
Abstract
Inflammatory diseases have become increasingly prevalent with industrialization. To address this, numerous anti-inflammatory agents and molecular targets have been considered in clinical trials. Among molecular targets, protease-activated receptors (PARs) are abundantly recognized for their roles in the development of chronic inflammatory diseases. In particular, several inflammatory effects are directly mediated by the sensing of proteolytic activity by PARs.
PARs belong to the seven transmembrane domain G protein-coupled receptor family, but are unique in their lack of physiologically soluble ligands. In contrast with classical receptors, PARs are activated by N-terminal proteolytic cleavage. Upon removal of specific N-terminal peptides, the resulting N-termini serve as tethered activation ligands that interact with the extracellular loop 2 domain and initiate receptor signaling. In the classical pathway, activated receptors mediate signaling by recruiting G proteins. However, activation of PARs alternatively lead to the transactivation of and signaling through receptors such as co-localized PARs, ion channels, and toll-like receptors.
In this review we consider PARs and their modulators as potential therapeutic agents, and summarize the current understanding of PAR functions from clinical and in vitro studies of PAR-related inflammation.
Similar content being viewed by others


Introduction
The four mammalian members of the protease-activated receptor (PAR) family PAR1, PAR2, PAR3, and PAR4 are encoded by the genes F2R [1], F2RL1 [2], F2RL2 [3], and F2RL3 [4], respectively. Human PAR1 was discovered in 1991 as a key thrombin receptor on platelets [5, 6]. Although human and mouse PAR2 genes are homologous to PAR1 genes, PAR2 is not responsive to thrombin [2, 7, 8]. Unexpected responses of platelets to thrombin in PAR1 knockout mice lead to the discovery of the thrombin receptors PAR3 and PAR4 [4, 9, 10]. PAR regulation varies between species and tissues, with differing expression levels, protease cleaving activities, dimerization with other receptors, compartimentalization, trafficking, posttranslational modifications, and co-localization with co-receptors, as shown in Fig. 1.

Mechanisms of PAR activation. PAR activation is regulated by a direct proteolytic cleavage at the N-terminus, b homo- or heterodimerization with other PARs and transactivation through the cleaved tethered ligand, c compartmentalization on the cell surface, d degradation or recycling by endosomal trafficking, e posttranslational modifications such as glycosylation, phosphorylation, and ubiquitination, and f co-localization with other receptors and cofactors
Studies of PAR activation under physiological conditions are crucial for the understanding of the pathophysiological roles of PARs, such as those in inflammatory disorders.
Cleavage and activation of PARs and signal transduction
PARs are specifically cleaved and irreversibly activated by various endogenous proteases, and by exogenous proteases from bacteria, plants, fungi, and insects. Proteases, soluble or cell membrane associated (bound to co-receptors or specific membrane compartments), cleave specific N-terminal peptides of PARs, resulting in exposure of new N-terminal peptides that serve as tethered activation ligands, which bind a conserved region on extracellular loop 2 (ECL2) [5, 11]. This interaction initiates conformational changes and alters affinity for intracellular G proteins [12]. Various N-terminal cleavage sites have been described, and these have various active conformations with specific G protein preferences. Multiple cleavage site-specific cellular responses are generally referred to as biased signaling, and the ensuing models describe how distinct proteases with distinct cleavage sites induce protease-specific responses via the same PAR [13, 14].
In contrast with PAR-activating proteases, other proteases cleave PARs at cleavage sites that are not related to signaling. Under these conditions, shedding of the PAR1 terminus, which removes the thrombin activation site, was first recognized as a mechanism for rendering platelets irresponsive to thrombin [15]. These truncated PARs can no longer be proteolyticaly activated, but remain activated by ligands from adjacent PARs [16]. Alternatively, truncated PARs bind soluble peptides with affinity for ECL2 by mimicking the tethered ligand. Both mechanisms result in receptor activation [17, 18]. Multiple ECL2-binding agonist peptides have been described and shown to induce signaling from truncated and uncleaved PARs (see agonist peptides in Tables 5, 6, 7).
PAR activation by proteolytical cleavage
PAR-cleaving proteases are a focus of many current studies. Whereas some PAR-cleaving proteases produce N-terminal components with regulatory roles, others render the receptors irresponsive to further protease exposure as shown in Fig. 2 and summarized in Tables 1, 2, 3 and 4. Important proteases are discussed below.

Proteolytic PAR cleavage. a N-terminal sequences of human PARs (PAR1–4) containing potential cleavage sites. b Proteolytic cleavage of PARs by soluble exogenous proteases exposes new N-terminal sequences that serve as tethered ligands for G protein dependent activation of receptors. Alternatively, proteolytic cleavage at other sites destroys the function of the receptor to prevent intracellular signal transduction
Mammalian proteases
Serine proteases
Thrombin, the key protease of coagulation, is generated by proteolytic cleavage of zymogen prothrombin. Although thrombin production predominantly occurs on platelets and subendothelial vascular walls, extravascular thrombin has been detected in synovial fluid [19] and around tumors [20]. Thrombin has long been known to activate platelets, and the discovery of PAR1 initiated research into the underlying molecular mechanisms. PAR1 contains a hirudin-like domain, which has a high affinity thrombin binding site and recruits thrombin via exosite I. This interaction enables thrombin to specifically and efficiently activate PAR1 [6]. Similarly, PAR3 contains a hirudin-like thrombin recruitment site, which results in cleavage [9, 21]. In other studies, mouse PAR3 maintained thrombin recruitment activity but lost its receptor function, as discussed above [22,23,24]. Thrombin also cleaves and activates PAR4, which, in contrast with PAR1, lacks a hirudin-like domain. Thus, higher concentrations of thrombin activate PAR4 and initiate intracellular signaling [10]. PAR2 is considered the only PAR that resists cleavage or activation by thrombin [4, 25], although emerging evidence suggests that at very high concentrations (100–500 nM), thrombin may directly cleave and activate PAR2 [26, 27].
