
Abstract
Background
Androgen receptor (AR) and polycomb repressive complex 2 (PRC2) are known to co-occupy the loci of genes that are downregulated by androgen-stimulus. Long intergenic non-coding RNA (lincRNA) PVT1 is an overexpressed oncogene that is associated with AR in LNCaP prostate cancer cells, and with PRC2 in HeLa and many other types of cancer cells. The possible involvement of PVT1 in mediating androgen-induced gene expression downregulation in prostate cancer has not been explored.
Methods
LNCaP cell line was used. Native RNA-binding-protein immunoprecipitation with anti-AR or anti-EZH2 was followed by RT-qPCR with primers for PVT1. Knockdown of PVT1 with specific GapmeRs (or a control with scrambled GapmeR) was followed by differentially expressed genes (DEGs) determination with Agilent microarrays and with Significance Analysis of Microarrays statistical test. DEGs were tested as a tumor risk classifier with a machine learning Random Forest algorithm run with gene expression data from all TCGA-PRAD (prostate adenocarcinoma) tumors as input. ChIP-qPCR was performed for histone marks at the promoter of one DEG.
Results
We show that PVT1 knockdown in androgen-stimulated LNCaP cells caused statistically significant expression upregulation/downregulation of hundreds of genes. Interestingly, PVT1 knockdown caused upregulation of 160 genes that were repressed by androgen, including a significantly enriched set of tumor suppressor genes, and among them FAS, NOV/CCN3, BMF, HRK, IFIT2, AJUBA, DRAIC and TNFRSF21. A 121-gene-set (out of the 160) was able to correctly predict the classification of all 293 intermediate- and high-risk TCGA-PRAD tumors, with a mean ROC area under the curve AUC = 0.89 ± 0.04, pointing to the relevance of these genes in cancer aggressiveness. Native RIP-qPCR in LNCaP showed that PVT1 was associated with EZH2, a component of PRC2. PVT1 knockdown followed by ChIP-qPCR showed significant epigenetic remodeling at the enhancer and promoter regions of tumor suppressor gene NOV, one of the androgen-repressed genes that were upregulated upon PVT1 silencing.
Conclusions
Overall, we provide first evidence that PVT1 was involved in signaling a genome-wide androgen-dependent transcriptional repressive program of tumor suppressor protein-coding genes in prostate cancer cells. Identification of transcriptional inhibition of tumor suppressor genes by PVT1 highlights the pathway to the investigation of mechanisms that lie behind the oncogenic role of PVT1 in cancer.
Video Abstract
Similar content being viewed by others


Background
Prostate cancer (PCa) is the second most diagnosed type of cancer in men in the world [1]. Development of PCa is highly dependent on androgen receptor (AR) [2, 3], a transcription factor that induces expression activation or repression of prostate-specific genes through AR interaction with hundreds of coactivators or corepressors [4]. The ability of AR to repress transcription in LNCaP prostate cancer cells is related among other factors to cooperation with EZH2 histone-modifying enzyme, a component of the polycomb repressor complex 2 (PRC2) [5]. Specific AR-associated coregulators provide expression fine-tuning of hundreds of androgen-responsive target genes [6, 7], although the full complement of factors underlying expression activation or repression in the prostate have not yet been fully defined.
Long non-coding RNAs (lncRNAs) are pervasively transcribed in the human genome [8] and are recognized as an important layer of gene expression regulation [9]. Only a small number of lncRNAs have so far been functionally characterized, and their diverse roles include modification of chromatin states to increase or suppress transcriptional activation [10]. In 2004, we showed that in prostate cancer the expression of a set of antisense intronic non-coding RNAs correlates to the degree of tumor differentiation [11]. Over the past years, knowledge about lncRNAs involvement in prostate cancer has continuously progressed, and some three dozen lncRNAs have had their molecular mechanisms characterized in PCa [12, 13]. Understanding the complex network of gene regulation involving lncRNAs, and identifying their gene targets, will allow for their use in new strategies for diagnosis, prognosis and cancer therapy [14, 15].
Regarding the possible interplay between AR and lncRNAs, two studies have demonstrated AR association with HOTAIR [16], PCGEM and PRCNR1 [17] lncRNAs in PCa. Our group has recently shown that in LNCaP cells the AR is significantly associated with over six hundred other lncRNAs [18]. Interestingly, the oncogenic lncRNA PVT1 was one of the RNAs that was associated with AR, a finding that was not explored in that work [18]. PVT1 is a long intergenic non-coding RNA (lincRNA) that is overexpressed in twenty-five different tumor tissues [19] including prostate adenocarcinoma [19, 20]. Clinical studies demonstrate that increased PVT1 expression is correlated with shorter disease-free survival in prostate cancer [21] and with shorter overall survival in renal cell and colorectal carcinoma [22].
Knockdown of PVT1 in prostate cancer cells and in other types of cancer cells resulted in decreased cell viability, induction of apoptosis and reduction in tumor volume [22], however the mechanisms of action of PVT1 are largely unknown. PVT1 has recently been shown in prostate cancer cells to act as a sponge for microRNA-186, thus increasing the expression of Twist1 oncogene and promoting cell invasion and metastasis [23]. In fact, one of the mechanisms of PVT1 function in prostate tumors, as well as in many other tumors, has been to act as a miRNA sponge in the cytoplasm [22]. Nevertheless, PVT1 has also been described in prostate cancer to act in the nucleus to down-regulate miR-146a expression by inducing the methylation of CpG island in its promoter [24]. PVT1 has been described in other types of cancer to act in the nucleus; thus, in ovarian cancer PVT1 represses miR-214 expression by recruiting EZH2 to its promoter [25], and in non-small cell lung cancer (NSCLC) PVT1 inhibits the expression of Large Tumor Suppressor Kinase 2 (LATS2) by recruiting EZH2 to the LATS2 promoter [26]. In other tissues such as breast cancer [27], hepatocellular carcinoma [28], gastric cancer [29], and cholangiocarcinoma [30], PVT1 has been shown to interact with EZH2 and epigenetically inhibit the expression of some target genes. In fact, no description of PVT1 involvement with large-scale repression of gene expression has been published. PVT1 has first been detected as one of the hundreds of lincRNAs physically associated in cervical cancer (HeLa cells) with the Polycomb repressive complex 2 (PRC2) proteins SUZ12 and EZH2 [31]. In that work, knockdown of six PRC2-associated lincRNAs, including HOTAIR and TUG1, has been shown to increase the expression of hundreds of genes known to be repressed by PRC2, suggesting that these lincRNAs function as expression silencers by regulating the epigenetic landscape in the cell [31]; however PVT1 was not included as one of the six lincRNAs that were functionally characterized [31]. Here, we show that in LNCaP cells PVT1 was associated with PRC2, besides being associated with AR. Also, using PVT1 knockdown we show for the first time that in LNCaP cells PVT1 was involved with the genome-wide regulation of gene expression. Interestingly, a set of androgen repressed genes increased their expression levels after PVT1 knockdown, and tumor suppressor genes were enriched among them. We show that besides the known role of PVT1 on regulating miRNA levels, this lincRNA acted to repress the transcription of hundreds of mRNAs in prostate cancer cells.
