
Zuo S, Xue Y, Tang S, Yao J, Du R, Yang P, Chen X. 14-3-3 epsilon dynamically interacts with key components of mitogen-activated protein kinase signal module for selective modulation of the TNF-alpha-induced time course-dependent NF-kappaB activity. J Proteome Res. 2010;9:3465–78.
Choi HK, Park SY, Oh HJ, Han EJ, Lee YH, Lee J, Jun WJ, Choi KC, Yoon HG. PKA negatively regulates PP2Cbeta to activate NF-kappaB-mediated inflammatory signaling. Biochem Biophys Res Commun. 2013;436:473–7.
Zhao Y, Liang L, Fan Y, Sun S, An L, Shi Z, Cheng J, Jia W, Sun W, Mori-Akiyama Y, et al. PPM1B negatively regulates antiviral response via dephosphorylating TBK1. Cell Signal. 2012;24:2197–204.
Biacchesi S, Merour E, Chevret D, Lamoureux A, Bernard J, Bremont M. NV proteins of fish novirhabdovirus recruit cellular PPM1Bb protein phosphatase and antagonize RIG-I-mediated IFN induction. Sci Rep. 2017;7:44025.
Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE, Vande Woude GF, O’Connor PM, Appella E. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci USA. 1997;94:6048–53.
Lu X, Nguyen TA, Moon SH, Darlington Y, Sommer M, Donehower LA. The type 2C phosphatase Wip1: an oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008;27:123–35.
Le Guezennec X, Bulavin DV. WIP1 phosphatase at the crossroads of cancer and aging. Trends Biochem Sci. 2010;35:109–14.
Oghabi Bakhshaiesh T, Majidzadeh AK, Esmaeili R. Wip1: a candidate phosphatase for cancer diagnosis and treatment. DNA Repair. 2017;54:63–6.
Fujimoto H, Onishi N, Kato N, Takekawa M, Xu XZ, Kosugi A, Kondo T, Imamura M, Oishi I, Yoda A, et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006;13:1170–80.
Lowe J, Cha H, Lee MO, Mazur SJ, Appella E, Fornace AJ Jr. Regulation of the Wip1 phosphatase and its effects on the stress response. Front Biosci. 2012;17:1480–98.
Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, Timofeev ON, Dudgeon C, Fornace AJ, Anderson CW, et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell. 2006;23:757–64.
Shi L, Tian Q, Feng C, Zhang P, Zhao Y. The biological function and the regulatory roles of wild-type p53-induced phosphatase 1 in immune system. Int Rev Immunol. 2020;39:280–91.
Shen XF, Zhao Y, Cao K, Guan WX, Li X, Zhang Q, Zhao Y, Ding YT, Du JF. Wip1 deficiency promotes neutrophil recruitment to the infection site and improves sepsis outcome. Front Immunol. 2017;8:1023.
Liu G, Hu X, Sun B, Yang T, Shi J, Zhang L, Zhao Y. Phosphatase Wip1 negatively regulates neutrophil development through p38 MAPK-STAT1. Blood. 2013;121:519–29.
Schito ML, Demidov ON, Saito S, Ashwell JD, Appella E. Wip1 phosphatase-deficient mice exhibit defective T cell maturation due to sustained p53 activation. J Immunol. 2006;176:4818–25.
Choi J, Nannenga B, Demidov ON, Bulavin DV, Cooney A, Brayton C, Zhang Y, Mbawuike IN, Bradley A, Appella E, et al. Mice deficient for the wild-type p53-induced phosphatase gene (Wip1) exhibit defects in reproductive organs, immune function, and cell cycle control. Mol Cell Biol. 2002;22:1094–105.
Uyanik B, Grigorash BB, Goloudina AR, Demidov ON. DNA damage-induced phosphatase Wip1 in regulation of hematopoiesis, immune system and inflammation. Cell Death Discov. 2017;3:17018.
Sun B, Hu X, Liu G, Ma B, Xu Y, Yang T, Shi J, Yang F, Li H, Zhang L, et al. Phosphatase Wip1 negatively regulates neutrophil migration and inflammation. J Immunol. 2014;192:1184–95.
Zhen H, Zhao L, Ling Z, Kuo L, Xue X, Feng J. Wip1 regulates blood-brain barrier function and neuro-inflammation induced by lipopolysaccharide via the sonic hedgehog signaling signaling pathway. Mol Immunol. 2018;93:31–7.
Lowe JM, Cha H, Yang Q, Fornace AJ Jr. Nuclear factor-kappaB (NF-kappaB) is a novel positive transcriptional regulator of the oncogenic Wip1 phosphatase. J Biol Chem. 2010;285:5249–57.
Chew J, Biswas S, Shreeram S, Humaidi M, Wong ET, Dhillion MK, Teo H, Hazra A, Fang CC, Lopez-Collazo E, et al. WIP1 phosphatase is a negative regulator of NF-kappaB signalling. Nat Cell Biol. 2009;11:659–66.
Ishida A, Kameshita I, Fujisawa H. A novel protein phosphatase that dephosphorylates and regulates Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1998;273:1904–10.
Li P, Fan JB, Gao Y, Zhang M, Zhang L, Yang N, Zhao X. miR-135b-5p inhibits LPS-induced TNFalpha production via silencing AMPK phosphatase Ppm1e. Oncotarget. 2016;7:77978–86.
Ishida A, Sueyoshi N, Kameshita I. Functions and dysfunctions of Ca(2+)/calmodulin-dependent protein kinase phosphatase (CaMKP/PPM1F) and CaMKP-N/PPM1E. Arch Biochem Biophys. 2018;640:83–92.