In contrast with thrombin, the anticoagulant protease activated protein C (aPC) binds to the co-receptor endothelial protein C receptor (EPCR) to promote the cleavage and activation of co-localized PAR1 [28, 29] and induce anti-apoptotic and protective effects on endothelial barrier permeability [29,30,31,32,33]. Compartmentalization of PAR1 and co-localization with EPCR in calveolae is crucial for efficient cleavage by aPC [13]. Moreover, aPC cleaves PAR3 in humans and mice [21, 34, 35] and acts as a PAR3 shedding protease that prevents thrombin-induced barrier disruption [21]. However, the dependency of aPC cleavage of PAR3 on EPCR remains controversial [21, 35]. Similar to aPC, coagulation factor Xa binds EPCR and mediates proteolytic activation of PAR1 and PAR3 [21, 28, 36,37,38,39]. In addition, EPCR-bound factor Xa reportedly cleaves PAR2 and initiates inflammatory signaling [40]. PAR2 was also shown to be activated by tissue factor (TF)-bound coagulation factor VIIa [40,41,42]. Yet recent studies suggest that the TF-VIIa complex does not directly activate PAR2, and rather activates matriptase, which cleaves and activates PAR2 [42,43,44]. Anti-inflammatory signaling was also previously related to PAR1 cleavage by EPCR-bound VIIa [45, 46]. Taken together, these studies indicate that TF-Xa–VIIa complexes activate PAR1 and PAR2 [47].
Trypsins are PAR-activating proteases with roles as major digestive enzymes in the duodenum [48]. Trypsin is also secreted by epithelial cells, nervous system cells [49], and tumor cells [50, 51]. Trypsins may also be involved in cell growth and coagulation, as suggested by secretion from human vascular endothelial cells [52]. Trypsin cleaves human PAR1 and PAR4 at putative protease cleavage sites, and thereby prevents thrombin signaling in endothelial cells and platelets [4, 53]. Trypsin is the major PAR2 cleaving protease that initiates inflammatory signaling [2, 7].
Tryptase is the main protease of mast cells, and activates PAR2 by proteolytic cleavage to induce calcium signaling and proliferation [54,55,56,57]. The source tissue of tryptase reportedly plays an important role in the cleavage and induction of tryptase-activated PAR signaling, reflecting differences in posttranslational modifications, such as glycosylation and sialic acid modifications [54, 58]. Tryptase induces calcium signaling via PAR1 when PAR2 is co-expressed, but cannot activate human platelets, suggesting that tryptase does not directly cleave PAR1 [54,55,56,57]. Chymase is a mast cell serine protease that also cleaves PAR1 in human keratinocytes and fibroblasts, and thus prevents thrombin sensitivity [59]. Moreover, the epithelial serine protease matriptase cleaves and initiates inflammatory responses in human and mouse keratinocytes and in Xenopus oocytes overexpressing human PAR2 [44, 60,61,62,63].
PARs have been identified as substrates of kallikreins, which are serine proteases that have been related to various inflammatory and tumorigenic processes [64]. Kallikrein-4 increases intracellular calcium levels via PAR1 and PAR2, but activates PAR1 most efficiently [65]. Kallikrein-14 induces calcium signaling via PAR1, PAR2, and PAR4, but can also shed PAR1 to prevent signaling. Rat platelets are activated by kallikrein-14 via the proteolytic cleavage of PAR4, but are not activated by kallikrein-5 and kallikrein-6 [66]. Instead, neurotoxic effects of kallikrein-6 were inhibited by blocking PAR1 and PAR2, indicating a direct proteolytic role in PAR activation [67].
Neutrophils are mobilized to sites of inflammation and infection, where they modulate inflammatory signaling, in part by secreting PAR-cleaving proteases. The neutrophil serine protease cathepsin G prevents thrombin-induced effects by cleaving PAR1 into non-functional parts [68, 69]. In contrast, cathepsin G reportedly induced chemoattractant signaling via PAR1, further supporting the role of cathepsin G in PAR1 activation [70]. Another unexpected observation of cathepsin G was that cleavage sites differ between recombinant and native human PAR2 [26, 71, 72]. These discrepancies may reflect the influence of cell types and posttranslational modifications on PAR cleavage. Studies in mice and humans show that platelet activation by cathepsin G is dependent on PAR3 and PAR4 [71, 73, 74]. Cathepsin G also cleaves and activates PAR4 on endothelial cells [75]. The neutrophil proteases elastase and proteinase-3 cleave recombinant PAR1 and PAR2 at various sites [26, 72]. Recently, rat elastase was shown to cleave and activate PAR1, although sequences of rat and human PAR1 have low homology [76]. In contradiction with neutrophil proteases that prevent PAR signaling at sites of inflammation, monocytes secret the protease cathepsin S, which initiates inflammatory signaling by cleaving PAR2 [72, 77, 78]. Low concentrations of the fibrinolytic protease plasmin prevent platelet activation by cleaving PAR1, whereas high concentrations of plasmin lead to the cleavage and activation of PAR1 [79]. Plasmin also cleaves PAR2 and prevents subsequent activation by trypsin [26, 80].