Materials and methods
Cell line and standard culture medium
Androgen-dependent LNCaP human prostate carcinoma cells (American Type Culture Collection, USA) were hormone-starved for 48 h in RPMI medium (Gibco, USA) supplemented with charcoal-stripped fetal bovine serum (CSS, Sigma-Aldrich, USA), and subsequently treated as described in the sections below. See Additional file 1: Methods for additional procedures and details.
Native RNA-binding protein immunoprecipitation (RIP)
For native RIP [32] with LNCaP cells, CSS-supplemented medium was renewed, 10 nM synthetic androgen analog R1881 (Methyltrienolone, Sigma-Aldrich, USA) or vehicle (ethanol) were added, cells were incubated for an additional 24 h, and processed as described in the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, USA). See Additional file 1: Methods for additional procedures and details.
Cell fractionation
For LNCaP cells fractionation, CSS-supplemented medium was renewed, 10 nM R1881 (or vehicle) was added, cells were incubated for an additional 24 h, and processed for subcellular fractionation by differential centrifugation [33]. See Additional file 1: Methods for additional procedures and details.
PVT1 knockdown
Androgen-starved cells were treated with lipofectamine 3000 complexed with a pool of PVT1_2 and PVT1_5 antisense LNA GapmeRs (150 pmol each, Exiqon-Qiagen, USA) designed against PVT1 (Additional file 2: Table S1), or with a negative control scrambled GapmeR (300 pmol). For this, CSS-supplemented medium was renewed (5 mL medium), 30 μL lipofectamine 3000 complexed with 300 pmol LNA GapmeRs was added, and cells were incubated for 24 h. Subsequently, 1.0 nM R1881 or vehicle (ethanol) was added without changing the culture medium and cells were incubated for an additional 24 h. Cells were harvested with TRizol; total RNA was purified using RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions.
Dose-dependent effect of hormone on gene expression levels
CSS-supplemented medium was renewed and either 0.1, 1 and 10 nM R1881 or vehicle (ethanol) were added. After 24 h incubation, cells were harvested with TRizol; RNA was purified using RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions.
Chromatin immunoprecipitation (ChIP)
PVT1 was silenced by knockdown as described above, except that 900 pmol GapmeR was used (a pool of PVT1_2 and PVT1_5 (450 pmol each), or scrambled GapmeR at 900 pmol). After 24 h incubation, 10 nM R1881 was added, cells were incubated for additional 24 h, and processed with the Magna ChIP A/G kit (17-10085, Millipore) according to the manufacturer’s instructions. See Additional file 1: Methods for additional procedures and details.
Reverse transcription and cDNA synthesis
In the RIP assay, 10 μL RNA sample from the immunoprecipitate were used for reverse transcription (in the expression assays, 1 μg total RNA was used instead) with SuperScript IV First Strand Synthesis System (Invitrogen, USA) and oligo-dT-(20) or random hexamer primers, according to the manufacturer's recommendations.
Real-time quantitative PCR
qPCR was performed with specific primer pairs (Additional file 2: Table S1) and cDNA from the RIP or expression assays or DNA from the ChIP assay. See Additional file 1: Methods for additional procedures and details.
Genome-wide gene expression analysis in LNCaP cells under PVT1 silencing
Agilent SurePrint G3 Human Gene Expression v3 (8 × 60 k, G4851C) microarrays were used. Total RNA (200 ng) was obtained as described above in item PVT1 knockdown, and RNA samples with a minimum RNA Integrity Number (RIN) > 8 were used. See Additional file 1: Methods for additional procedures and details.
Statistical analyses
Significance Analysis of Microarrays (SAM) statistical test [34] was used with cutoff q-value ≤ 0.01 (i.e., a False Discovery Rate ≤ 1%) [35]. Genes with q-value ≤ 0.01 and |log2fold-change|> 1 were considered significantly differentially expressed. Gene Ontology analyses were performed with DAVID [36].
Gene expression correlations in the TCGA dataset
The TCGA prostate adenocarcinoma (TCGA-PRAD) dataset was used and PVT1-related disease-free survival analysis was done with TANRIC tool [37]. To test the 121-gene-set as a tumor risk classifier, we implemented a machine learning Random Forest algorithm in python Scikit-Learn (v.0.20.2) [38] with gene expression data from all TCGA-PRAD tumors as input. See Additional file 1: Methods for additional procedures and details.
Results
PVT1 associated with AR and with PRC2 in LNCaP cells
Human PVT1 lincRNA (Accession NR_003367.3) is transcribed from chromosome 8 (q24.21) within a 400 kb-long locus (Fig. 1a), in a so-called “gene-desert” region where no protein-coding genes are transcribed [39].

LincRNA PVT1 associated both with AR and EZH2 in hormone-starved or in androgen-stimulated LNCaP cells. a Snapshot of the PVT1 genomic locus on human Chromosome 8 showing the PVT1 lincRNA, along with the pair of PCR primers (blue arrowheads) that was used for its quantification. The two lower insets show the locations of two antisense LNA GapmeR oligos (red and green blocks) that were used for PVT1 knockdown in the experiments of Fig. 2. b–e RIP with anti-AR or nonspecific antibody (IgG), followed by RT-qPCR for genes PVT1 (b, target), PCGEM (c, positive control), DRAIC (d), and U1-19 (e, negative control). f–i, RIP with anti-EZH2 or nonspecific antibody (IgG), followed by RT-qPCR for PVT1 (f, target), for two different isoforms of SFPQ (g, h, positive controls) and U1-19 (i, negative control). j–m RIP with anti-SUZ12 or nonspecific antibody (IgG), followed by RT-qPCR for PVT1 (j, target), for two different isoforms of SFPQ (k, l, positive controls) and U1-19 (m, negative control). LNCaP cells under hormone-starved (-R1881, blue) or androgen-stimulated conditions (+ R1881, red) were tested. Data shown are mean ± s.e.m. of four biological replicates. T-test, *P < 0.05, **P < 0.01, ***P < 0.005
We performed native RIP-qPCR in order to test if PVT1 associated with AR and with PRC2 in LNCaP cells. PVT1 was detected as significantly enriched in the anti-AR fraction relative to the non-specific IgG control in hormone-starved or androgen-stimulated cells (Fig. 1b, blue and red bars), similar to the positive control lncRNA PCGEM [17] (Fig. 1c, blue and red bars). Also interesting, prostate tumor suppressor lincRNA DRAIC [40], previously detected by us with RIP-seq as one of the other RNAs associated with AR [18], was confirmed here by RIP-qPCR (Fig. 1d, blue and red bars), whereas negative control snRNA U1-19 was not associated with AR (Fig. 1e).
