
Abstract
Background
In the event of amino acid starvation, the cell activates two main protective pathways: Amino Acid starvation Response (AAR), to inhibit global translation, and autophagy, to recover the essential substrates from degradation of redundant self-components. Whether and how AAR and autophagy (ATG) are cross-regulated and at which point the two regulatory pathways intersect remain unknown. Here, we provide experimental evidence that the mammalian target of rapamycin (mTOR) complex 1 (mTORC1) specifically located at the lysosome level links the AAR with the autophagy pathway.
Methods
As an inducer of the AAR, we used halofuginone (HF), an alkaloid that binds to the prolyl-tRNA synthetase thus mimicking the unavailability of proline (PRO). Induction of AAR was determined assessing the phosphorylation of the eukaryotic translation initiation factor (eIF) 2α. Autophagy was monitored by assessing the processing and accumulation of microtubule-associated protein 1 light chain 3 isoform B (LC3B) and sequestosome-1 (p62/SQSTM1) levels. The activity of mTORC1 was monitored through assessment of the phosphorylation of mTOR, (rp)S6 and 4E-BP1. Global protein synthesis was determined by puromycin incorporation assay. mTORC1 presence on the membrane of the lysosomes was monitored by cell fractionation and mTOR expression was determined by immunoblotting.
Results
In three different types of human cancer cells (thyroid cancer WRO cells, ovarian cancer OAW-42 cells, and breast cancer MCF-7 cells), HF induced both the AAR and the autophagy pathways time-dependently. In WRO cells, which showed the strongest induction of autophagy and of AAR, global protein synthesis was little if any affected. Consistently, 4E-BP1 and (rp)S6 were phosphorylated. Concomitantly, mTOR expression and activation declined along with its detachment from the lysosomes and its degradation by the proteasome, and with the nuclear translocation of transcription factor EB (TFEB), a transcription factor of many ATG genes. The extra supplementation of proline rescued all these effects.
Conclusions
We demonstrate that the AAR and autophagy are mechanistically linked at the level of mTORC1, and that the lysosome is the central hub of the cross-talk between these two metabolic stress responses.

Similar content being viewed by others


Background
Mammalian cells have developed adaptive mechanisms to sense and counteract amino acid deprivation in order to maintain a homeostatic intracellular pool of amino acids [1]. According to the availability of amino acids, mammalian cells regulate protein synthesis and autophagy [2, 3]. Autophagy is a lysosomal degradation process through which the cell eliminates the redundant or damaged cytosolic material to recover the basic components to secure the macromolecular turnover and cell homeostasis. Autophagy promptly raises up when the cell is subjected to an adverse nutrient condition, such as in the case of amino acid deprivation [4].
A first sensor of amino acids levels is provided by the general control nonderepressible 2 kinase (GCN2) that detects the uncharged tRNAs resulting from the lack of amino acids (1, 5). In this situation, GCN2 phosphorylates the Serine 51 of the α-subunit of eukaryotic translation initiation factor (eIF) 2α. Such phosphorylation causes a reduction in translation initiation and protein synthesis. Also, phosphorylated eIF2α promotes the translation of specific mRNAs containing in their 5′ leader unique upstream open reading frames, such as the activating transcription factor 4 (ATF4) mRNA. In turn, ATF4 triggers the transcriptional amino acid response pathway (AAR) by inducing the expression of several target genes, including activating transcription factor 3 (ATF3), CEBP homologous protein (CHOP) and asparagines synthetase (ASNS) [1, 5,6,7]. Of note, recent works indicate that the deprivation of different individual amino acids may trigger distinct AARs [1, 8].
A second sensor of amino acids levels is provided by the mammalian target of rapamycin (mTOR) complex 1 (mTORC1). The complex includes mTOR, the 40 kDa Pro-rich Akt substrate (PRAS40), the mammalian lethal with SEC13 protein 8 (mLST8), the DEP domain-containing mTOR-interacting protein (DEPTOR) and the regulatory-associated protein of mTOR (RAPTOR) [3]. When active, mTORC1 promotes cell growth by stimulating the protein synthesis through the phosphorylation of the eIF4E-binding protein 1 (4E-BP1) and of p70S6 kinase 1 that in turn phosphorylates the ribosomal protein S6 (S6). Particularly, the phosphorylation of Thr37/46, Thr70 and Ser65 in 4E-BP1 frees eIF4E that can then bind to eIF4G allowing the initiation of cap-dependent translation.
Moreover, active mTORC1 inhibits autophagy by phosphorylating the autophagy-related (ATG) proteins ATG13 and Unc-51 Like Autophagy Activating Kinase 1 (ULK1). The activity of mTORC1 is regulated by several signals, including growth factors, cellular energy level, oxygen level and nutrients, particularly amino acids [3, 9, 10]. Upon amino acid deprivation, mTORC1 is inactivated with the resulting inhibition of protein synthesis and activation of autophagy. Subcellular control of mTORC1 by amino acids levels occurs via the Rag GTPases that are held on the membranes of the late endosomes/lysosomes (LEL) by the Ragulator (LAMTOR) complex. In presence of amino acids, the Rags positively regulate mTORC1 by recruiting the complex on the LEL membranes [11, 12].
Clearly, the AAR and the autophagy processes must be coordinated by the availability of amino acids. Whether and how these processes are cross-regulated and at which point the two regulatory pathways intersect remain unknown. Here, we investigated on these issues taking advantage of the molecular mechanism of action of the febrifugine-derivative halofuginone (HF). This drug was reported to mimic an AAR in Th17 lymphocytes by interfering with the utilization of proline [13,14,15]. Here, we show that in several cancer cell lines HF induces the AAR and concomitantly triggers the autophagy response by promoting the proteasome-mediated degradation of mTOR and the nuclear translocation of the autophagy transcription factor TFEB. An excess of proline could prevent all these events, proving that the unavailability of one single (particular) amino acid can trigger both the AAR and autophagy. Interestingly, we found that HF had a little impact on global protein synthesis and stimulated mTORC2 activity. Our data provide the first demonstration that the AAR and autophagy are mechanistically linked and suggest that the therapeutic properties of HF could be mediated by autophagy.
Methods
Reagents
Unless otherwise specified, culture media, antibiotics, antibodies and analytical grade chemicals were from Sigma-Aldrich Corp., St. Luis, MO, USA. Primary antibodies were obtained from the following sources: rabbit monoclonal anti-ATG7 (04–1055, EMD Millipore Corporation, Billerica, MA, USA), mouse monoclonal anti-eIF2α (2103, Cell Signaling Technology Inc., Danvers, MA, USA), rabbit monoclonal anti-phospho-eIF2α Ser 51 (3398, Cell Signaling Technology Inc.), mouse monoclonal anti-Golgin 97 (sc-59,820, Santa Cruz Biotechnology Inc., Dallas, TX, USA), mouse monoclonal anti-LAMP-1 (555,798, Becton, Dickinson and Company, New Jersey, NJ, USA), rabbit polyclonal anti-LC3B (L7543, Sigma-Aldrich Corp.), rabbit monoclonal anti-p62/SQSTM1 (D5E2) (8025, Cell Signaling Technology Inc.), rabbit polyclonal S6 ribosomal protein (5G10) (2217, Cell Signaling Technology Inc.), rabbit monoclonal anti-phospho-S6 ribosomal protein (Ser235/236) (4856, Cell Signaling Technology Inc.), rabbit monoclonal anti-mTOR (2983, Cell Signaling Technology Inc.), rabbit polyclonal anti-phospho-mTOR Ser 2448 (2971, Cell Signaling Technology Inc.), rabbit polyclonal anti-phospho-mTOR Ser 2481 (2974, Cell Signaling Technology Inc.), mouse monoclonal anti-β-Tubulin (T5293, Sigma-Aldrich Corp.), rabbit monoclonal anti-RAPTOR (2280, Cell Signaling Technology Inc.), rabbit polyclonal TFEB (4240, Cell Signaling Technology Inc.), Rabbit monoclonal anti-4E-BP1 (53H11) (9644, Cell Signaling), Rabbit monoclonal anti-phospho- 4E-BP1 Thr 37/46 (236B4) (2855, Cell Signaling) Rabbit polyclonal anti-phospho-Akt Ser 473 (9271, Cell Signaling), Rabbit monoclonal anti-Akt (pan) (11E7) (4685, Cell Signaling), Mouse monoclonal anti-β-actin clone AC-15 (A5441, Sigma-Aldrich), Mouse monoclonal anti-puromycin clone 12D10 (MABE343, Merck Millipore, Darmstadt, Germany). Secondary antibodies employed for immunoblotting were purchased from the following sources: Horse Radish Peroxidase-conjugated goat anti-mouse IgG (170–6516, Bio-Rad, Hercules, CA, USA), Horse Radish Peroxidase-conjugated goat anti-rabbit IgG (170–6515, Bio-Rad, Hercules, CA, USA). Secondary antibodies employed for immunofluorescence were purchased from the following sources: IRIS 2 goat anti-rabbit IgG (2WS-08, Cyanine Technologies S.p.A., Torino, Italy), IRIS 3 goat anti-mouse IgG (3WS-07, Cyanine Technologies S.p.A., Torino, Italy).