The serine proteases granzyme A and granzyme B induce intracellular signaling pathways that lead to neuronal death via PAR1 [81, 82]. Recently, granzyme K was also shown to activate PAR1 and promote inflammatory endothelial signaling [83, 84]. Few studies show activation of PAR1 by proteases of the granzyme family, and the details of this interaction remain poorly characterized.
Cysteine proteases
Calpain-1 is a calcium-dependent cysteine protease that has been associated with inflammatory disorders, and initiates calcium signaling pathways by activating PAR1 [26]. At very high concentrations, calpain-2 was also shown to cleave PAR2, and the authors suggested that this cleavage event inactivated PAR2 [26]. Recently, calpain-1 was shown to be induced by thrombin-activated PAR1, and subsequently regulated the internalization of PAR1 [85].
Metalloproteases
Matrix metalloproteases (MMPs) are known to be involved in various inflammatory- and cancer-related conditions. MMP-1 cleaves human PAR1 and initiates platelet activation [86,87,88,89]. MMP-1 also regulates cancer cell activities depending on PAR1 availability [90]. Similarly, MMP-2 cleaves human PAR1 and enhances platelet activation [91], and MMP-3, MMP-8, and MMP-9 were shown to induce platelet activation via PAR1 [92]. Whether these three MMPs cleave PAR2 is not clear, although PAR2 activation by trypsin induced secretion of MMP-9 in human airways, suggesting that MMP-9 is a PAR2-activating protease [93]. In mice, PAR1 expression was regulated by MMP-12, and activated PAR1 increased MMP-12 secretion [94, 95]. A similar feedback loop involving MMP-12 and PAR2 has been reported in mice [96]. Moreover, MMP-13 was shown to activate PAR1 and induce intracellular signaling [87], and thrombin-induced activation of PAR1 and PAR3 was associated with increased levels of MMP-13 in human chondrocytes [24].
In addition to coagulation and inflammation, PAR activation may play roles in human germ cells, where the serine protease testisin activates PAR2 and induces calcium signaling and ERK1/2 activation. This interaction may play roles in the regulation of ovarian and testicular cancer, as suggested previously [97, 98].
Non-mammalian proteases
Exogenous proteases from various species that modulate PAR activation are disscues in the following section and are summarized in Fig. 3.

Non-mammalian exogenous proteases induce PAR-driven pathological effects. Various proteases are secreted from bacteria, amoebae, insects, plants, fungi, and snakes, and can cleave PARs and modulate signal transduction, leading to inflammation, thrombosis, or pain
Bacterial proteases
Endogenous mammalian proteases are not the only regulators of PAR activation. Indeed, both pathogenic and commensal bacteria secret various proteases that cleave PARs and act as inflammatory modulators [99]. In this section, we describe bacterial proteases that either activate PARs, and thus allow bacteria to penetrate host barriers, or inactivate PARs to prevent inflammatory signaling by the host.
The human pathogen Pseudomonas aeruginosa secrets two PAR-cleaving proteases with contrasting effects. The exoprotease LepA cleaves and activates PAR1, PAR2, and PAR4, and subsequently induces nuclear factor kappa B (NFκB) promoter activity [100], whereas cleavage by elastase EPa inactivates PAR2 to prevent inflammation in lungs [101].
The streptococcal pyrogenic exotoxin B (SpeB) of Group A Streptococcus also inactivates PAR1 by cleaving it, and thereby renders human platelets unresponsive to thrombin [102]. In mice, proteases of Streptococcus pneumoniae cleaved PAR2 and facilitated the spread of the pathogen from the airways into the blood stream [103]. PAR1 has also been associated with S. pneumonia-mediated sepsis in mice, although direct cleavage of PAR1 was not shown [104, 105]. Pulmonary inflammation from S. pneumoniae infections is reduced in PAR4 knockout mice [106], further supporting this causal link.
Inflammation-associated periodontal diseases are predominantly induced by the Porphyromonas gingivalis cysteine protease gingipain R, which activates PAR2 [107, 108]. Subsequently, gingipain R activates PAR1 and PAR4, and thereby, human platelets [109,110,111]. This mechanism may also explain associations between periodontitis and cardiovascular events [112].
In addition, supernatants from Propionibacterium acnes cultures initiated inflammatory signaling in human keratinocytes via PAR2 [92]. The virulence of P. acnes was also reduced in PAR2 knockout mice [113], further suggesting that PAR2 is involved in bacterial infections.
Serralysin is a matrix metalloprotease expressed by Serratia marcescens, and induced inflammation in human airway cells via PAR2 in vitro [114].
Finally, Bacillus thermoproteolyticus rokko secretes the metalloprotease thermolysin, which cleaves and inactivates PAR1 to prevent thrombin-induced signaling in rat astrocytes [115, 116]. The in vitro effects of PAR2-cleavage by thermolysin, however, vary between cell lines [116].
Amoeba proteases
In acanthamoebic keratitis, PAR2 triggers inflammation following secretion of the plasminogen activator (aPA) by Acanthamoeba strains, leading to induction of IL-8 in human corneal epithelial cells [117].