Next, we found PVT1 as significantly enriched in the anti-EZH2 fraction relative to the non-specific IgG control, and again, the association with EZH2 occurred under hormone-starved and androgen-stimulated conditions (Fig. 1f, blue and red bars). Positive control lncRNA SFPQ isoforms 1 and 2 [31] was significantly associated with EZH2 (Fig. 1g, h), whereas negative control snRNA U1-19 was not (Fig. 1i).
RIP-qPCR was repeated against SUZ12, another component of PRC2, and PVT1 showed a significant enrichment in the anti-SUZ12 fraction relative to the non-specific IgG control under hormone-starved conditions (Fig. 1j, blue bar). Under androgen stimulation (Fig. 1j, red bars), PVT1 enrichment was non-significant. Similarly, positive control lncRNA SFPQ isoforms 1 and 2 was significantly associated with SUZ12 only under hormone-starved conditions (Fig. 1k, l, blue bars), and not under androgen stimulation (Fig. 1k, l, red bars). Negative control snRNA U1-19 was not associated with SUZ12 (Fig. 1m).
Association of PVT1 with the two histone modifying enzymes raised the question about the subcellular localization of this lincRNA. We cultured LNCaP cells in the absence or presence of androgen, submitted them to subcellular fractionation, and we determined that PVT1 was 98.5–98.8 localized in the nuclear fraction, and 1.2–1.5% in the cytoplasm (Additional file 2: Fig. S1).
PVT1 knockdown affected the expression of hundreds of genes in LNCaP cells
Given the finding that PVT1 was associated with both AR and PRC2 in LNCaP, we verified if PVT1 might be related to a genome-wide repression of gene expression in these cells under androgen stimulation. Initially, LNCaP cells were treated with androgen to establish the androgen-responsive genes. Expression-microarrays analyses identified 1155 genes significantly downregulated and 883 genes upregulated in androgen-stimulated compared with hormone-starved cells (Fig. 2a, Additional file 2: Table S2).

Androgen repressed the transcription of hundreds of genes and PVT1 knockdown partially reversed this repression in LNCaP cells. LNCaP cells in culture were deprived of androgen (-R1881) or stimulated with 1 nM androgen (+ R1881), RNA was extracted from four biological replicates, purified and used. a Detection with microarrays of genome-wide statistically significant gene expression changes (one replicate in each column, one gene in each line; z-core color scale on the left) (q-value < 0.01, fold-change < 0.5 or fold-change > 2). b Validation with RT-qPCR of changes in expression of known androgen-responsive genes. c PVT1 knockdown with a pool of two antisense LNA GapmeRs (PVT1 KD) was efficient both in the presence (red) or in the absence (blue) of androgen, compared with a scrambled GapmeR (CTRL KD). d RNA was extracted from LNCaP cells after PVT1 knockdown or CTRL knockdown and used for detection with microarrays of genome-wide statistically significant gene expression changes induced by PVT1 knockdown (q-value < 0.01, fold-change < 0.5 or fold-change > 2). e Venn diagram shows that 160 genes were both downregulated by androgen and upregulated after PVT1 knockdown under androgen stimulation. T-test, *P < 0.05, **P < 0.01, ****P < 0.001; mean ± s.e.m., four biological replicates
Control validation by RT-qPCR (Fig. 2b) showed that known androgen-induced genes FASN, NDRG1, PSA and TMPRSS2 [5, 41] had their expression significantly upregulated by androgen (Fig. 2b, upper panel). Similarly, DRAIC, OPRK1 and SI, known to have their expression repressed under androgen stimulation [5, 40, 41], were significantly downregulated by androgen (Fig. 2b, lower panel). Importantly, PVT1 expression was not affected by androgen stimulation (Fig. 2b, lower panel).
Next, PVT1 knockdown was induced in LNCaP cells with a pool of two antisense LNA GapmeRs, PVT1_2 (Fig. 1a, red block) and PVT1_5 (Fig. 1a, green block). This pool was able to effectively reduce PVT1 expression to 30% or 10% of its endogenous level in hormone-starved (Fig. 2c, blue bars) or androgen-stimulated cells (Fig. 2c, red bars), respectively.
Finally, genome-wide expression-microarrays analyses showed that PVT1 knockdown in androgen-stimulated LNCaP cells caused a significant upregulation of 654 genes and downregulation of 510 genes compared with their expression in control androgen-stimulated cells treated with a scrambled GapmeR (Fig. 2d, Additional file 2: Table S3).
A Gene Ontology (GO) analysis of the 654 upregulated genes showed a significant enrichment of “Regulation of RNA biosynthetic process” and “Regulation of gene expression” categories (Additional file 2: Fig. S2a). Noteworthy, GO categories related to tumor suppressor processes such as “Apoptotic process” and “Programmed cell death” were significantly enriched among the upregulated genes (Additional file 2: Fig. S2a), along with GO categories involved with “Negative regulation of cell proliferation” and “Negative regulation of developmental process” (Additional file 2: Fig. S2a).
Conversely, GO enrichment analysis of the 510 genes downregulated upon PVT1 knockdown showed that “Protein kinase activity” along with “Regulation of GTPase activity” were the two most significantly enriched categories (Additional file 2: Fig. S2b), suggesting that PVT1 in LNCaP cells might cause upregulation of genes involved in signaling pathways.
In hormone-starved cells, PVT1 knockdown also caused a significant expression change of hundreds of genes (396 upregulated and 436 downregulated), compared with their expression in control hormone-starved cells treated with a scrambled GapmeR (Additional file 2: Fig. S3a, Table S4). “Cell proliferation” was one of the top most significantly enriched GO categories among the 396 upregulated genes (Additional file 2: Fig. S3b), also including “Positive regulation of cell cycle arrest”; this again suggests that endogenous PVT1 in LNCaP cancer cells might repress the expression of genes related to cell cycle arrest and favor cell proliferation, even under hormone starvation. Among the 436 downregulated genes (Additional file 2: Fig. S3c), “Protein kinase” was one of the most significantly enriched GOs, again showing that endogenous amounts of PVT1 in hormone-starved LNCaP cells might upregulate genes involved in signaling pathways.
Since PVT1 associated with PRC2, we focused on the androgen-repressed genes and asked if PVT1 knockdown would be able to increase the expression of a subset of those genes.