Cell cultures and treatments
The following tumor-derived human cell lines available from the ATCC (Rockville, MD, USA) were used: WRO (thyroid carcinoma), MCF-7 (breast adenocarcinoma), and OAW-42 (ovarian carcinoma). The cells were cultured under standard conditions (37 °C, 5% CO2) in RPMI (WRO) or Minimum Essential Medium (MCF-7 and OAW-42). Media were supplemented with 10% fetal bovine serum (Lonza, Basel, Switzerland), 2 mM glutamine and 1% penicillin-streptomycin solution. Where indicated, cells were exposed to 100 nM halofuginone (HF) in presence or absence of 10 mM ammonium chloride (NH4+), or 30 μM chloroquine (CQ), or 10 μM MG132, or 35 μM cycloheximide (CHX) in complete culture medium or Earle’s Balanced Salt Solution (EBSS) for the indicated time. EBSS contains 1% glucose and is widely used as a culture medium to starve the cells of amino acids and serum growth factors (E2888). In rescue experiments, extra proline was added to WRO complete culture medium. Proline was supplied 10 times more concentrated than the original culture medium concentration (2 mM).
siRNA and plasmid transfection
siRNA and plasmid transfections were performed following manufacturer’s protocols with Lipofectamine 2000 (Life Technologies Ltd., Paisley, UK). Treatments were performed 36 h after the transfection. siRNA sequences: control duplex siRNA 5′-AGG UAG UGU AAU CGC CUU GTT-3′; ATG7 siRNA 5′-GGG UUA UUA CUA CAA UGG UGT T-3′. The origin and use of the GFP-FYVE and GFP-LC3 plasmids have been reported previously [16,17,18].
Immunoblotting
Cells were harvested in RIPA Buffer supplemented with protease inhibitor cocktail, and phosphatase inhibitors (sodium fluoride and sodium orthovanadate) and homogenized using an ultrasonic cell disruptor XL (Misonix, Farmingdale, NY, US). Protein concentration was assessed with a Bradford assay and equal amounts of protein (30 μg of total cell homogenates) were separated by SDS-PAGE and transferred onto PVDF membrane. After blocking with 5% non-fat milk (Santa Cruz Biotechnology Inc.), the filter was probed with designated primary and secondary antibodies, developed with the enhanced chemiluminescence method (PerkinElmer Inc., Waltham, MA, USA). Bands were imaged and subjected to densitometry using the VersaDOC Imaging System apparatus (Bio-Rad) equipped with the software Quantity One (Bio-Rad). Representative western blotting of at least three independent experiments are shown.
Immunofluorescence
WRO cells were plated on coverslips at 25,000 cells/cm2 and let adhere 24 h before the designated treatments. The cells were then washed in PBS, fixed over night with 4% paraformaldehyde at 4 °C, permeabilized with 0.2% Triton X-100 in PBS for 10 min and processed for immunostaining with indicated primary antibodies and corresponding secondary antibodies. Images were captured with a Leica DMI6000 fluorescence microscope (Leica Microsystems AG, Wetzlad, DE) equipped with the software Leica Application Suite V. 3.8 (Leica Microsystems AG). Representative images of at least three independent experiments are shown.
Subcellular fractionation
WRO cells were cultured in presence or absence of 100 nM halofuginone for 8 h, washed twice with ice-cold PBS and harvested in homogenization buffer (0.25 M sucrose, 2 mM Hepes Buffer, PBS) supplemented with protease inhibitor cocktail, and phosphatase inhibitors (sodium fluoride and sodium orthovanadate). Cell suspensions were homogenized using a 2 ml dounce tissue grinder and then centrifuged at 1000x g for 10 min at 4 °C in order to obtain post-nuclear supernatants (PNS). The PNS were loaded on 11 ml of 15 to 65% discontinuous sucrose gradient prepared in homogenization buffer and centrifuged at 20,0000x g for 16 h at 4 °C using a SW-41 swing rotor (Beckman Coulter, Inc., Brea, CA, USA). Twelve fractions (1 ml each) were collected from the top of the gradient and processed for immunoblotting analysis with the indicated antibodies.
Puromycin incorporation assay
WRO cells were seeded on p35 petri at 40,000 cells/cm2 and let adhere 24 h before to perform the treatment. After exposure to 100 nM HF in presence/absence of 2 mM proline in complete medium or incubation with EBSS for the indicated time, the cell pellets were washed with PBS 1X, supplemented with 5 μg/ml puromycin (P7255, Sigma-Aldrich) and incubated at 37 °C for 10 min. The cell pellets were then washed with PBS1X and incubated at 37 °C for 5 min. Cells were lysed in RIPA Buffer supplemented with protease inhibitor cocktail and phosphatase inhibitors and the homogenates used for western blotting.
Data analysis and statistics
All data were replicated at least three times in separate experiments. Densitometric analyses of immunoblot bands were performed with Quantity One (Bio-Rad laboratories) software. Differences between indicated protein ratios were analyzed by Student t test. A p value ≤ of 0.05 was considered significant. Immunofluorescence intensity density was determined with ImageJ 1.48v (http://imagej.nih.gov/ij/) software. GraphPad Prism was employed for statistical analysis (GraphPad Software Inc.).
Results
Halofuginone induces the amino acid response pathway in human cancer cell types
In a first set of experiments, three different types of human cancer cells, namely thyroid cancer WRO cells, ovarian cancer OAW-42 cells, and breast cancer MCF-7 cells, were exposed to 100 nM HF and eIF2α phosphorylation, chosen to monitor the induction of the AAR, was assessed at 4, 8 and 24 h. Increased phosphorylation of eIF2α was clearly detectable in all the cell lines starting from 4 h of treatment with HF (Fig. 1). The level of phosphorylated eIF2α was differently modulated along the incubation time in the three different cell lines (Fig. 1). Our data confirm that HF induces the AAR pathway in all the investigated cancer cell lines.

Halofuginone induces the amino acid starvation response (AAR) pathway. Thyroid cancer WRO cells, breast cancer MCF-7 cells, and ovarian cancer OAW-42 cells were exposed to 100 nM halofuginone (HF) for 4, 8 and 24 h. Total and phosphorylated (Ser 51) eIF2α protein levels were assessed by immunoblotting. As loading control, filters were stripped and probed with anti β-Tubulin as loading control. Representative immunoblots from three independent experiments are shown along with P-eIF2α/eIF2α band intensity ratios as index of AAR
The amino acid starvation response induced by halofuginone is paralleled by up-regulation of basal autophagy
Next, we checked whether autophagy was induced along with the AAR by HF. Processing and accumulation of lipidated microtubule-associated protein 1 light chain 3 isoform B (LC3B) were assumed as an index of the autophagosomes present in the cells [19].
A typical pattern of LC3B-I and of LC3B-II present in the cells is shown in Fig. 2a. The conversion of LC3B-I to LC3B-II, which is indicative of autophagosome formation [20], increased in all cell lines upon exposure to HF (Fig. 2a, LC3B-II/I ratio). Like for eIF2α phosphorylation, autophagy was differently modulated in the cell lines tested during the incubation time, likely reflecting the different genetic and proteome background and intracellular pool of amino acids. A significant increase of LC3B-II/I ratio was observed by 8 h in WRO and MCF7 cells, and by 24 h in OAW-42, indicating an increase in the autophagosome formation following exposure to HF. In the case of WRO cells, we also noted that the chronic (24 h) exposure to HF causes some 45% cell detachment and apoptosis (data not shown). From now on, we chose the WRO cells as representative to investigate more in depth the mechanisms linking the AAR with autophagy, limiting the exposure to HF at 8 h to avoid mis-interpretation due to cell toxicity.