Reptile proteases
Following snakebites, coagulation disorders in humans and mice occur due to the presence of venom proteases. In Proatheris superciliaris bites, venom proteases activate platelets by activating PAR1 and PAR4 [118]. Bothrops atrox and B. jararaca are snake species of the family viperidae. These snakes secrete the serine proteases PA-BJ and thrombocytin, which activate human platelets via PAR1 and PAR4 [119].
Insect proteases
Several cysteine and serine proteases from insects induce inflammation-associated diseases such as asthma. For example, dust mite allergens contain the serine proteases DerP2, DerP3, and DerP9 [120] and the cysteine protease DerP1. DerP1 induces PAR2-dependent signaling, whereas thrombin-induced PAR1-signaling is prevented by these proteases in human epithelial cells [121]. DerP3 was also recently shown to activate PAR4, and this process was associated with allergies to dust mites [122].
Similar to proteases from house dust mites, three serine proteases (E1–E3) from cockroach extracts activate PAR2 and induce inflammatory signaling in mice and humans [123,124,125].
Fungal proteases
Pen C is a serine protease from Penicillium citrinum that induces IL-8 in human airway cells by activating PAR1 and PAR2 [126]. Proteases from Aspergillus fumigatus have also been shown to prevent PAR2-dependent inflammation [127]. Moreover, serine proteases from Alternaria alternate induced calcium signaling in human bronchial cells and induced inflammation in mice by secreting IL-33 following PAR2 activation [128,129,130].
Plant proteases
Bromelain is a mixture of cysteine proteases that is extracted from pineapple which is used as a PAR-independent anti-inflammatory agent [131]. Bromelain cleaves PAR2 and thereby prevents the associated inflammatory signaling [132]. In another study, however, bromelain, ficin, and papain activated PAR2 and PAR4 by proteolytic cleavage, leading to increased intracellular calcium levels [133]. Thus, further studies are required to further clarify the modes of action of pineapple proteases.
Cleavage-independent PAR activation by agonist peptides
Independent of proteolytic cleavage, PARs can be activated by synthetic soluble ligands corresponding with cleaved N-terminal sequences, or can be transactivated by cleavage-generated N-terminal regions of homo- or heterodimer partners.
Synthetic peptides that mimic the first six amino acids of tethered N-terminal ligands can act as agonist peptides that activate PARs in the absence of cleavage events [11, 18, 134]. Specific activation of PARs by a soluble agonist peptide was first shown for human PAR1 with the peptide SFLLRN [6, 18]. However, this peptide also activated PAR2 [135,136,137] and therefore various peptides were tested for specific PAR1 activation. Yet, PAR1 was the most specifically and efficiently activated by TFLLRN [138]. In addition to thrombin agonist peptides, other PAR1 agonist peptides have been identified. In particular, the peptide NPNDKYEPF reproduced the effects of aPC [28], and PRSFFLRN corresponds with the N-terminal peptide generated by MMP-1 [86]. SLIGKV corresponds with the trypsin cleaved N-terminal region of human PAR2. However, the corresponding rat N-terminus SLIGRL is a more specific and efficient PAR2 agonist in rodents and humans [136, 139], and only the synthetic peptide LIGRLO achieved this effect more efficiently than SLIGRL in humans [140]. The roles of ECL-2 in specific PAR activation have been shown using labeled PAR2 agonist peptides [141, 142]. Because the thrombin generated PAR3 peptide does not activate the G protein autonomously, no such agonist peptides have been identified to date [9, 143]. GYPGKF corresponds with the thrombin-cleaved human PAR4 and has weak activity as an agonist [144]. But replacement of the first amino acid glycine (G) with alanine (A) induced PAR4 by 10-fold. This peptide may be suitable as a platelet activator in humans and mice [145].
Several models of PAR–PAR interactions have been proposed and extensively studied based on PAR transactivation by agonist peptides [146]. When PAR1 is blocked on endothelial cells, however, thrombin, and not the PAR1-specific agonist peptide TFLLRN, induces signaling, reportedly by facilitating the heterodimerization of PAR1 and PAR2 [147]. Thrombin activation of the PAR1–PAR2 heterodimer leads to constitutive internalization and activation of β-arrestin by the PAR1 C-tail [146]. Accordingly, the required co-localization of PAR1 and PAR2 was shown in a human overexpression system, in mice studies of sepsis, and in PAR1–PAR2-driven cancer growth in a xenograft mouse model [148, 149]. In other studies, stable heterodimerization of human PAR1 and PAR4 was shown in platelet cells, and thrombin accelerated platelet activation under these conditions [150, 151]. Similar studies of mouse platelets showed efficient activation of platelets by thrombin in the presence of PAR3–PAR4 heterodimers [143]. Consistent with the thrombin-cleaved PAR3 peptide, which is not self-activating, PAR3 signaling was observed in the presence of PAR1 or PAR2 [22, 23, 34, 152]. Yet, heterodimerization influenced signal transduction and PAR membrane delivery due to enhanced glycosylation [153].