PVT1 knockdown restored the expression of a set of genes that were repressed by androgen in LNCaP cells
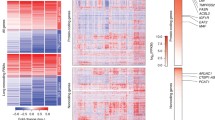
To answer this question, we looked for overlap between androgen-responsive downregulated genes and genes whose expression was upregulated by PVT1 knockdown under androgen stimulation. We identified 160 genes within the intersection between the two datasets (Fig. 2e, Additional file 2: Table S5). An expression heatmap shows that these 160 genes were all downregulated by androgen with endogenous levels of PVT1 (Fig. 3a) and upon PVT1 knockdown they were upregulated (Fig. 3a). It indicates that PVT1 acted as a transcriptional repressor of these target genes, possibly involving AR and PRC2.

Genes de-repressed by PVT1 knockdown in androgen-stimulated LNCaP cells were enriched in tumor suppressor functions. a Gene expression heatmap of the 160 genes (one in each line) that were significantly downregulated by androgen with endogenous PVT1 (CTRL KD, + R1881) and were de-repressed (upregulated) by PVT1 knockdown in androgen-stimulated cells (PVT1 KD, + R1881) (q-value < 0.01, fold-change < 0.5 or fold-change > 2). For each condition indicated at the top, four biological replicates are shown (one in each column); z-score color scale is shown at left. b Gene Ontology terms significantly enriched (Benjamini–Hochberg corrected P < 0.05) with the 160 genes that were de-repressed by PVT1 knockdown in androgen-stimulated cells. c Androgen dose-dependence of FAS gene expression. d Validation by RT-qPCR of the de-repression of FAS expression upon PVT1 knockdown in androgen-starved (blue) or androgen-treated cells (red). e Androgen dose-dependence of NOV gene expression. f Validation by RT-qPCR of the de-repression of NOV expression upon PVT1 knockdown in androgen-starved (blue) or androgen-treated cells (red). T-test, *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001; mean ± s.e.m., three biological replicates
Genes repressed by PVT1 in LNCaP cells were related to tumor suppressor functions
GO enrichment analysis of the 160 genes whose expression was de-repressed by PVT1 knockdown showed that a number of categories related to tumor suppressor functions were significantly enriched (Fig. 3b). The most significantly enriched GO was “Cell death”; also noteworthy were “Regulation of cell death”, “Single organism cell adhesion” and “Apoptotic process”.
Next, we validated by RT-qPCR the expression changes caused by PVT1 knockdown in eight selected genes that are involved with either apoptosis or cell adhesion, out of the 160 genes repressed by androgen and de-repressed by PVT1 knockdown. In parallel, to demonstrated that these eight genes could have a physiological relevance in the context of the androgen regulatory program, we stimulated LNCaP cells with different concentrations of synthetic androgen (0.1, 1 and 10 nM R1881) for 24 h (in the presence of endogenous levels of PVT1) and observed a dose-dependent target repression for all eight tested genes (Fig. 3c, e; Additional file 2: Fig. S4a to f).
FAS expression was repressed by androgen in the presence of endogenous PVT1 (Fig. 3c) and after PVT1 knockdown (Fig. 3d) its expression was restored both under hormone-starved (blue bars) and androgen-stimulated conditions (red bars). FAS tumor suppressor gene encodes a receptor protein that initiates the caspases cascade of cell death signals [42].
NOV expression was markedly repressed by androgen in LNCaP with endogenous PVT1 (Fig. 3e). PVT1 knockdown caused a two- to threefold increase in NOV expression (Fig. 3f), both in hormone-starved (blue bars) and in androgen-stimulated cells (red bars). NOV (nephroblastoma overexpressed, or NOV/CCN3) is a tumor suppressor gene encoding an extracellular-matrix protein that increases cell adhesion [43, 44].
The other six tumor suppressor genes tested by RT-qPCR were all de-repressed by PVT1 knockdown (Additional file 2: Fig. S4a to f), namely AJUBA (or JUB), BMF, DRAIC, IFIT2, TNFRSF21 and HRK, which indicates that PVT1 acted as a mediator of transcriptional repression of a number of androgen-sensitive genes in LNCaP cells. Importantly, TP53 tumor suppressor gene [45] was also found among the 654 genes that were upregulated upon PVT1 knockdown in androgen-treated LNCaP (Additional file 2: Table S3), although TP53 expression was not repressed by androgen.
Finally, in order to check if all of the above effects took place under unchanged AR, EZH2 and SUZ12 expression levels, i.e. to check if these genes were not themselves PVT1 targets, we measured the mRNA levels of these genes in LNCaP cells after PVT1 silencing (Additional file 2: Fig. S5a to d). Only AR isoform 2 showed a significant 30% expression reduction after PVT1 knockdown under androgen stimulation (Additional file 2: Fig. S5b), and the other genes tested here by RT-qPCR were not affected. Therefore, upregulation of the above 160 genes upon PVT1 knockdown was not related to an increase in AR or to a decrease in PRC2 components EZH2 and SUZ12. Most likely, it was related to a change in occupancy of these epigenetic modifiers at target gene loci upon PVT1 knockdown.
The genes de-repressed by PVT1 knockdown in LNCaP cells had their expression inversely correlated to patients’ prostate tumor risk classification
To evaluate the relevance of PVT1 transcriptional repression in prostate cancer we analyzed RNA-seq data from 497 prostate adenocarcinoma samples (TCGA-PRAD). First, we determined that high PVT1 levels were a statistically significant predictor of shorter disease-free survival when comparing patient tumors with high (33% highest) and low (33% lowest) PVT1 expression levels (Fig. 4a).

Genes de-repressed by PVT1 knockdown in LNCaP cells had their expression in the TCGA prostate adenocarcinoma dataset correlated to tumor risk. a Kaplan–Meier disease-free survival analysis for TCGA-PRAD patients based on the PVT1 expression level in the tumors of the first 3-quantile low-PVT1 samples (blue, n = 163) and third 3-quantile high-PVT1 samples (red, n = 167). b Cumulative density distribution of Spearman rho correlation between the expression levels of PVT1 and each of the 121 genes upregulated after PVT1 depletion in LNCaP cells. Correlation was calculated for each gene in the 121-gene-set across all TCGA-PRAD tumors from patients separated into three groups comprised of samples with low-, medium- or high-PVT1 expression (orange, blue and green lines, respectively). The Kolmogorov–Smirnov test (two-tail ks-test) was applied to calculate the significance of the difference between the groups’ distribution (orange***, P value < 4.5 × 10–6; blue***, P value < 9.0 × 10–4). c PVT1 expression level in TCGA-PRAD tumor samples with high (n = 174) and intermediate risk (n = 119). Significance was calculated using a two-tail t-test, **P < 0.01. d Classification of intermediate- and high-risk TCGA-PRAD tumors using a Random Forest machine-learning model. The ROC curves show the area under the curve (AUC) for the Random Forest model classification performance in a 5-way cross-validation (crossVal) scheme. Expression of the 121-gene-set across all intermediate- and high-risk TCGA-PRAD tumors were used as the input features for the machine-learning model. The 5-way cross-validation mean is indicated by the blue line, and the gray shade represents the validation standard deviation. e Rank of the top 10 most predictive genes in the Random Forest tumor classification analysis. Each blue point and error bars represent the mean ± S.D. of the Gini importance score for that gene, obtained in the 5-way cross-validation scheme
Next, in the TCGA-PRAD dataset we identified 121 annotated genes (Additional file 2: Table S5) out of the 160 genes de-repressed by PVT1 knockdown in LNCaP, and we retrieved their expression levels in all patient tumors. Patient tumors were clustered into three groups with low, medium and high levels of PVT1 (33% quantiles), and the mean PVT1 expression was significantly different among the groups (Additional file 2: Fig. S6a). We calculated the correlation between the expression of PVT1 and the expression of each gene, across all patient tumors in each of the three groups. Expression of the 121-gene set was significantly more negatively correlated with PVT1 in the high-PVT1 tumors (Fig. 4b, green) compared with medium- and low-PVT1 tumors (Kolmogorov–Smirnov test).