Halofuginone induces autophagy. a Cells were exposed to halofuginone (HF) as in Fig. 1 and LC3B protein levels were assessed by immunoblotting. As loading control, filters were stripped and probed with ant β-Tubulin as loading control. Representative immunoblots are shown along with LC3B-II/I band intensity ratios as index of autophagosome formation. Histograms show mean (±SD) LC3B-II/I band intensity ratios of three different experiments. Statistically significant differences between LC3B-II/I ratios after to before HF are shown (*, p ≤ 0.05). b WRO cells were exposed to halofuginone (HF) for 8 h in the presence or in the absence of 30 μM chloroquine (CQ) and LC3B protein levels were assessed by immunoblotting. As loading control, filters were stripped and probed with anti β-Tubulin as loading control. Representative immunoblots are shown along with LC3B-II/I band intensity ratios as index of autophagosome formation. Relative amount of newly formed autophagosomes is expressed as LC3B-II/I ratios difference in the presence or absence of CQ (APH biogenesis). Histograms show mean (±SD) LC3B-II/I band intensity ratios of 3 different experiments. Statistically significant differences between LC3B-II/I ratios after to before HF are shown (*, p ≤ 0.05)
Next, we assessed the accumulation of LCB-II in the presence or absence of 30 μM chloroquine (CQ) by immunoblotting to confirm that basal autophagy is up-regulated following HF. Chloroquine alkalinizes the acidic compartments, preventing the autophagosome-lysosome fusion and impairing the degradation of autophagosomes and of their content [19, 21]. Assuming the net conversion of LC3B-I into LC3B-II (ratio LC3B-II/I) in the presence vs absence of CQ as the relative amount of newly formed autophagosomes (APH biogenesis), basal autophagy was clearly up-regulated following 8 h of exposure to HF (Fig. 2b).
We then assessed LC3B-II in WRO cells transfected with the siRNA targeting the essential autophagy protein ATG7 or with the control RNA duplex to confirm that the increase of LC3B-II following HF results from the induction of autophagy. LC3B-II/I ratio increased upon incubation with HF as expected, and such increase was annulled when ATG7 was silenced (Fig. 3a).

Halofuginone induces autophagosome formation and eventually interferes with its fusion with lysosomes. a WRO cells were transiently transfected with ATG7 siRNA or control duplex (Ct. Du.) siRNA. After 36 h, the cells were exposed to 100 nM halofuginone (HF) for 8 h. The expression of ATG7, LC3B and β-Tubulin was analyzed by immunoblotting of cell homogenates. Representative immunoblots are shown along with LC3B-II/I band intensity ratios as index of autophagosome formation. b WRO cells plated on coverslips were treated with 100 nM halofuginone (HF) in the presence or absence of 10 mM ammonium chloride (NH4+). After 8 h the cells were fixed, processed for LC3 (green) and LAMP-1 (red) immunostaining and imaged by fluorescence microscopy. Nuclei were stained with DAPI. Scale bars: 10 μm. c Bars indicate the average yellow fluorescence intensity density of immunofluorescences shown in b. Data are from 5 different images for each condition. Error bars: standard deviation. Statistically significant differences between fluorescence intensity densities before and after NH4+ are shown (*, p ≤ 0.05). The images shown are representative of four separate experiments. d WRO cells were plated on coverslips and transiently transfected with vectors expressing GFP-FYVE or GFP-LC3. After 36 h, the cells were exposed to 100 nM halofuginone (HF) for 8 h. Following HF, cells were imaged by fluorescence microscopy. Scale bars: 10 μm
We investigate the induction of autophagy also by LC3B immunofluorescence in WRO cells exposed for 8 h to HF, NH4+, or both (Fig. 3b, c). Like chloroquine, NH4+ prevents the autophagosome-lysosome fusion, and impairs the degradation of autophagosomes and of their content including the LC3B-II bound to the inner membrane of the autolysosomes. Cells were stained for LC3B that results in a punctate fluorescence corresponding to the lipidated LC3B-II either bound to the autophagosomes or autolysosomes, and for LAMP-1 (lysosomal associated protein-1) that labels both endosomes/lysosomes and autolysosomes. In WRO cells exposed to HF an increase of LC3B puncta, indicative of lipidated LC3B either bound to the autophagosomes (in green) or autolysosomes (in yellow) is clearly evident and suggests the up-regulation of autophagy. Exposure to NH4+ alone led to the accumulation of vesicles that reflect the basal level of autophagy. The vesicle accumulating following NH4+ are mainly autolysosomes (in yellow), which result from the impairment of LC3B degradation in the autolysosome, together with few autophagosomes (in green), which result from the impairment of the autophagosome-lysosomes fusion. When the cells were exposed to HF in the presence of NH4+ the amount of LC3B-II only labeled (green) vesicles was even greater, indicating that more autophagosomes not fused with lysosomes were accumulating in the cells. Moreover, the increase in yellow-labeled vesicles following NH4+ was greater in cells not exposed to HF, indicating a suboptimal fusion between autophagosome and lysosomes in HF-treated cells. We confirmed the induction of autophagy by HF in WRO cells transiently expressing either GFP-FYVE or GFP-LC3. Transfected cells showed a punctate GFP fluorescence following exposure to HF that indicates the induction of autophagy and formation of autophagosomes (Fig. 3d).
Taken together, the above data support the view that HF has a dual-effect on the autophagy system: initially it induces the formation of autophagosomes, and later it slows-down their fusion with lysosomes and their degradation.
Halofuginone, not amino acid starvation, allows protein synthesis along with autophagy
We investigated on the functional relationship between autophagy, AAR and global protein synthesis in the cells exposed to HF. In a first set of experiments, we included the conditions of amino acid and serum starvation (EBSS), which is known to induce both AAR and autophagy, and of mRNA translation inhibition by cycloheximide (CHX), which is known to block the elongation step.
The three treatments induced autophagy, as monitored by the conversion of LC3B-I into LC3B-II (Fig. 4a). As an additional marker monitoring the autophagy flux, we determined the actual level in the cells of sequestosome-1 (p62/SQSTM1), which tags the autophagy substrates and is degraded along with them [19]. Following HF, p62 levels decreased of about 50 to 70% compared to the control at 8 h (Fig. 4a). Based on p62 level, it is evident that the three treatments also stimulated the autophagosome degradation, besides inducing their biogenesis, being both these effects more prominent in EBSS condition.

Halofuginone and amino acid starvation both induce AAR yet only the former still allows mTOR-dependent protein synthesis. a, b WRO cells were exposed for 4 and 8 h to 100 nM halofuginone (HF), 35 μM cycloheximide (CHX) or amino acid/serum growth factors deprivation (EBSS). Thereafter, the cell homogenates were assessed for markers of autophagy (LC3B, p62) (panel a), of AAR (P-eIF2α, phosphorylated in Ser 51) (panel b), and mTOR ability to promote protein synthesis (P-S6, phosphorylated in Ser 235/236, and P-4E-BP1, phosphorylated in Thr 37/46) (panel b). Filters were, then stripped and probed with antibodies for total eIF2α or S6 or 4E-BP1 or β-Tubulin, as indicated. Densitometry analysis of the protein bands for p62/Tubulin, LC3B-II/I and P-(rp)-S6/(rp)-S6 is shown. (c) WRO cells were treated with 100 nM HF, in standard medium supplemented or not with 2 mM proline (PRO), or with 100 nM rapamycin (Rap) for the time indicated. The expression of total and phosphorylated (Ser 473) Akt, total and phosphorylated (Thr 37/46) 4E-BP1, and total and phosphorylated (Ser 2448) mTOR were assessed by immunoblotting. Filters were stripped and probed with anti-β-Tubulin as loading control. d, e WRO cells were exposed for 4 and 8 h to 100 nM HF in presence or absence of 2 mM proline (PRO) (panel d) or incubated for 8 h in EBSS (panel e) and the presence of puromycin incorporated in neosynthesized proteins was revealed by immunoblotting of cell homogenates. Filters were stripped and probed with anti-β-Actin as loading control. Blots shown in this Figure are representative of three independent experiments with reproducible data
Under the same experimental conditions, we assayed the signaling pathways governing the AAR and protein synthesis. As shown in Fig. 4b, both HF and EBSS promptly and strongly induced the phosphorylation of eIF2α, which slightly declined by 8 h, while on opposite it increased by 8 h in CHX-treated cells. These data indicate that the three treatments effectively induced an AAR. Then, we assayed the signaling pathways downstream mTORC1 governing protein synthesis. 4E-BP1 is the most studied and ubiquitously expressed member of a family of eIF4E binding proteins. Phosphorylation of 4E-BP1 at multiple sites (primed at Thr 37/46) reduces its affinity for eIF4E that becomes free and available to complex with eIF4G thus allowing the initiation of cap-dependent translation. The level of phosphorylation identifies three bands with different migratory rate, respectively named from top γ (the most phosphorylated), β (intermediate) and α (the least phosphorylated). Data in Fig. 4b show that HF and CHX maintain the hyper-phosphorylation of 4E-BP1 (indicated by the prominence of the γ band) while in EBSS is detectable only the hypo-phosphorylated isoform (α band). Next, we looked at the phosphorylation of (rp)S6, which correlates with the translational rate. We found that S6 was phosphorylated at any time in the cells incubated with HF while it was completely de-phosphorylated in the cells incubated in EBSS (Fig. 4b). S6 was phosphorylated also in the cells exposed to CHX, in which elongation of mRNA translation is blocked.