In addition to activation by heterodimerization, PARs interact with other receptors, such as ion channels, other G protein-coupled receptors (GPCRs), receptor tyrosine kinases (RTKs), receptor serine/threonine kinases (RSTKs), NOD-like receptors, and TLRs [154]. In particular, PAR2 initiated inflammatory signaling pathways, resulting in pain due to transactivation of the ion channels TRPV1 and TRPV4 in humans and mice [155,156,157,158,159]. Similar inflammatory effects follow transactivation of the RTKs EGFR and VEGFR by PAR2 and PAR4 [160,161,162,163]. Bacterial interactions with PARs suggest important roles of PARs in infectious disease. In agreement, TLRs recognize bacteria-derived molecules and contribute to innate immunity [164, 165]. Moreover, direct interactions of PAR2 with TLR3 and TLR4 were necessary for inflammatory responses to LPS in human cell lines and knockout mice and rats [166,167,168,169,170,171].
PAR signaling
Activation pathways
PARs belong to a large family of GPCRs and induce multiple signaling pathways after coupling with heterodimeric G proteins. Activation of the Gα-subunit due to the exchange of a guanine from GDP to GTP results in dissociation of the Gβγ-dimer and activation of downstream pathways [172, 173].
Following proteolytic cleavage or induction of agonist peptides, the engaged signaling pathways vary between tissues, cell lines, and the availability of co-receptors for transactivation. Depending on the ligand, specific α-subunits are activated, and these regulate subsequent cellular functions as summarized in Fig. 4. For example, thrombin-stimulated PAR1 activates the small GTPase protein RhoA via ERK1/2 kinases, but not via Rac1, whereas aPC-stimulated PAR1 induces Rac1 via Akt kinase, but not via RhoA [13, 174,175,176]. Moreover, in accordance with PAR1 cleavage sites, aPC prevents thrombin-induced RhoA signaling [16]. However, in contrast with thrombin-induced RhoA activation on platelets and endothelial cells, PAR1-agonist peptides and thrombin activated the inhibitory G protein Gi which leads to the inhibition of adenylyl cyclase in human fibroblasts [177, 178]. Other studies indicate that PAR2 activation is less tissue specific than PAR1 activation, and trypsin and VIIa cleaved PAR2 and activated Gαq and Gi, resulting in calcium influx, MAPK activation, and inflammatory signaling [8, 179].

G protein-coupled signaling induced by PAR activation. Depending on the tethered ligand, activated PAR couples with G protein α-subtypes. Gαq activates phospholipase Cβ (PLCβ), which mobilizes calcium. This further activates MAPKs (ERK1/2) and induces Ras signaling. Primarily, Gα12/12 and Gaq activate the Rho pathway. Gαi inhibits the activation of adenylyl cyclase, which leads to reduced production of cAMP. In contrast, the βγ-subunit functions as a negative regulator when bound to the α-subunit. After receptor activation, subunits separate, and the βγ-subunit interacts with other proteins, thereby activating or inhibiting signaling
Signaling by tethered ligands can differ from that generated by corresponding soluble agonist peptides. For example, thrombin-cleaved PAR1 activated Gα12/13 and Gαq and induced Rho and Ca2+ signaling, whereas the PAR1-agonist peptide activated only Ga12/13 and downstream RhoA-dependent pathways that affected endothelial barrier permeability [180]. Similar observations of human platelets suggested that platelet activation followed coupling of thrombin-activated PAR1 with multiple heterotrimeric G protein subtypes, including Gα12/13 and Gαq [181,182,183]. Moreover, trypsin and the PAR2-agonist peptide induced ERK1/2 signaling and inflammation by activating PAR2 [29, 180, 184,185,186]. β-arrestins also play major roles in PAR-induced signaling independently of G protein activation. For instance, aPC-activated PAR1 induces cytoprotective effects by recruiting β-arrestin in endothelial cells. Thus, aPC cleavage fails to protect β-arrestin deficient cells from the effects of thrombin [187, 188]. In addition, multiple studies show that activated PAR2 co-localizes with β-arrestin-1 and arrestin-2 and induces ERK1/2 signaling [77, 189,190,191].
Desensitization and termination
PAR activation is regulated by internalization and proteolytic desensitization, which limits the duration of signaling. For instance, PAR1 is constitutively internalized and recycled or agonist-induced internalized and degraded as described in [192, 193] and shown in the scheme of Fig. 5. As discussed above, some PAR-cleaving proteases abolish receptor responses by removing (shedding) or destroying the tethered ligands. For example, PAR1 is inactivated following cleavage by cathepsin G, and thrombin activation is hence prevented, allowing the formation of clotting under inflammatory conditions.

PAR trafficking. Activation-independent constitutive or agonist-induced internalization regulates PAR1 signaling
Depending upon proteolytic cleavage, PAR1 rapidly internalizes or accumulates on the cell surface [194, 195]. Activated PAR1 is internalized via clathrin- and dynamin-dependent mechanisms, and is sorted from early endosomes to lysosomes for degradation [196,197,198,199]. Although the mechanisms that terminate PAR1 signaling are not clearly understood, this process is known to involve phosphorylation, ubiquitination, and recruitment of β-arrestin [200,201,202,203,204]. In contrast with PAR1, activated PAR2 is not constitutively internalized [205]. Thus, to prevent persistent signaling upon activation, PAR2 is phosphorylated and ubiquitinated and then binds β-arrestin before being internalized and degraded [206,207,208]. Under these conditions, the activated and internalized PAR2 is not recycled and instead induces β-arrestin-dependent endosomal ERK1/2 signaling in the cytoplasm [189, 191, 209]. Thus, large cytoplasmic stores of newly generated PAR2 are required for rapid externalization and activation on cell membranes [210]. Although less is known about how PAR4 signaling is terminated, recent observations suggest that PAR4 internalization is independent of β-arrestin and slowly occurs via clathrin- and dynamin-dependent pathways [211]. In agreement, human platelets internalized PAR4 much slower than PAR1, and exhibited prolonged PAR4 signaling activity [212]. Moreover, growing evidence indicates that PAR–PAR heterodimerization is important for internalization, and that the underlying mechanisms include PAR2-dependent glycosylation of PAR4, thus affecting membrane transport [153]. Upon internalization, endosomal PAR4 dimerizes with the purinergic receptor P2Y12 and induces Akt signaling by recruiting β-arrestin within endosomes [213].