We identified in the TCGA-PRAD cohort the intermediate-risk tumors (n = 119; see Methods and Additional file 1: Methods) and the high-risk tumors (n = 174; see Methods and Additional file 1: Methods), and found that the expression of PVT1 in the high-risk group was significantly (t-test p = 0.01) higher than in the intermediate-risk group (Fig. 4c).
In order to test the 121-gene-set as a tumor risk classifier, we trained a machine learning algorithm using the 121-genes expression data from 80% of all 293 intermediate- and high-risk TCGA-PRAD tumor samples. Next, we classified the tumors of the remaining 20% patients in this independent validation group, repeating the procedure in a 5-way cross-validation test, each with different patients in the 20% validation group (and therefore, each with a different training set corresponding to the 80% remaining samples). The 121-gene-set was able to correctly predict all 293 intermediate- and high-risk TCGA-PRAD tumors, with a mean ROC area under the curve AUC = 0.89 ± 0.04 (0.82–0.94) (Fig. 4d). The top 10 most important classifier genes (highest Gini scores) are shown in Fig. 4e; importance of all genes for the machine-learning classification is in Additional file 2: Fig. S6b.
Knockdown of PVT1 caused an epigenetic remodeling in the NOV gene enhancer and promoter regions
We have chosen the NOV gene locus for mapping the possible changes in epigenetic marks occurring upon PVT1 knockdown because NOV is one of the most strongly repressed genes by AR and EZH2 in LNCaP cells [46] and upon AR occupancy of the enhancer at 63-kb upstream of the transcriptional start site (TSS) of NOV, a DNA loop occurs, bringing the enhancer closer to the promoter region of the NOV gene [46]. Then, AR recruits the EZH2 that catalyzes the trimethylation of histone H3 in lysine 27 around the NOV promoter, leading to inhibition of expression via epigenetic silencing [46]. Our hypothesis was that if PVT1 lincRNA interacted with AR and EZH2, knockdown of PVT1 would change the epigenetic landscape in the NOV gene locus. In parallel, as a non-related gene whose expression was not affected by PVT1 knockdown, we looked at the epigenetic marks in the PSA gene locus. After PVT1 knockdown there was a significant increase in H3K27me3 occupancy at the NOV enhancer (Fig. 5a) and a significant increase in H3K27ac occupancy at the NOV promoter (Fig. 5c). No significant change in occupancy of these histone marks was observed in the negative control gene PSA (Fig. 5b, d). The histone mark H3K27ac is associated with increased activation of gene transcription, primarily localizing around the TSS of human genes [47]. The increase in H3K27ac mark occupancy is in line with the activation of NOV transcription when PVT1 was silenced (Fig. 3f).

Epigenetic marks at the enhancer and promoter genomic regions of the NOV gene were remodeled upon PVT1 knockdown. LNCaP cells in culture with androgen were treated with a pool of two antisense LNA GapmeRs targeting PVT1 (PVT1 KD) or with a scrambled GapmeR (CTRL KD), cells were then lysed and submitted to the Chromatin Immunoprecipitation (ChIP) protocol. a, b ChIP with anti-H3K27me3 Ab; or c, d with anti-H3K27ac Ab. The co-precipitated DNA was subjected to qPCR with primers for the enhancer (left two bars) and promoter (right two bars) genomic regions of the gene indicated at the bottom of each panel, either the NOV or the PSA gene. T-test, *P < 0.05, mean ± s.e.m. of three biological replicates
Discussion
This report is the first direct investigation of the genome-wide gene expression regulation effected by PVT1 in prostate cancer cells. First, we identified that PVT1 was associated in LNCaP cells both with AR and EZH2, a component of the polycomb complex, suggesting that PVT1 could be potentially important for the androgen-induced repression program. In fact, PVT1 could act as a scaffold for protein complexes, possibly acting in a similar way as HOTAIR lincRNA [48], which is important for driving two repressive complexes, histone methyltransferase PRC2 and histone demethylase LSD1 to their specific targets [48]. Next, we showed that PVT1 knockdown in LNCaP cells affected the expression of over one thousand genes, indicating that a complex change in the gene expression program occurred upon PVT1 silencing. In fact, the majority of the affected genes (56%) were upregulated upon PVT1 silencing, indicating that PVT1 predominantly acted as a transcriptional repressor. PVT1 could possibly be one of the mediators of the previously reported role of AR as a global transcriptional repressor in LNCaP cells [5], where AR and EZH2 co-occupy the regulatory and promoter regions of androgen-repressed genes [5].
The smaller set of downregulated genes upon PVT1 silencing (44% of the affected genes) may have their expression modulated through the PVT1-miRNA-mRNA axis, whereby the well documented miRNAs sponging role of PVT1 would control the levels of critical miRNAs that target specific mRNAs in the cell [22]. The novel genome-wide transcriptional repressive role of PVT1 lincRNA on protein-coding genes, which was uncovered by our experimental results, points to an additional mechanism of action whose future detailed characterization is warranted.
Most interestingly, among the genes upregulated by PVT1 knockdown, a number of enriched GO categories were related to apoptosis and programmed cell death, either looking at the entire set of 654 upregulated genes or at the subset of 160 genes that had been repressed by androgen-stimulation and were de-repressed by PVT1 silencing. “Cell adhesion” was another relevant enriched GO, among the genes upregulated by PVT1 silencing. This indicates that the endogenous level of PVT1 in LNCaP cells resulted in the repression of tumor suppressor genes involved with apoptosis and cell adhesion, which is in line with the known oncogenic role of PVT1 [19]. Overall, these results suggest that increased levels of endogenous PVT1, known to occur in prostate cancer [19, 20], could lead to downregulation of tumor suppressor genes and favor cell proliferation.