mTOR is the catalytic subunit of two distinct complexes, mTORC1 and mTORC2, that differ for composition, functionality and substrates. mTORC1 regulates, among others, the protein synthesis and autophagy processes, while mTORC2 phosphorylates, among other substrates, AKT at Serine 473. To better understand the effect of HF on mTOR-dependent protein synthesis we incubated the cells in the presence of HF with or without an excess of free proline. Rapamycin was also included in this experiment as an inhibitor of mTORC1 activity. Rapamycin effectively switched off the activity of mTOR as indicated by phosphorylation of mTOR (S2448) itself and of its downstream 4E-BP1 (Fig. 4c). HF confirmed to maintain hyper-phosphorylated 4E-BP1, while slightly reducing the phosphorylation of mTOR that was however rescued by the excess of proline (Fig. 4c). Interestingly, HF stimulated the activity of mTORC2, as testified by S473 phosphorylation of AKT, an effect that was totally reversed by proline, while Rapamycin did not affect mTORC2 activity as expected (Fig. 4c). Finally, we assessed global protein synthesis in these conditions through the puromycin incorporation assay. Data in Fig. 4d-e suggest that HF does not impair protein translation (or it does to a very little extent) while EBSS largely impairs it. Taken together, these data indicate that HF induces AAR while still allowing protein synthesis despite some inhibition of mTOR activity. In contrast, EBSS induces AAR along with total inhibition of mTOR-dependent protein synthesis.
Halofuginone effectively links the proline-deficient AAR to autophagy
In theory, autophagy and AAR could be two un-related processes contemporarily induced by HF. The AAR induced by HF follows the inhibition of prolyl-tRNA synthetase activity and can be reverted by extra supplementation of proline [14]. We exploited this fact to prove definitively the ability of HF to link AAR and autophagy. The supplementation of standard culture medium with 2 mM proline was sufficient to rescue the activation of AAR pathway by HF, as indicated by the restoration of the steady state levels of eIF2α phosphorylation at 8 h (Fig. 5a). At the same time, proline supplementation almost completely abrogated the induction of autophagy by HF, as indicated by the level of LC3B protein (Fig. 5a) and by the relative amounts of LC3B-positive vesicles (green- and yellow-stained) (Fig. 5b, c) in the cells. Interestingly, extra supplementation with the amino acid leucine was unable to rescue the AAR induced by HF, demonstrating the specific action of this drug in mimicking the starvation by proline (data not shown).

Excess of proline rescues the induction of AAR and of autophagy by halofuginone. a WRO cells were treated with 100 nM halofuginone (HF) in standard medium supplemented or not with 2 mM proline (PRO) as indicated. A representative immunoblotting of P-eIF2α versus total eIF2α (marker of AAR) and of LC3B (marker of autophagy) is shown. Densitometry analysis of the protein bands was performed and the LC3B-II/ I band density ratios are shown. A similar pattern of protein expression was observed in two other separate experiments. b WRO cells were plated on coverslips and treated as in (a). After 8 h the cells were fixed, processed for LC3 (green) and LAMP-1 (red) immunostaining and imaged by fluorescence microscopy. Nuclei were stained with DAPI. Scale bars: 10 μm. c Bars indicate the average yellow fluorescence intensity density of immunofluorescences shown in (b). Data are from five different images for each condition. Error bars: standard deviation. Statistically significant differences between fluorescence intensity densities after to before HF, or after to before PRO in the presence of HF are shown (*, p ≤ 0.05)
From these data, we conclude that proline starvation, as mimicked by HF, links the AAR with autophagy.
Halofuginone induces the detachment from lysosomes and subsequent proteasome-mediated degradation of mTOR
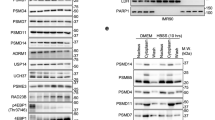
In the presence of amino acids, active mTORC1 is recruited on the membrane of lysosomes and phosphorylates its substrates to promote protein synthesis and to inhibit autophagy. Conversely, amino acid starvation releases mTORC1 from the lysosome membrane and this results in the activation of the ULK1 complex and of the autophagy interactome [3, 22, 23]. We reasoned that to raise up basal autophagy, HF would cause the detachment of mTORC1 from the lysosomes. Thus, we checked the abundance of mTORC1 in the lysosome fraction of WRO cells treated for 8 h with HF. Subcellular fractions were separated by discontinuous sucrose gradient and characterized by western blotting for organelle enrichment using LAMP-1, as a marker of lysosome, and Golgin-97, as a marker of the Golgi Complex. Tubulin, a component of the cytoskeleton, was used as a marker of the cytosolic fraction. RAPTOR, a unique component of mTORC1, was used to discriminate mTORC1 from mTORC2 [24, 25]. A typical pattern of the subcellular localization of mTOR (both total and phosphorylated at Ser 2481) and of RAPTOR is shown in Fig. 6a. This experiment evidenced that upon treatment with HF the abundance of mTOR and of phospho-mTOR is greatly reduced in the lighter lysosome-containing fraction 4 (possibly corresponding to unloaded small lysosomes). Concurrently, RAPTOR protein level decreased only in the same fraction in which mTOR also decreased upon HF treatment (i.e., fraction 4). Noteworthy, in untreated cells, fraction 4 is the one, among all the LAMP-1 positive fractions [4 to 8], containing the largest amount of mTOR (both total and phosphorylated at Ser 2481) and of RAPTOR (Fig. 6a). Interestingly, fractions 2 and 3, enriched in cytoplasmic (Tubulin) and Golgi-associated (Golgin-97) proteins and poor of lysosomes (the relative amount of LAMP-1 in these fractions is negligible), are positive for mTOR and RAPTOR. It is to be noted that the relative amount of mTORC1 proteins in these fractions was not increased upon HF treatment (Fig. 6a).

Halofuginone induces the detachment of mTORC1 from the lysosomes and partial proteasome degradation of mTOR. a WRO cells were exposed or not to 100 nM halofuginone for 8 h and subcellular fractions were separated by discontinuous sucrose gradient from 15 to 55% sucrose density. Nine fractions were collected and characterized by immunoblotting with the indicated antibodies. A strong reduction in the content of mTOR and RAPTOR proteins is clearly appreciable in fraction 4 of WRO cells treated with HF. A similar trend was observed in two other independent experiments and with diverse sucrose gradient. Post-nuclear supernatant (PNS) and gradient loading fraction (L) were loaded as controls. FR.N.: fraction number. b WRO cells were exposed or not to 100 nM of halofuginone (HF), or 10 μM of the proteasome inhibitor MG132, or both. After 8 h, the cells were collected and homogenates processed for immunoblotting to assess mTOR protein level. As loading control, the filter was stripped and probed with anti β-Tubulin antibody. The pattern of protein expression shown was reproduced in three separate experiments. Densitometry of the protein bands was performed and average mTOR/Tubulin ratios are shown in the histogram graph. Error bars: standard deviation. Statistically significant differences between mTOR protein levels are shown (*, p ≤ 0.05)
Thus, upon HF treatment the mTORC1 is no more associated with (small) lysosomes though apparently it does not relocate to the lighter subcellular fractions.
Indeed, we found that the absolute amount of mTOR in cell homogenate and in the cytoplasmic fraction obtained by differential centrifugation was reduced in the cells treated with HF (data not shown). We thus hypothesized that HF could induce the proteolysis of mTOR soon after provoking its detachment from the lysosome. The proteasome would be the candidate for performing such proteolysis. We therefore assessed the protein level of mTOR in WRO cells exposed to HF for 8 h in the absence or the presence of the proteasome inhibitor MG132. As shown in Fig. 6b, the cellular amount of mTOR protein was lower in HF-treated than in control cells, and the concomitant treatment with MG132 prevented the loss of mTOR induced by HF. These data indicate that upon treatment with HF mTOR is indeed degraded, though not completely, by the proteasome.