Susila A, Chan H, Loh AX, Phang HQ, Wong ET, Tergaonkar V, Koh CG. The POPX2 phosphatase regulates cancer cell motility and invasiveness. Cell Cycle. 2010;9:179–87.
Koh CG, Tan EJ, Manser E, Lim L. The p21-activated kinase PAK is negatively regulated by POPX1 and POPX2, a pair of serine/threonine phosphatases of the PP2C family. Curr Biol. 2002;12:317–21.
Hoon JL, Li HY, Koh CG. POPX2 phosphatase regulates cell polarity and centrosome placement. Cell Cycle. 2014;13:2459–68.
Zhang S, Guo T, Chan H, Sze SK, Koh CG. Integrative transcriptome and proteome study to identify the signaling network regulated by POPX2 phosphatase. J Proteome Res. 2013;12:2525–36.
Grimm TM, Dierdorf NI, Betz K, Paone C, Hauck CR. PPM1F controls integrin activity via a conserved phospho-switch. J Cell Biol. 2020;219:e202001057.
Weng T, Koh CG. POPX2 phosphatase regulates apoptosis through the TAK1-IKK-NF-kappaB pathway. Cell Death Dis. 2017;8:e3051.
Zhang C, Han X, Yang L, Fu J, Sun C, Huang S, Xiao W, Gao Y, Liang Q, Wang X, et al. Circular RNA circPPM1F modulates M1 macrophage activation and pancreatic islet inflammation in type 1 diabetes mellitus. Theranostics. 2020;10:10908–24.
Shinoda Y, Fujita K, Saito S, Matsui H, Kanto Y, Nagaura Y, Fukunaga K, Tamura S, Kobayashi T. Acyl-CoA binding domain containing 3 (ACBD3) recruits the protein phosphatase PPM1L to ER-Golgi membrane contact sites. FEBS Lett. 2012;586:3024–9.
Li MG, Katsura K, Nomiyama H, Komaki K, Ninomiya-Tsuji J, Matsumoto K, Kobayashi T, Tamura S. Regulation of the interleukin-1-induced signaling pathways by a novel member of the protein phosphatase 2C family (PP2Cepsilon). J Biol Chem. 2003;278:12013–21.
Wang B, Zhou Q, Bi Y, Zhou W, Zeng Q, Liu Z, Liu X, Zhan Z. Phosphatase PPM1L prevents excessive inflammatory responses and cardiac dysfunction after myocardial infarction by inhibiting IKKbeta activation. J Immunol. 2019;203:1338–47.
Henmi T, Amano K, Nagaura Y, Matsumoto K, Echigo S, Tamura S, Kobayashi T. A mechanism for the suppression of interleukin-1-induced nuclear factor kappaB activation by protein phosphatase 2Ceta-2. Biochem J. 2009;423:71–8.
Hop HT, Arayan LT, Reyes AWB, Huy TXN, Min W, Lee HJ, Son JS, Kim S. Simultaneous RNA-seq based transcriptional profiling of intracellular Brucella abortus and B. abortus-infected murine macrophages. Microb Path. 2017;113:57–67.
Kohn M. Turn and face the strange: a new view on phosphatases. ACS Cent Sci. 2020;6:467–77.
Cerny O, Kamanova J, Masin J, Bibova I, Skopova K, Sebo P. Bordetella pertussis adenylate cyclase toxin blocks induction of bactericidal nitric oxide in macrophages through cAMP-dependent activation of the SHP-1 phosphatase. J Immunol. 2015;194:4901–13.
Lu R, Pan H, Shively JE. CEACAM1 negatively regulates IL-1beta production in LPS activated neutrophils by recruiting SHP-1 to a SYK-TLR4-CEACAM1 complex. PLoS Pathog. 2012;8:e1002597.
Pereira JM, Chevalier C, Chaze T, Gianetto Q, Impens F, Matondo M, Cossart P, Hamon MA. Infection reveals a modification of SIRT2 critical for chromatin association. Cell Rep. 2018;23:1124–37.
Chatterjee J, Beullens M, Sukackaite R, Qian J, Lesage B, Hart DJ, Bollen M, Kohn M. Development of a peptide that selectively activates protein phosphatase-1 in living cells. Angew Chem Int Ed Engl. 2012;51:10054–9.
Krzyzosiak A, Sigurdardottir A, Luh L, Carrara M, Das I, Schneider K, Bertolotti A. Target-based discovery of an inhibitor of the regulatory phosphatase PPP1R15B. Cell. 2018;174:1216–28.
Schapira M, Calabrese MF, Bullock AN, Crews CM. Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov. 2019;18:949–63.
Acknowledgements
The authors thank members of the Hauck lab and members of the graduate program InViTe for stimulating discussions and suggestions.
Funding
Open Access funding enabled and organized by Projekt DEAL. CHTS acknowledges funding by the graduate program InViTe supported by the Baden-Württemberg Ministry of Science, Research and Art and CRH was supported by the German Research foundation (DFG, Grant number Ha2856/10–1). TMG was supported by a fellowship of the DFG-funded Konstanz Research School Chemical Biology (KoRS-CB).
Author information
Authors and Affiliations
Contributions
CHTS and TMG wrote the initial draft of the manuscript; CRH expanded and revised the manuscript; CHTS generated all figures. All three authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors concur with the submission and potential publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Seumen, C.H.T., Grimm, T.M. & Hauck, C.R. Protein phosphatases in TLR signaling. Cell Commun Signal 19, 45 (2021). https://doi.org/10.1186/s12964-021-00722-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-021-00722-1