Knockdown of PVT1 in prostate cancer cell lines has been shown to increase the abundance of activated cleaved caspase-9 and -3 proteins and to induce cell apoptosis [21]. Here, we identified with microarrays and confirmed by RT-qPCR that PVT1 knockdown increased the expression of FAS, which is the pro-apoptotic gene encoding the membrane receptor that triggers the caspases activation cascade [42]. Low FAS expression has been described in LNCaP and PC3 cells [49, 50], however correlation between FAS and PVT1 expression was not known until now.
We also showed here that PVT1 knockdown did upregulate the expression of NOV/CCN3, the tumor suppressor gene encoding a protein that induces apoptosis in fibroblasts [51]. The NOV/CCN3 gene regulates cell adhesion, migration, proliferation, differentiation and survival [52, 53]. NOV/CCN3 decreased expression was already described in various tumor types and cancer cell lines including prostate cancer LNCaP cells [43, 44, 46, 54]. We observed that after PVT1 knockdown there was a significant increase in H3K27me3 occupancy at the NOV/CCN3 enhancer and a significant increase in H3K27ac occupancy at the NOV/CCN3 promoter. Using NOV/CCN3 as a model gene, Wu et al. [46] demonstrated that in LNCaP and VCaP cells, AR directly binds the enhancers of target genes and controls the promoter through DNA looping with enhancer. In addition, they showed that, while the enhancer elements are important in recruiting AR, the promoter dictates gene repression through repressive chromatin remodeling that is mediated by the histone H3 lysine 27 (H3K27) methyl-transferase EZH2 [46]. The increase in activating H3K27ac mark occupancy at the NOV/CCN3 promoter that we observed here upon PVT1 reduction, in parallel with an increased H3K27me3 occupancy at the NOV/CCN3 enhancer, suggest that the lincRNA could drive the DNA looping, and that in its absence the promoter became free from the EZH2 deposition of methylation marks at the promoter region. In this context, it is interesting to note that upon PVT1 decrease, the NOV/CCN3 locus with increased H3K27me3 at the enhancer has assumed a poised enhancer configuration [55, 56]. It is tempting to speculate that a wider epigenetic reprograming has taken place, which could as well include DNA methylation that is known to reversibly regulate H3K27ac and H3K27me3 at tissue-specific enhancers [57]. Identification of additional genome-wide changes in epigenetic marks induced by PVT1 knockdown will be an interesting line for future investigation.
While NOV/CCN3 is known as a matricellular, secreted protein, a cytoplasmic form of the NOV/CCN3 protein has been shown in prostate cancer LNCaP cells to interact with the N-terminal domain of the AR protein to inhibit nuclear translocation of AR, thereby acting in a negative feedback loop to block AR function [58]. In LNCaP cells, NOV/CCN3 physically interacts with the AR protein to retain it in the cytoplasm, thus reducing AR nuclear translocation, and NOV/CCN3 overexpression strongly suppresses LNCaP growth program [58]. Of clinical relevance, NOV/CCN3 depletion is a main driver of AR signaling and androgen-independent Castration Resistant Prostate Cancer (CRPC) progression and drug resistance; a significant decrease of NOV/CCN3 expression is observed in CRPC compared with localized prostate cancer in various prostate cancer patient cohorts [58]. We suggest that increased levels of PVT1 in advanced prostate cancer tumors could modulate an enhanced epigenetic inhibitory effect of AR on NOV/CCN3 expression, through the AR-EZH2-PVT1 axis.
Other pro-apoptotic genes that were found upregulated upon PVT1 knockdown were BMF, which encodes a pro-apoptotic BCL2-family protein [59], IFIT2, which triggers apoptosis through a BCL2-dependent mitochondrial pathway [60], TNFRSF21, an apoptosis activator gene in bladder cancer [61], and HRK, which encodes a pro-apoptotic protein that interacts with BCL2 and Bcl-XL apoptosis-repressor proteins [62]. PVT1 knockdown also upregulated the expression of AJUBA (or JUB), which encodes a cell–cell junction protein [63] whose low expression is observed in metastatic prostate cancer [64], of DRAIC lncRNA, a tumor suppressor gene that represses migration and cell invasion in LNCaP cells [40], and of tumor suppressor TP53, a master regulator of stress-induced FAS-dependent apoptosis [65]. Overall, our data indicate that in LNCaP cells PVT1 caused transcriptional repression of an important set of tumor suppressor genes, and suggest that the increased expression of PVT1 in advanced prostate cancer types [19, 20] may contribute to the aggressive phenotype by reducing apoptosis and cell–cell adhesion.
Our data show correlation of high-PVT1-expression tumors with shorter disease-free patient survival in the 497-patients TCGA-PRAD cohort, confirming the results of a previous publication [21] with a 152-Chinese-patients cohort. Also, we detected an inverse correlation in the TCGA-PRAD tumors between the expression of PVT1 and the expression of the 121-gene-set (out of the 160 genes that were repressed by androgen and de-repressed by PVT1 knockdown in LNCaP cells), which points to the relevance of further characterizing the interaction between PVT1 and tumor suppressor genes in determining an aggressive phenotype in prostate cancer. In fact, the ability of the 121-gene-set expression profile to correctly predict tumor risk in the 293-patients intermediate- and high-risk TCGA-PRAD cohort (AUC = 0.89 ± 0.04), indicates that the role of this set of genes in prostate cancer progression should be the subject of further investigation.
Conclusions
In prostate cancer cells, we show a novel genome-wide androgen-related role of PVT1 lincRNA in signaling the transcriptional repression of protein-coding genes, and we show that the PVT1-targeted repressed gene set was enriched in tumor suppressor functions. This may lie behind the known aggressive phenotype of tumors expressing high levels of PVT1 oncogene, which highlights the pathway towards further investigation of the mechanisms that link PVT1 and its tumor suppressor targeted genes. We also show that the expression profile of a repressed 121-gene-set can be used with high confidence as a predictor of prostate-cancer patients’ high-risk tumors.
Availability of data and materials
The microarray gene expression datasets generated for this study can be found in the GEO DataSets (https://www.ncbi.nlm.nih.gov/gds/) under accession number GSE133372.