The excess of proline prevents the detachment of mTOR from lysosomes and its degradation induced by halofuginone
We have previously shown that the extra supplementation of proline could rescue the effects of HF in inducing the AAR and autophagy. At this point, it was mandatory to close the circle and prove that mTOR links the AAR with autophagy, and that its degradation induced by HF is indeed the mechanism for inducing autophagy. Thus, the level of total and phosphorylated (active) mTOR protein was determined by western blotting in cells exposed to HF in the absence or presence of additional proline. As shown in Fig. 7a, proline supplementation rescued the reduction of total mTOR protein levels provoked by HF. Phosphorylated mTOR at Ser 2448 and Ser 2481 were also decreased in HF-treated cells, likely reflecting the decreased amount of the total protein rather than a specific impairment of mTOR phosphorylation. Again, proline supplementation rescued the levels of both phosphorylated isoforms of mTOR in the cells co-treated with HF. We have hypothesized that upon HF treatment mTOR is degraded soon after its detachment from the lysosomes. We tested this hypothesis by looking at the lysosomal localization of mTOR in the cells treated with HF in the absence or presence of extra proline. To this end, LAMP-1 and mTOR were identified by immunofluorescence staining and their co-localization assessed by fluorescence microscopy. The images in Fig. 7b and c show a general reduction of labeled mTOR (in green) and more specifically of mTOR co-stained with LAMP-1 (in yellow) in the cells treated with HF, which are consistent with the detachment from lysosomes and degradation of the protein. Noteworthy, proline supplementation rescued the subcellular localization of mTOR at the level of the lysosomes observed in not treated cells, indicating that the excess of proline could counteract the effects of HF on mTOR localization and degradation.

AAR pathway induced by halofuginone triggers mTOR degradation. a WRO cells were exposed or not to 100 nM halofuginone (HF) in the presence or in the absence of additional 2 mM proline (PRO). After 8 h, the cell homogenates were subjected to immunoblotting to assess total mTOR protein level and its phosphorylation status (at Ser 2448 and Ser 2481). The same set of samples was loaded in three different gels in order to avoid multiple stripping and re-probing of the same filter. Each filter was probed with anti β-Tubulin antibody as loading control. The pattern of protein expression shown was reproduced in three separate experiments. Densitometry of the protein bands was performed and mTOR/Tubulin ratios are included. b WRO cells adherent to coverslips and treated as in (a) were fixed, processed for mTOR (green) and LAMP-1 (red) immunostaining and imaged by confocal fluorescence microscopy. Nuclei were stained with DAPI. Scale bars: 10 μm. The images shown are representative of three separate experiments. c Bars indicate the average yellow fluorescence intensity density of immunofluorescences shown in (b). Data are from five different images for each condition. Error bars: standard deviation. Statistically significant differences between fluorescence intensity densities after to before HF, or after to before PRO in the presence of HF are shown (*, p ≤ 0.05)
Chronic treatment with halofuginone induces the nuclear relocation of the autophagy transcription factor TFEB
In nutrient rich conditions, lysosomal-associated mTORC1 phosphorylates the transcription factor EB (TFEB). As a result, TFEB remains sequestered in the cytoplasm and is transcriptionally inactive. Upon activation, as in the case of amino acid starvation, mTORC1 is released from the lysosomes and TFEB, not phosphorylated by mTORC1, relocates to the nucleus and can initiate the transcription of ATG genes [26]. We assessed by immunofluorescence the subcellular localization of TFEB in WRO cells after exposure to HF. Partial relocation of TFEB from the cytosol to the nucleus was clearly detectable when the cells were treated with HF for 24 h (Fig. 8a, b).

TFEB relocates into the nucleus upon chronic induction of AAR by HF. a WRO cells plated on coverslips were treated or not with 100 nM halofuginone. After 8 and 24 h, the cells were fixed, processed for TFEB (green) immunostaining and imaged by confocal fluorescence microscopy. Nuclei were stained with DAPI. Scale bars: 10 μm. The images shown are representative of three separate experiments. b Bars indicate the average green + blue fluorescence intensity density of immunofluorescences shown in (a). Data are from five different images for each condition. Error bars: standard deviation. Statistically significant difference between fluorescence intensity densities after to before HF is shown (*, p ≤ 0.05)
Discussion
When subjected to nutrient starvation the cell activates an array of protective responses to face the metabolic stress. Yet, if chronically deprived of nutrient and oxygen, the cell eventually succumbs despite of, or because of, the prolonged activation of these metabolic stress responses. In particular, amino acid starvation triggers two main stress responses: the AAR, which inhibits protein synthesis, and autophagy, which degrades the redundant self-structures in an attempt to provide the substrates needed for survival. This study was designed to dissect the cross-talk between these two metabolic stress pathways triggered in response to amino acid starvation. The AAR is a pathway finalized to inhibit protein synthesis when amino acids are not available. The first step in this pathway is the phosphorylation of eIF2α at Serine 51 [6]. In parallel, ATF4, a transcription factor of the cAMP response element binding protein (CREB), is synthesized [27]. In turn, ATF4 promotes the transcription of genes involved in biosynthesis, transport and metabolism of amino acids, including the aminoacyl-tRNA synthetases. Importantly, the eIF2α-ATF4 pathway has been linked to the stress-induced expression of ATG genes [28]. Autophagy is per se triggered by amino acid starvation via the mTORC1 pathway. A complex regulatory network modulates Rags activity, and in turn mTORC1, by sensing amino acids levels in the cytosol or inside the LEL lumen. Cytosolic proteins have been reported to be involved in sensing leucine (leucyl-tRNA synthetase, LRS; Sestrin 2) [29, 30], or arginine (cellular Arginine sensor for mTORC1, CASTOR1) [31, 32], or amino acids (cytoslic folliculin, FLCN; folliculin interacting protein, FNIP) [33] levels in the cytosol and regulate Rags activity. Transmembrane proteins have been identified to sense alanine, proline and glycine (SLC36A1, PAT1) [34], or arginine (SLC38A9, SNAT9) [35,36,37], or amino acids (vacuolar-adenosine triphosphatase proton pump, v-ATPase) [23] levels inside the LEL lumen and to regulate Rags. Other works support a model in which the LEL are not the only subcellular hub for amino acids-dependent mTORC1 regulation [10]. In fact, the transmembrane protein SLC36A4 (PAT4) has been reported to sense glutamine and serine levels inside the trans-Golgi network lumen and to modulate the activity of mTORC1 localized on the Golgi membranes [38].
HF has been previously reported to induce autophagy [39,40,41]. In nutrient-rich conditions, HF induces autophagy by inactivating mTORC1 and the resulting dephosphorylation of ULK1 at Ser 757 [42]. Here, we show that mTORC1 specifically located at the lysosome level links the AAR induced by HF with the autophagy pathway. More in depth, our data show that the induction of the AAR pathway triggers autophagy by promoting the detachment from the lysosomes and the proteasome degradation of mTOR. Subcellular fractionation analyses showed that it is the lysosome-associated, not the Golgi Complex-associated pool of mTOR that is degraded by the proteasome. On long term, the sustained AAR and autophagy led to the nuclear translocation of TFEB, a transcription factor of many ATG genes. In homeostatic conditions, TFEB is stabilized in the cytoplasm by mTORC1-mediated phosphorylation. Thus, only when LEL-associated mTOR is extensively degraded the pool of (un-phosphorylated) TFEB can translocate into the nucleus and direct the synthesis of pro-autophagy genes.
As expected following the undocking of mTOR from the lysosomes and in line with recent works [39, 41], we observed the induction of ATG by HF in all the analyzed cell lines. On long term sustained AAR and autophagy, as induced by HF, led to apoptosis. It was previously reported that the chronic up-regulation of ATF4 leads to the transcription of DNA-damage- inducible transcript 3, which in turn promotes the transcription of pro-apoptotic genes [43]. Thus, AAR and autophagy pathways intersect at two stages of the stress response, in the early stage to coordinate the block of protein synthesis and the degradation of the intracellular pool of protein, and in the later stage to coordinate the programmed cell death. It remains to clarify whether apoptosis in HF-treated cells ensued in spite of or because of the hyper-activation of autophagy.