Depending on stimuli, PAR expression patterns are regulated by complex combinations of cell surface presentation, endocytosis, vesicle born or recycled (i.e., re-exocytosed) receptors, and trafficking modes that are linked to posttranslational modifications of PAR.
Role of PARs in inflammation
With the current increases in the prevalence of inflammatory diseases, published in in vitro and in vivo studies of the roles of PARs in inflammation have become more numerous. These are reviewed below.
Systemic inflammation and inflammatory cells in the cardiovascular system
PARs are critical for the interplay between clotting proteases of platelets, endothelial cells, and vascular smooth muscle cells that regulate hemostasis, vascular barrier function, vascular tone, vascular homeostasis, cell adhesion, and inflammatory responses [150]. The roles of PARs in these processes vary significantly between species. Specifically, whereas functional PAR1 and PAR4 are expressed in human platelets [214], PAR1, PAR3, and PAR4 have been found in guinea pig platelets [215]. Whereas mouse and rat platelets lack PAR1, they are activated at low concentrations of thrombin, which is recruited by PAR3 onto the surface of platelets and then efficiently activates PAR4 [4]. Due to interspecies differences in PAR expression, mouse and rat studies of PARs are difficult to translate to humans. PARs in endothelial cells contribute positive regulatory signals for endothelial adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and E-selectin [216, 217], all of which promote vascular barrier function. As a counterpart of intravascular cells, PAR4 induces leukocyte migration [75], and PAR2 expressed on macrophages promotes inflammatory modulators such as interleukin-8 (IL-8) [218]. These modes of signaling all contribute to a complex PAR-mediated interplay of endothelial cells that is orchestrated by intravascular cells and cytokine secretion. In addition, PARs, particularly PAR1, regulate vascular barrier function, and hence, extravasation of macromolecules such as complement proteins and antibodies. In addition, thrombin-mediated activation of PAR1 increases endothelial barrier permeability by activating mitogen-activated protein kinases (MAPKs) [219]. Although this effect is reversed by activated protein C (aPC)-mediated activation of PAR1 [28, 174, 175, 220]. Thrombin further promotes prostaglandin 2 (PGE2) secretion, and consequent endothelial barrier permeability [221]. Similarly, PAR1 activation increased vascular leakage in a murine model [222]. Inflammatory mediators, such as tumor necrosis factor alpha (TNFα), were shown to regulate the expression of endothelial PAR2, and the authors suggested that these data were indicative of barrier protective effects of PAR2 [223]. Several other studies show that PAR2 activation induces endothelium-dependent relaxation in blood vessels of mice and in arteries of rats [224,225,226,227,228]. In contrast, dual activities of PAR2 on blood vessels were reported in a study of rats [229]. In this line, thrombin-activated PAR1 induced the expression of vascular endothelial growth factor in smooth muscle cells [230], thus revealing the relationship between coagulation and vascular growth. Although the roles of PARs in the development of arteriosclerosis are yet to be elucidated, PAR2 and PAR4 were induced in human arteries under inflammatory conditions [223], suggesting important roles of PARs in vascular inflammation.
Chronic inflammation of the gastrointestinal tract
In the gut lumen, human and bacterial proteases are both present at high concentrations. Similar to endothelial barriers, proteases regulate intestinal barrier permeability via PARs, all four of which are expressed by cells of the gastrointestinal tract [9, 224, 231, 232]. Trypsins and tryptases are prominent intestinal proteases, suggesting likely involvement of PAR2 as a major receptor of intestinal inflammation. In accordance, intestinal tight junctions are disrupted by PAR2-activating proteases, leading to inflammatory signaling in humans and rats [139, 206, 233, 234]. Although the roles of PARs in irritable bowel syndrome (IBS) and inflammatory bowel diseases remain unclear, roles of PARs in intestinal barrier function have been described. Specifically, PAR1 and PAR2 regulated permeability and chloride secretion, which are involved in diarrhea and constipation in IBS patients [234,235,236]. In addition, activated endosomal PAR2 caused persistent pain in a mouse model of IBS [209].