Abbreviations
- AR:
-
Androgen receptor
- AUC:
-
Area under the curve
- ChIP-qPCR:
-
Chromatin immunoprecipitation followed by quantitative PCR
- crossVal:
-
Cross-validation test
- CSS:
-
Charcoal-stripped fetal bovine serum
- DEG:
-
Differentially expressed gene
- FDR:
-
False Discovery Rate
- GapmeR:
-
16-Nucleotides-long oligonucleotide enriched with LNA in the flanking regions and DNA in an LNA-free central gap
- GO:
-
Gene Ontology
- KD:
-
Knockdown
- lincRNA:
-
Long intergenic non-coding RNA
- LNA:
-
Locked nucleic acid
- lncRNA:
-
Long non-coding RNA
- PCa:
-
Prostate cancer
- PRC2:
-
Polycomb repressive complex 2
- qPCR:
-
Quantitative PCR
- RIN:
-
RNA Integrity Number
- RIP-qPCR:
-
RNA-binding-protein immunoprecipitation followed by quantitative PCR
- ROC:
-
Receiver operating characteristic
- RT-qPCR:
-
Reverse transcription followed by quantitative PCR
- SAM:
-
Significance Analysis of Microarrays
- TCGA-PRAD:
-
The Cancer Genome Atlas-Prostate adenocarcinoma
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Debes JD, Tindall DJ. The role of androgens and the androgen receptor in prostate cancer. Cancer Lett. 2002;187:1–7.
Zhou Y, Bolton EC, Jones JO. Androgens and androgen receptor signaling in prostate tumorigenesis. J Mol Endocrinol. 2015;54:R15-29.
Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28:778–808.
Zhao JC, Yu J, Runkle C, Wu L, Hu M, Wu D, Liu JS, Wang Q, Qin ZS, Yu J. Cooperation between Polycomb and androgen receptor during oncogenic transformation. Genome Res. 2012;22:322–31.
McNair C, Urbanucci A, Comstock CE, Augello MA, Goodwin JF, Launchbury R, Zhao SG, Schiewer MJ, Ertel A, Karnes J, et al. Cell cycle-coupled expansion of AR activity promotes cancer progression. Oncogene. 2017;36:1655–68.
Murthy S, Wu M, Bai VU, Hou Z, Menon M, Barrack ER, Kim SH, Reddy GP. Role of androgen receptor in progression of LNCaP prostate cancer cells from G1 to S phase. PLoS ONE. 2013;8:e56692.
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–89.
Marchese FP, Raimondi I, Huarte M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017;18:206.
Ransohoff JD, Wei Y, Khavari PA. The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol. 2018;19:143–57.
Reis EM, Nakaya HI, Louro R, Canavez FC, Flatschart AV, Almeida GT, Egidio CM, Paquola AC, Machado AA, Festa F, et al. Antisense intronic non-coding RNA levels correlate to the degree of tumor differentiation in prostate cancer. Oncogene. 2004;23:6684–92.
Das R, Feng FY, Selth LA. Long non-coding RNAs in prostate cancer: Biological and clinical implications. Mol Cell Endocrinol. 2019;480:142–52.
Mitobe Y, Takayama KI, Horie-Inoue K, Inoue S. Prostate cancer-associated lncRNAs. Cancer Lett. 2018;418:159–66.
Balas MM, Johnson AM. Exploring the mechanisms behind long noncoding RNAs and cancer. Noncoding RNA Res. 2018;3:108–17.
Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29:452–63.
Zhang A, Zhao JC, Kim J, Fong KW, Yang YA, Chakravarti D, Mo YY, Yu J. LncRNA HOTAIR enhances the androgen-receptor-mediated transcriptional program and drives castration-resistant prostate cancer. Cell Rep. 2015;13:209–21.
Yang L, Lin C, Jin C, Yang JC, Tanasa B, Li W, Merkurjev D, Ohgi KA, Meng D, Zhang J, et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature. 2013;500:598–602.
daSilva LF, Beckedorff FC, Ayupe AC, Amaral MS, Mesel V, Videira A, Reis EM, Setubal JC, Verjovski-Almeida S. Chromatin landscape distinguishes the genomic loci of hundreds of androgen-receptor-associated LincRNAs from the loci of non-associated LincRNAs. Front Genet. 2018;9:132.
He RQ, Qin MJ, Lin P, Luo YH, Ma J, Yang H, Hu XH, Chen G. Prognostic significance of LncRNA PVT1 and its potential target gene network in human cancers: a comprehensive inquiry based upon 21 cancer types and 9972 cases. Cell Physiol Biochem. 2018;46:591–608.
Xiao M, Feng Y, Liu C, Zhang Z. Prognostic values of long noncoding RNA PVT1 in various carcinomas: an updated systematic review and meta-analysis. Cell Prolif. 2018;51:e12519.
Yang J, Li C, Mudd A, Gu X. LncRNA PVT1 predicts prognosis and regulates tumor growth in prostate cancer. Biosci Biotechnol Biochem. 2017;81:2301–6.
Derderian C, Orunmuyi AT, Olapade-Olaopa EO, Ogunwobi OO. PVT1 signaling is a mediator of cancer progression. Front Oncol. 2019;9:502.
Chang Z, Cui J, Song Y. Long noncoding RNA PVT1 promotes EMT via mediating microRNA-186 targeting of Twist1 in prostate cancer. Gene. 2018;654:36–42.
Liu HT, Fang L, Cheng YX, Sun Q. LncRNA PVT1 regulates prostate cancer cell growth by inducing the methylation of miR-146a. Cancer Med. 2016;5:3512–9.
Chen Y, Du H, Bao L, Liu W. LncRNA PVT1 promotes ovarian cancer progression by silencing miR-214. Cancer Biol Med. 2018;15:238–50.
Wan L, Sun M, Liu GJ, Wei CC, Zhang EB, Kong R, Xu TP, Huang MD, Wang ZX. Long noncoding RNA PVT1 promotes non-small cell lung cancer cell proliferation through epigenetically regulating LATS2 expression. Mol Cancer Ther. 2016;15:1082–94.
Guo GC, Wang F, Han ML, Gu YT, Duan X, Li L. Long non-coding RNA PVT1 facilitates cell proliferation by epigenetically regulating FOXF1 in breast cancer. Rsc Advances. 2018;8:2740–50.
Guo J, Hao C, Wang C, Li L. Long noncoding RNA PVT1 modulates hepatocellular carcinoma cell proliferation and apoptosis by recruiting EZH2. Cancer Cell Int. 2018;18:98.
Kong R, Zhang EB, Yin DD, You LH, Xu TP, Chen WM, Xia R, Wan L, Sun M, Wang ZX, et al. Long noncoding RNA PVT1 indicates a poor prognosis of gastric cancer and promotes cell proliferation through epigenetically regulating p15 and p16. Mol Cancer. 2015;14:82.
Yu Y, Zhang M, Liu J, Xu B, Yang J, Wang N, Yan S, Wang F, He X, Ji G, et al. Long non-coding RNA PVT1 promotes cell proliferation and migration by silencing ANGPTL4 expression in cholangiocarcinoma. Mol Ther Nucleic Acids. 2018;13:503–13.
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA. 2009;106:11667–72.
Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell. 2010;40:939–53.