Here, we used HF as a strong inducer of AAR. HF mimics a reduced cellular pool of available proline by competing with it for the prolyl-tRNA synthetase active site [14]. The resulting accumulation of uncharged tRNAPRO leads to the activation of the AAR pathway. AAR induced by HF reached a peak at different time of incubation in the different cell lines tested, probably reflecting the different pool of free proline available in the cell. HF indeed acted much alike CHX (an inhibitor of protein synthesis) and EBSS (a condition of amino acid and serum starvation) in inducing the AAR and autophagy pathways. The extra supplementation of proline, not of leucine (known to be a major regulator of mTOR), rescued all the phenotypic features triggered by HF, including the effects on mTOR degradation. Interestingly, HF stimulated the phosphorylation of AKT, an effect that was reversed by proline. We may speculate that HF by disrupting mTORC1 from the lysosomal membrane frees mTOR which is partly degraded by the proteasome and partly made available to complex in mTORC2. However, this hypothesis must be proven experimentally.
More importantly, we found differences between HF and amino acid starvation (EBSS) with respect to mTOR-dependent protein synthesis control. The signaling pathways governing protein synthesis and their alterations in cancer cells have been the subject of recent excellent reviews [44,45,46]. Briefly, in the presence of amino acid and of active PI3K-AKT, mTORC1 can phosphorylate p70S6k and 4E-BP1. P70S6k will then phosphorylate (S235/236) S6, whose level correlates with the rate of mRNA translation. Phosphorylation of 4E-BP1 reduces its affinity for eIF4F, which can then bind to eIF4G and allows the initiation of translation. We found that HF could maintain 4E-BP1 highly phosphorylated (γ isoform), while in EBSS only α-4E-BP1 (faintly phosphorylated form) was apparent. This has been associated with ongoing and repressed protein synthesis situations, respectively. In fact, while α and β isoforms (i.e., the least and intermediate phosphorylated forms) of 4E-BP1 associate with eIF4E, the appearance of the γ isoform (the most phosphorylated one) reflects the release of eIF4E [47, 48]. Notably, in EBSS the total amount of 4E-BP1 appeared reduced. It has been shown that in cancer cells hyper-phosphorylation of 4E-BP1 at multiple sites plays an important role in its stabilization and overexpression [49], which may explain the higher level of the γ isoform observed in control and HF-treated cells. We also found that in HF-treated, but not in EBSS-treated cells, S6 was phosphorylated, despite HF reduced to some extent mTOR phosphorylation. Consistent with the signaling, the puromycin incorporation assay showed that protein synthesis was largely suppressed in the cells incubated in EBSS while it was little if any affected when exposed to HF in complete medium. Thus, compared to complete starvation, HF treatment may present the advantage to stimulate the removal of potentially dangerous material by autophagy while allowing protein synthesis. In other words, HF permits the recycling of the basic components resulting from the degradation of autophagic cargo in new essential cellular constituents, while this process is precluded during starvation. The effects seen with HF resemble those reported for resveratrol (RV), a nutraceuticals that acts as a protein restriction mimetic [50, 51]. In a separate work, we have shown that culturing ovarian cancer cells in complete medium containing RV or in EBSS inhibited protein synthesis, as indicated by down-phosphorylation of mTOR and of its downstream target eIF4E-binding protein 1 (4E-BP1) and of S6 and concomitant hyper-phosphorylation of eIF2α, while inducing autophagy in parallel [51]. However, EBSS was more effective in inhibiting protein synthesis while RV was more effective in inducing autophagy [51]. In a recent study, Xia and colleagues have shown that HF-induced autophagy suppresses MCF-7 migration and invasion by down-regulating STMN1 and p53, indicating that HF-induced autophagy may play an important role in the anti-cancer effect of HF [41]. Intriguingly, RV too was shown to suppress the migration of ovarian cancer cells by raising the level of autophagy [52].
HF is a racemic halogenated derivate of febrifugine, an alkaloid extract of the plant Blue Evergreen Hydrangea (Dichroa febrifuga Lour) known for its antiprotozoal activity and used as antimalarial remedy in traditional Chinese medicine [14, 53]. The mechanism of its antimalarial property remains obscure. Our data suggest that this property is related to its effects on the autophagy-lysosome system. The lysosome is an indispensable station for the maturation cycle of the plasmodium, and drugs able to buffer the acidity in the lysosome, such as chloroquine, disrupts this cycle. Chloroquine is now known to also impair the last step of the autophagy process. Here, we found that HF initially induces autophagosome formation and later impairs the autophagosome-lysosome fusion step, much alike chloroquine. Of note, our work is the first to show a dual-effect of HF on autophagy as the other studies investigating HF-induced autophagy reported only the induction of autophagy by the HF and not an impairment of the autophagosome-lysosome fusion step [39, 41]. We hypothesize that the effect of HF on the autophagosome-lysosome fusion step may differ depending on the cell line, HF concentration and incubation time. More investigation is needed to clarify the effect of HF on the autophagosome-lysosome fusion step since in this work we focused only on short term incubation time to avoid mis-interpretation due to cell toxicity. HF elicits also a general anti-inflammatory response by inhibiting the differentiation of inflammatory Th17 cells, and this effect was clearly sue to the induction of the AAR [13,14,15]. HF is currently employed in clinical trials due to its therapeutic potential in fibrotic diseases and cancer [42, 54,55,56,57,58,59] (https://clinicaltrials.gov/). The data here reported strongly support the view that the therapeutic potential of HF is linked to its ability to induce autophagy, and eventually cell death, besides its modulatory effect on protein synthesis.
Conclusions
The cartoon shown in Fig. 9 summarizes the main findings reported in this study. Overall, our data demonstrate that the AAR and autophagy are mechanistically linked by mTORC1, and that the lysosome is the central hub of the cross-talk between these two metabolic stress responses. Our results also suggest that the therapeutic potential of HF is linked to its ability to trigger autophagy.

Schematic representation of the main findings of the study. Under normal conditions (left), mTORC1 is recruited on the membrane of the lysosomes and downregulates autophagy by inhibiting ULK1 complex. HF (right) triggers the Amino Acid starvation Response (AAR) by inhibiting the prolyl-tRNA synthetase and induces the detachment of mTORC1 from the lysosomes. As result, mTORC1 no longer inhibits ULK1, TFEB detaches from mTORC1 and relocates into the nucleus, and autophagy is induced. Following HF, mTOR is partially inactivated and degraded by the proteasome. Extra supplementation of proline rescues the induction of AAR, mTORC1 detachment from the lysosomes, mTOR degradation and autophagy induction
Abbreviations
- AAR:
-
Amino Acid starvation Response
- ATF4:
-
activating transcription factor 4
- ATG:
-
autophagy
- CHX:
-
cycloheximide
- CQ:
-
chloroquine
- EBSS:
-
Earle’s Balanced Salt Solution
- eIF2α:
-
eukaryotic translation initiation factor 2α
- HF:
-
halofuginone
- LAMP-1:
-
lysosomal associated protein-1
- LC3B:
-
microtubule-associated protein 1 light chain 3 isoform B
- LEL:
-
late endosomes/lysosomes
- mTOR:
-
mammalian target of rapamycin
- mTORC1:
-
mammalian target of rapamycin (mTOR) complex 1
- NH4 + :
-
ammonium chloride
- p62/ SQSTM1:
-
sequestosome-1
- PRO:
-
proline
- RAPTOR:
-
regulatory-associated protein of mTOR
- S6:
-
ribosomal protein S6
- TFEB:
-
transcription factor EB
References
Bröer S, Bröer A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem J 2017;474(12):1935–1963. doi: https://doi.org/10.1042/BCJ20160822. Review. PubMed PMID: 28546457; PubMed Central PMCID: PMC5444488.
Tan VP, Miyamoto S. Nutrient-sensing mTORC1: integration of metabolic and autophagic signals. J Mol Cell Cardiol 2016. PubMed PMID: 26773603.
Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011;12(1):21–35. PubMed PMID: 21157483. Pubmed Central PMCID: 3390257.
Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res 2014;24(1):42–57. PubMed PMID: 24343578. Pubmed Central PMCID: 3879708.
Deval C, Chaveroux C, Maurin AC, Cherasse Y, Parry L, Carraro V, et al. Amino acid limitation regulates the expression of genes involved in several specific biological processes through GCN2-dependent and GCN2-independent pathways. FEBS J 2009;276(3):707–718. PubMed PMID: 19120448.
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 2003;11(3):619–633. PubMed PMID: 12667446.
Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 2006;34(Pt 1):7–11. PubMed PMID: 16246168.
Brasse-Lagnel C, Lavoinne A, Husson A. Control of mammalian gene expression by amino acids, especially glutamine. FEBS J 2009;276(7):1826–1844. PubMed PMID: 19250320.
Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol 2013;15(6):555–564. PubMed PMID: 23728461. Pubmed Central PMCID: 3743096.
Goberdhan DC, Wilson C, Harris AL. Amino acid sensing by mTORC1: intracellular transporters mark the spot. Cell Metab 2016;23(4):580–589. PubMed PMID: 27076075.