Inflammatory diseases of the respiratory system
It has long been suggested that PARs are involved in the pathophysiology of respiratory disorders, reflecting observations of elevated levels of PAR-activating proteases, such as thrombin and tryptase, in bronchoalveolar lavage fluid from patients with pulmonary inflammation [237, 238]. In a sheep asthma model and in asthmatic patients, tryptase inhibitors reduced inflammation [239, 240], further indicating important roles of PAR2 in respiratory disease. These roles of PARs are also suggested by the prominence of a variety of non-mammalian PAR-activating proteases, such as those of house dust mites and cockroaches [120, 123, 124]. Expression of PAR1, PAR2, and PAR4 on bronchial epithelial and smooth muscle cells induced inflammatory signaling in multiple studies [55, 121, 241,242,243,244,245]. PAR2 is also upregulated in epithelial cells of patients with asthma and chronic obstructive pulmonary syndrome (COPD) [246, 247]. Whether PAR2 activation results in bronchoconstriction or dilatation remains controversial, in part owing to interspecies differences and tissue dependencies [242, 248, 249]. In humans, however, PAR1-agonist peptides with thrombin, and a PAR2-agonist peptide with trypsin and tryptase, induced bronchoconstriction by inducing Ca2+ signaling in airway smooth muscle cells [241, 244]. Moreover, the long-term activation of PAR1 and PAR2 led to pulmonary fibrosis in mice models [250].
Inflammatory skin diseases
High concentrations of exogenous proteases are present on the skin of various species, and these may activate PARs to regulate epidermal permeability and barrier function [251]. Indeed, epidermal inflammation has been linked to PAR1 and PAR2 activation in keratinocytes, which comprise the epidermal barrier with sub-epidermal skin fibroblasts [179, 252, 253]. Subsequent release of IL-8, IL-6, and granulocyte macrophage colony-stimulating factor (GM-CSF) was also observed previously [254], potentially involving NFκB activation [255]. In addition, the inflammatory roles of PAR2 have been demonstrated in mice models of atopic dermatitis due to elevated tryptase and PAR2 expression levels [256, 257]. Similar to studies in mouse models, PAR2 was upregulated in patients with atopic dermatitis, and PAR2 agonists increased itch, causing irresponsiveness of sensory nerves to therapy with antihistamines [258].
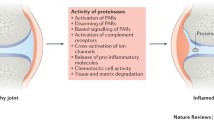
Rheumatic disease
“Rheumatic disease” is a common term for autoimmune diseases that affect joints, bones, and muscles. Although rheumatic disorders are numerous, some of the common underlying symptoms include chronic joint inflammation, stiffness, and pain [259]. Currently, PAR2 is the only PAR that has been associated with the development of rheumatic diseases [260]. Direct roles of PAR2 in rheumatic diseases were first indicated in 2003 in a mouse study by Ferrell et al. [261]. In their study, a PAR2-agonist peptide induced strong inflammatory effects in wt mice, causing joint swelling and synovial hyperemia, whereas joint swelling was absent in PAR2 deficient mice [261]. Similarly, in patients with rheumatoid arthritis, PAR2 is upregulated in inflamed tissues [262]. Further increases in PAR2 expression were noted in monocytes, and the PAR2-agonist peptide upregulated IL-6. In contrast, PAR2 expression was decreased after treatments with antirheumatic drugs [263], further supporting the role of PAR2 in rheumatic disease.
PAR modulators as targets for therapy
The complexity of PAR regulation is indicated by the culmination of specific proteolytic cleavage modes (inactivating or activating), protease inhibitors, and cofactors, and with the effects of PAR glycosylation and dimerization (Fig. 1). In this section we discuss classes of agonists and antagonists that have been tested as PAR modulators for use as therapeutic agents as summarized in Fig. 6 and Tables 5, 6 and 7.

PAR modulators. Pharmacological substances, such as 1) peptides and peptidomimetics, 2) blocking antibodies, 3) small molecules, 4) pepducins, and 5) parmodulins are used as therapeutic agents that affect PAR activities
Peptide agonists and antagonists are short synthetic peptides that mimick the PAR-tethered ligand that is liberated by proteolytic cleavage, as described above. These peptides either induce signal transduction or prevent cleavage-dependent signaling following PAR rapid internalization, and some C- or N-terminal modifications of soluble ligand sequences have resulted in increased activation efficiency [18]. Peptidomimetic antagonists are small protein-like chains that mimick the tethered ligands of PARs, and were recently used as PAR modulators for the first time [264].Soon after PARs were discovered, PAR1 blocking antibodies were reported [265], and these blocked protease binding and or the cleavage site of the receptor. Non-peptide small molecules, such as the PAR1 antagonists vorapaxar [266] and atopaxar [267], also interact with PARs, mainly via ECL2.Only two classes of intracellular PAR antagonists have been developed to date. Pepducins are cell penetrating palmitoylated peptides that were derived from the intracellular loop of PAR, and these interfere with G protein binding [268]. Parmodulins, in contrast, are small molecules that bind PARs at the G protein binding pocket of the C-tail to compete with Gαq subunits, but not with other Gα subunits [269].