Ayupe AC, Tahira AC, Camargo L, Beckedorff FC, Verjovski-Almeida S, Reis EM. Global analysis of biogenesis, stability and sub-cellular localization of lncRNAs mapping to intragenic regions of the human genome. RNA Biol. 2015;12:877–92.
Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–21.
Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc Ser B Stat Methodol. 1995;57:289–300.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Li J, Han L, Roebuck P, Diao L, Liu L, Yuan Y, Weinstein JN, Liang H. TANRIC: an interactive open platform to explore the function of lncRNAs in cancer. Cancer Res. 2015;75:3728–37.
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. Scikit-learn: machine learning in Python. J Mach Learn Res. 2012;12:2825–30.
Nusbaum C, Mikkelsen TS, Zody MC, Asakawa S, Taudien S, Garber M, Kodira CD, Schueler MG, Shimizu A, Whittaker CA, et al. DNA sequence and analysis of human chromosome 8. Nature. 2006;439:331–5.
Sakurai K, Reon BJ, Anaya J, Dutta A. The lncRNA DRAIC/PCAT29 locus constitutes a tumor-suppressive nexus. Mol Cancer Res. 2015;13:828–38.
Jin HJ, Kim J, Yu J. Androgen receptor genomic regulation. Transl Androl Urol. 2013;2:157–77.
Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27:6194–206.
Yeger H, Perbal B. CCN family of proteins: critical modulators of the tumor cell microenvironment. J Cell Commun Signal. 2016;10:229–40.
Zuo GW, Kohls CD, He BC, Chen L, Zhang W, Shi Q, Zhang BQ, Kang Q, Luo J, Luo X, et al. The CCN proteins: important signaling mediators in stem cell differentiation and tumorigenesis. Histol Histopathol. 2010;25:795–806.
Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–83.
Wu L, Runkle C, Jin HJ, Yu J, Li J, Yang X, Kuzel T, Lee C, Yu J. CCN3/NOV gene expression in human prostate cancer is directly suppressed by the androgen receptor. Oncogene. 2014;33:504–13.
Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903.
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–93.
Gao S, Lee P, Wang H, Gerald W, Adler M, Zhang L, Wang YF, Wang Z. The androgen receptor directly targets the cellular Fas/FasL-associated death domain protein-like inhibitory protein gene to promote the androgen-independent growth of prostate cancer cells. Mol Endocrinol. 2005;19:1792–802.
Lima L, Morais A, Lobo F, Calais-da-Silva FM, Calais-da-Silva FE, Medeiros R. Association between FAS polymorphism and prostate cancer development. Prostate Cancer Prostatic Dis. 2008;11:94–8.
Ren Z, Hou Y, Ma S, Tao Y, Li J, Cao H, Ji L. Effects of CCN3 on fibroblast proliferation, apoptosis and extracellular matrix production. Int J Mol Med. 2014;33:1607–12.
Lin CG, Chen CC, Leu SJ, Grzeszkiewicz TM, Lau LF. Integrin-dependent functions of the angiogenic inducer NOV (CCN3): implication in wound healing. J Biol Chem. 2005;280:8229–37.
Lin CG, Leu SJ, Chen N, Tebeau CM, Lin SX, Yeung CY, Lau LF. CCN3 (NOV) is a novel angiogenic regulator of the CCN protein family. J Biol Chem. 2003;278:24200–8.
Maillard M, Cadot B, Ball RY, Sethia K, Edwards DR, Perbal B, Tatoud R. Differential expression of the ccn3 (nov) proto-oncogene in human prostate cell lines and tissues. Mol Pathol. 2001;54:275–80.
Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107:21931–6.
Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011;21:1273–83.
King AD, Huang K, Rubbi L, Liu S, Wang CY, Wang Y, Pellegrini M, Fan G. Reversible regulation of promoter and enhancer histone landscape by DNA methylation in mouse embryonic stem cells. Cell Rep. 2016;17:289–302.
Fong KW, Zhao JC, Kim J, Li S, Yang YA, Song B, Rittie L, Hu M, Yang X, Perbal B, Yu J. Polycomb-mediated disruption of an androgen receptor feedback loop drives castration-resistant prostate cancer. Cancer Res. 2017;77:412–22.
Puthalakath H, Villunger A, O’Reilly LA, Beaumont JG, Coultas L, Cheney RE, Huang DC, Strasser A. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293:1829–32.
Reich NC. A death-promoting role for ISG54/IFIT2. J Interferon Cytokine Res. 2013;33:199–205.
Pei N, Mao Y, Wan P, Chen X, Li A, Chen H, Li J, Wan R, Zhang Y, Du H, et al. Angiotensin II type 2 receptor promotes apoptosis and inhibits angiogenesis in bladder cancer. J Exp Clin Cancer Res. 2017;36:77.
Nakamura M, Shimada K, Konishi N. The role of HRK gene in human cancer. Oncogene. 2008;27(Suppl 1):S105-113.
Nola S, Daigaku R, Smolarczyk K, Carstens M, Martin-Martin B, Longmore G, Bailly M, Braga VM. Ajuba is required for Rac activation and maintenance of E-cadherin adhesion. J Cell Biol. 2011;195:855–71.
Jia L, Gui B, Zheng D, Decker KF, Tinay I, Tan M, Wang X, Kibel AS. Androgen receptor-regulated miRNA-193a-3p targets AJUBA to promote prostate cancer cell migration. Prostate. 2017;77:1000–11.
Bennett M, Macdonald K, Chan SW, Luzio JP, Simari R, Weissberg P. Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science. 1998;282:290–3.
Acknowledgements
The authors thank Gilbert O. Silveira, Instituto de Química, Universidade de São Paulo, for producing the illustrations. The results published here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Funding
This work was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) grant numbers 2014/03620-2 and 2018/23693-5 to SVA. AV received a fellowship from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) at the beginning of the work and subsequently from FAPESP/CAPES number 2015/00324-6, FCB received a fellowship from FAPESP number 2012/12005-4 and LFdS received a fellowship from CNPq. SVA received an established investigator career award fellowship from CNPq. The funding sources were not involved in the conduct of the research and/or preparation and submission of the article for publication.
Author information
Authors and Affiliations
Contributions
AV: conceptualization, data acquisition, analyses and interpretation, writing and revising; FCB: conceptualization, data acquisition, analyses and interpretation, writing and revising; LFdS: writing of software, data analyses; SVA: conceptualization, data analyses and interpretation, writing of original draft, revising, project administration. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary Methods.
Additional file 2:
Supplementary Figures and Tables.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Videira, A., Beckedorff, F.C., daSilva, L.F. et al. PVT1 signals an androgen-dependent transcriptional repression program in prostate cancer cells and a set of the repressed genes predicts high-risk tumors. Cell Commun Signal 19, 5 (2021). https://doi.org/10.1186/s12964-020-00691-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-020-00691-x