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. PubMed PMID: 20381137. Pubmed Central PMCID: 3024592.
Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320(5882):1496–1501. PubMed PMID: 18497260. Pubmed Central PMCID: 2475333.
Carlson TJ, Pellerin A, Djuretic IM, Trivigno C, Koralov SB, Rao A, et al. Halofuginone-induced amino acid starvation regulates Stat3-dependent Th17 effector function and reduces established autoimmune inflammation. J Immunol 2014;192(5):2167–2176. PubMed PMID: 24489094. Pubmed Central PMCID: 3936195.
Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol 2012;8(3):311–317. PubMed PMID: 22327401. Pubmed Central PMCID: 3281520.
Sundrud MS, Koralov SB, Feuerer M, Calado DP, Kozhaya AE, Rhule-Smith A, et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324(5932):1334–1338. PubMed PMID: 19498172. Pubmed Central PMCID: 2803727.
Castino R, Fiorentino I, Cagnin M, Giovia A, Isidoro C. Chelation of lysosomal iron protects dopaminergic SH-SY5Y neuroblastoma cells from hydrogen peroxide toxicity by precluding autophagy and Akt dephosphorylation. Toxicological sciences : an official journal of the Society of Toxicology. 2011;123(2):523–41. PubMed PMID: 21742779. Pubmed Central PMCID: 3218672.
Castino R, Bellio N, Follo C, Murphy D, Isidoro C. Inhibition of PI3k class III-dependent autophagy prevents apoptosis and necrosis by oxidative stress in dopaminergic neuroblastoma cells. Toxicological sciences : an official journal of the Society of Toxicology 2010;117(1):152–162. PubMed PMID: 20525898.
Trincheri NF, Follo C, Nicotra G, Peracchio C, Castino R, Isidoro C. Resveratrol-induced apoptosis depends on the lipid kinase activity of Vps34 and on the formation of autophagolysosomes. Carcinogenesis. 2008;29(2):381–389. PubMed PMID: 18048384.
Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. PubMed PMID: 26799652.
Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–545. PubMed PMID: 17611390.
Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. PubMed PMID: 20144757. Pubmed Central PMCID: 2852113.
Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control Centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013;14(5):283–296. PubMed PMID: 23609508. Pubmed Central PMCID: 4387238.
Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334(6056):678–683. PubMed PMID: 22053050. Pubmed Central PMCID: 3211112.
Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002 Jul 26;110(2):177–189. PubMed PMID: 12150926.
Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–175. PubMed PMID: 12150925.
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012;31(5):1095–1108. PubMed PMID: 22343943. Pubmed Central PMCID: 3298007.
Kilberg MS, Shan J, Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends in endocrinology and metabolism: TEM. 2009;20(9):436–43. PubMed PMID: 19800252. Pubmed Central PMCID: 3587693.
B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 2013;41(16):7683–7699. PubMed PMID: 23804767. Pubmed Central PMCID: 3763548.
Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell. 2012;149(2):410–424. PubMed PMID: 22424946.
Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351(6268):43–48. PubMed PMID: 26449471. Pubmed Central PMCID: 4698017.
Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, et al. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell. 2016;165(1):153–164. PubMed PMID: 26972053. Pubmed Central PMCID: 4808398.
Hallett JE, Manning BD. CASTORing new light on amino acid sensing. Cell. 2016;165(1):15–17. PubMed PMID: 27015302.
Tsun ZY, Bar-Peled L, Chantranupong L, Zoncu R, Wang T, Kim C, et al. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell 2013;52(4):495–505. PubMed PMID: 24095279. Pubmed Central PMCID: 3867817.
Ogmundsdottir MH, Heublein S, Kazi S, Reynolds B, Visvalingam SM, Shaw MK, et al. Proton-assisted amino acid transporter PAT1 complexes with rag GTPases and activates TORC1 on late endosomal and lysosomal membranes. PLoS One 2012;7(5):e36616. PubMed PMID: 22574197. Pubmed Central PMCID: 3344915.
Jung J, Genau HM, Behrends C. Amino acid-dependent mTORC1 regulation by the lysosomal membrane protein SLC38A9. Mol Cell Biol 2015;35(14):2479–2494. PubMed PMID: 25963655. Pubmed Central PMCID: 4475919.
Rebsamen M, Pochini L, Stasyk T, de Araujo ME, Galluccio M, Kandasamy RK, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature. 2015;519(7544):477–481. PubMed PMID: 25561175. Pubmed Central PMCID: 4376665.
Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science. 2015;347(6218):188–94. PubMed PMID: 25567906. Pubmed Central PMCID: 4295826.
Fan SJ, Snell C, Turley H, Li JL, McCormick R, Perera SM, et al. PAT4 levels control amino-acid sensitivity of rapamycin-resistant mTORC1 from the Golgi and affect clinical outcome in colorectal cancer. Oncogene. 2015 PubMed PMID: 26434594. Pubmed Central PMCID: 4705441.
Battu S, Afroz S, Giddaluru J, Naz S, Huang W, Khumukcham SS, et al. Amino acid starvation sensing dampens IL-1beta production by activating riboclustering and autophagy. PLoS Biol 2018;16(4):e2005317. PubMed PMID: 29621237. Pubmed Central PMCID: 5903674.
Qin P, Arabacilar P, Bernard RE, Bao W, Olzinski AR, Guo Y, et al. Activation of the Amino Acid Response Pathway Blunts the Effects of Cardiac Stress. Journal of the American Heart Association. 2017;6(5). PubMed PMID: 28487390. Pubmed Central PMCID: 5524058.
Xia X, Wang L, Zhang X, Wang S, Lei L, Cheng L, et al. Halofuginone-induced autophagy suppresses the migration and invasion of MCF-7 cells via regulation of STMN1 and p53. J Cell Biochem 2018;119(5):4009–4020. PubMed PMID: 29231257.
Chen GQ, Gong RH, Yang DJ, Zhang G, Lu AP, Yan SC, et al. Halofuginone dually regulates autophagic flux through nutrient-sensing pathways in colorectal cancer. Cell Death Dis 2017;8(5):e2789. PubMed PMID: 28492544. Pubmed Central PMCID: 5520722.
Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 2004;18(24):3066–3077. PubMed PMID: 15601821. Pubmed Central PMCID: 535917.
Loreni F, Mancino M, Biffo S. Translation factors and ribosomal proteins control tumor onset and progression: how? Oncogene. 2014;33(17):2145–2156. doi: https://doi.org/10.1038/onc.2013.153. Epub 2013 May 6. Review. PubMed PMID: 23644661.
Pelletier J, Graff J, Ruggero D, Sonenberg N. Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res 2015;75(2):250–263. doi: https://doi.org/10.1158/0008-5472.CAN-14-2789. Review. PubMed PMID: 25593033; PubMed Central PMCID: PMC4299928.
Biffo S, Manfrini N, Ricciardi S. Crosstalks between translation and metabolism in cancer. Curr Opin Genet Dev 2018;48:75–81. doi: https://doi.org/10.1016/j.gde.2017.10.011. Epub 2017 Nov 15. Review. PubMed PMID: 29153483.
Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem 1999;68:913–963. Review. PubMed PMID: 10872469.
Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A 2008;105(45):17414–17419. doi: https://doi.org/10.1073/pnas.0809136105. Epub 2008 Oct 27. PubMed PMID: 18955708; PubMed Central PMCID: PMC2582304.
Qin X, Jiang B, Zhang Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle 2016;15(6):781–786. doi: https://doi.org/10.1080/15384101.2016.1151581. Review. PubMed PMID: 26901143; PubMed Central PMCID: PMC4845917.
Villa-Cuesta E, Boylan JM, Tatar M, Gruppuso PA. Resveratrol inhibits protein translation in hepatic cells. PLoS One 2011;6(12):e29513. PubMed PMID: 22242130. Pubmed Central PMCID: 3248458.
Ferraresi A, Titone R, Follo C, Castiglioni A, Chiorino G, Dhanasekaran DN, et al. The protein restriction mimetic resveratrol is an autophagy inducer stronger than amino acid starvation in ovarian cancer cells. Mol Carcinog 2017;56(12):2681–2691. PubMed PMID: 28856729.
Ferraresi A, Phadngam S, Morani F, Galetto A, Alabiso O, Chiorino G, et al. Resveratrol inhibits IL-6-induced ovarian cancer cell migration through epigenetic up-regulation of autophagy. Mol Carcinog 2017;56(3):1164–1181. PubMed PMID: 27787915.
Coatney GR, Cooper WC, Culwell WB, White WC, Imboden CA, Jr. Studies in human malaria. XXV. Trial of febrifugine, an alkaloid obtained from Dichroa febrifuga lour., against the Chesson strain of Plasmodium vivax. Journal National Malaria Society. 1950;9(2):183–186. PubMed PMID: 15422372.
Chen Y, Liu W, Wang P, Hou H, Liu N, Gong L, et al. Correction: Halofuginone inhibits radiotherapy-induced epithelial-mesenchymal transition in lung cancer. Oncotarget. 2018;9(60):31787. PubMed PMID: 30167094. Pubmed Central PMCID: 6114976.
Elkin M, Ariel I, Miao HQ, Nagler A, Pines M, de-Groot N, et al. inhibition of bladder carcinoma angiogenesis, stromal support, and tumor growth by halofuginone. Cancer Res 1999 Aug 15;59(16):4111–4118. PubMed PMID: 10463616.
Koon HB, Fingleton B, Lee JY, Geyer JT, Cesarman E, Parise RA, et al. Phase II AIDS Malignancy Consortium trial of topical halofuginone in AIDS-related Kaposi sarcoma. Journal of acquired immune deficiency syndromes. 2011;56(1):64–8. PubMed PMID: 21068672. Pubmed Central PMCID: 3017346.
McGaha T, Kodera T, Phelps R, Spiera H, Pines M, Bona C. Effect of halofuginone on the development of tight skin (TSK) syndrome. Autoimmunity. 2002;35(4):277–282. PubMed PMID: 12482197.
Pines M, Nagler A. Halofuginone: a novel antifibrotic therapy. Gen Pharmacol 1998;30(4):445–450. PubMed PMID: 9522159.
Pines M, Snyder D, Yarkoni S, Nagler A. Halofuginone to treat fibrosis in chronic graft-versus-host disease and scleroderma. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2003;9(7):417–425. PubMed PMID: 12869955.
Acknowledgements
C.F. was supported with a “Liberato Di Lauro” post-doc fellowship granted by the “Associazione per la Ricerca Medica Ippocrate-Rhazi” (Novara, Italy). C.V. and A. F. were supported with a PhD fellowship from the Ministry of Education, University and Research (MIUR; Italy).
Funding
Not applicable.
Availability of data and materials
All data generated or analysed during this study are included in this research article.
Author information
Authors and Affiliations
Contributions
CF designed the experiments, performed the cell fractionation, and drafted the manuscript. CV and FM performed all experiments. AF and CS performed the imaging and statistical analyses. CI designed and supervised the experiments and finalized the manuscript. All authors reviewed the manuscript and approved its submission. Each of the authors confirms that this manuscript has not been previously published and is not currently under consideration by any other journal. Additionally, all of the authors have approved the contents of this paper and have agreed to the CCAS Journal’s submission policies.
Corresponding author
Ethics declarations
Authors’ information
Carlo Follo received his PhD degree in Biotechnologies for Human Health at the University of Piemonte Orientale (Novara, Italy) in 2009. In, 2003, Dr. Follo received his Master’s degree in Industrial Biotechnology at the Università degli Studi di Milano-Bicocca (Milano, Italy). He performed his PhD studies under the mentorship of Professor Ciro Isidoro. He spent one year as Postdoctoral Investigator at the Albert Einstein College of Medicine (Bronx, NY, USA), and thereafter as Postdoctoral Resercher for three years at Laboratory of Molecular Pathology of Professor Ciro Isidoro (University of Piemonte Orientale, Novara, Italy). From 2014, he is an Associate Specialist at University California San Francisco (UCSF, San Francisco, USA). His research is focused on the contribution of autophagy in mesothelioma.
He has co-authored thirty-two articles published in peer-reviewed journals.
Chiara Vidoni is a postdoctoral fellow at Università del Piemonte Orientale in the Laboratory of Molecular Pathology directed by Prof. Ciro Isidoro. In 2017, she received her PhD in Medical Sciences and Biotechnology at Università del Piemonte Orientale (Novara, Italy), working under the mentorship of Prof. Ciro Isidoro in the Laboratory of Molecular Pathology. She completed her Master’s degree in Medical Biotechnologies at Università del Piemonte Orientale in 2012. She received her Bachelor’s degree in Biotechnologies at Università del Piemonte Orientale in 2009. Her current research focused on the role and regulation of autophagy in neurodegenerative diseases, cancer and regenerative medicine.
She has co-authored eight articles published in peer-reviewed journals.
Federica Morani is currently a post-doctoral researcher at IRCSS Fondazione Stella Maris in Pisa, in Neurogenetics and Molecular Medicine Laboratory directed by Prof. F.M. Santorelli.
From March 2013 until September 2014 she obtained a postdoctoral fellow at Laboratory of Molecular Pathology directed by Prof. Ciro Isidoro (Novara, Italy). She received her PhD degree in Clinical and Experimental Medicine at Università del Piemonte Orientale (Novara, Italy) in 2013. Her work focused on the study of autophagy in several type of cancer (i.e. Glioblastoma, Neuroblastoma, Ovarian Cancer, Thyroid Cancer), particularly in the research of new molecular prognostic markers for Thyroid carcinoma. In 2008, she completed her Master’s degree in Biomolecular Sciences and Technologies cum laude at Università di Pisa (Pisa, Italy). In 2006, she completed her Bachelor’s degree in Molecular and Biological Sciences cum laude at Università di Pisa.
She has published twenty articles in peer-reviewed journals.
Alessandra Ferraresi is a PhD student in Medical Sciences and Biotechnology at Università del Piemonte Orientale (Novara, Italy), working under the mentorship of Prof. Ciro Isidoro in the Laboratory of Molecular Pathology. She completed her Master’s degree in Pharmaceutical Biotechnologies with honors at Alma Mater Studiorum-Università di Bologna in 2014. She received her Bachelor’s degree in Biotechnologies at Università di Parma in 2011. Her current research focused on the role and regulation of autophagy in cancer, particularly in cancer cell migration and resistance to metabolic stress.
She has co-authored seven articles published in peer-reviewed journals.
Christian Seca is a PhD student in Biomedical and Pharmaceutical Sciences at Laboratory of Cancer Signaling, GIGA-Molecular Biology of Diseases, University of Liège (Belgium). He worked as fellowship recipient at Università del Piemonte Orientale (Novara, Italy) in the Laboratory of Molecular Pathology under the mentorship of Prof. Ciro Isidoro. His work focused on the regulation of autophagy in cancer and nanobiotechnology and in vitro cellular imaging. He also spent 1 year as research intern in Stephenson Cancer Center, University of Oklahoma Health Sciences Center (USA) in Prof. Danny Dhanasekaran’s Lab.
In 2015, he completed his Master’s Degree in Medical Biotechnology with honors at Università degli Studi di Napoli “Federico II”. In 2012, he completed his Bachelor’s Degree in Biotechnology at Università degli Studi di Napoli “Federico II”.
He has co-authored two articles in peer-reviewed journals.
Ciro Isidoro (www.isidorolab.com) received his doctoral degree (summa cum laude) in Biological Sciences from the University of Torino (Italy) and his doctoral degree (summa cum laude) in Medicine and Surgery from the University of Piemonte Orientale (Novara, Italy). Currently, he is Professor of Pathology and of Experimental Oncology at the school of Medicine of University of Piemonte Orientale (Novara, Italy). He is also Visiting Professor at the Faculty of Medicine Siriraj Hospital of Mahidol University (Bangkok, Thailand) and at the Department of Cell Biology of the University of Oklahoma Collage of Medicine (Oklahoma City, United States). He is Professeur Honoraire at the Faculté de Medecine et de Phamacie de I. Université de Franche-Comté (Besancon, France). He is member of the scientific board of the “Centre for Integrative Cancer Research” of the Georgia Tech Institute, University of Georgia (Atlanta, US). He is Co-Editor in Chief of the J. of Traditional and Complementary Medicine, and Associate Editor of Autophagy, BMC Cancer, Molecular Carcinogenesis, J. of Ovarian Research, Genes and Cancers, J. of Molecular Signaling, and other journals. His fields of expertise include lysosome biogenesis and function, autophagy regulation in cancer and in neurodegenerative diseases, cell death pathways, mechanisms of action of natural products, epigenetic regulation of autophagy and of cell death.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Follo, C., Vidoni, C., Morani, F. et al. Amino acid response by Halofuginone in Cancer cells triggers autophagy through proteasome degradation of mTOR. Cell Commun Signal 17, 39 (2019). https://doi.org/10.1186/s12964-019-0354-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-019-0354-2