Examination of agonists and antagonists in vitro and in preclinical studies (Tables 5, 6 and 7)
Clinical studies
Despite the importance of PARs in various pathophysiological conditions, few PAR modulating tools have been tested in clinical studies, and even fewer have been established for treatment. Since the identification of PAR1 as a platelet thrombin receptor, an abundance of research has been conducted to identify PAR1 antagonists that can block platelet activation and prevent thrombotic cardiovascular events. The first clinically approved PAR1 antagonist was the small-molecule antagonist vorapaxar [266]. Phase II clinical trials of this agent showed reduced risks for myocardial infarction in patients treated with vorapaxar in combination with standard antiplatelet therapy. Moreover, the risks of bleeding complications were not significantly increased [270]. Subsequently, two large-scale phase III multicenter, randomized, double-blind, placebo-controlled studies of vorapaxar (ZONTIVITY, SCH530348) were performed. In the Thrombin Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events–Thrombolysis in Myocardial Infarction 50 (TRA 2°P-TIMI 50; details at www.ClinicalTrials.gov; NCT00526474) study, the rate of cardiovascular events at the second efficacy endpoint were significantly reduced by vorapaxar in combination with standard antiplatelet therapy [271]. Furthermore, in the Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome (TRACER; details at www.ClinicalTrials.gov; NCT00527943) study, vorapaxar reduced the hazard of first myocardial infarction of any type in patients who were treated within 24 h of having symptoms of a cardiovascular event. However, in the TRACER study, vorapaxar failed to prevent secondary ischemic events [272]. Because vorapaxar increased bleeding complications in the clinical setting, the alternative PAR1 antagonist atopaxar (E5555) [267] was tested in a phase II clinical trial called (Lessons From Antagonizing the Cellular Effects of Thrombin-Acute Coronary Syndromes (LANCELOT-ACS; details at www.ClinicalTrials.gov; NCT00548587) study [273]. Atopaxar inhibited platelet aggregation in ACS patients in a dose-dependent manner, and caused no side effects of abnormal platelet activation, such as bleeding [274, 275]. Yet, patients receiving atopaxar had dose-dependent increases in liver abnormalities [273].
To prevent the bleeding problems that arise from treatments with PAR1 antagonists, a new class of PAR1 antagonist was designed, and the member pepducin PZ-128 (P1-pal7) was tested in a phase I trial [276]. This study showed no reduction in platelet aggregation, but the platelet blocking effect of PZ-128 was reversible ex vivo in the presence of saturating concentrations of the PAR1 agonist peptide SFLLRN. Based on these promising findings, the new PAR1 blocking agent PZ-128 was considered in the coronary artery disease study Thrombin Receptor Inhibitory Pepducin-Percutaneous Coronary Intervention (TRIP-PCI). Data from this phase II trial are not yet available (details at www.ClinicalTrials.gov; NCT02561000).
As an alternative to PAR1 targeted antithrombotic drugs, the PAR4 small-peptide antagonist BMS-986120 reduced reversible thrombus formation ex vivo in a phase I trial [277]. Consequently, this promising anticoagulant PAR4 antagonist is currently being compared with a standard anticoagulant drug in a phase II study of stroke recurrence (details at www.ClinicalTrials.gov; NCT02671461).
Conclusion
Since the identification of PARs in the 1990s, studies of the complex mechanisms of PAR activation have been abundant, and these have clarified the roles of PARs in inflammatory disease. Various mammalian and non-mammalian proteases have also been recognized as PAR-mediated regulators of physiological and pathophysiological processes. Despite the development of various PAR modulators, few have been approved for therapeutic use. Obstacles to this therapeutic strategy include species differences in PAR expression and limited bioavailability of modulators in vivo and in clinical studies. Further research is needed to identify specific and efficient anti-inflammatory PAR modulators.
Change history
06 November 2019
Following the publication of this article [1], the authors reported an incorrect citation in the following sentence in the “Metalloproteases” sub-section:
Abbreviations
- AC:
-
Adenylyl Cyclase
- AKT:
-
Protein kinase B
- aPC:
-
Activated Protein C
- COPD:
-
Chronic Obstructive Pulmonary Syndrome
- COX:
-
Cyclooxygenase
- ECL:
-
Extracellular Loop
- EGFR:
-
Epidermal Growth Factor Receptor
- EPCR:
-
Endothelial Protein C Receptor
- ERK:
-
Extracellular Signal-regulated Kinase
- F2R:
-
Coagulation Factor 2 Receptor
- F2RL1:
-
Coagulation Factor 2 Receptor-Like 1
- F2RL2:
-
Coagulation Factor 2 Receptor-Like 2
- F2RL3:
-
Coagulation Factor 2 Receptor-Like 3
- FVIIa:
-
Activated Coagulation Factor VII
- FXa:
-
Activated Coagulation Factor X
- GM-CSF:
-
Granulocyte Macrophage Colony-stimulating Factor
- GPCR:
-
G Protein-Coupled Receptor
- IBD:
-
Inflammatory Bowel Disease
- IBS:
-
Irritable Bowel Syndrome
- ICAM:
-
Intercellular Adhesion Molecule
- IL:
-
Interleukin
- LepA:
-
Pseudomonas aeruginosa-Derived Large Extracellular Protease
- LPS:
-
Lipopolysaccharides
- MAPK:
-
Mitogen-Activated Protein Kinase
- MMP:
-
Matrix Metalloprotease
- NFκB:
-
Nuclear Factor kappa B
- PAR:
-
Protease-Activated Receptor
- penC:
-
Penicillium citrinum-Derived Alkaline Serine Protease C
- PGE2:
-
Prostaglandin E2
- PI3K:
-
Phosphatidylinositol-3-Kinase
- PLCβ:
-
Phospholipase C beta
- Rac1:
-
Ras-Related C3 Botulinum Toxin Substrate 1
- RhoA:
-
Ras homolog gene family, member A
- RSTK:
-
Receptor Serine/Threonine Kinase
- RTK:
-
Receptor Tyrosine Kinase
- SpeB:
-
Streptococcal Pyrogenic Exotoxin B
- TF:
-
Tissue Factor
- TLR:
-
Toll-Like Receptor
- TM:
-
Thrombomodulin
- TRPV:
-
Transient Receptor Potential Channels Vanilloid Subtype
- VCAM:
-
Vascular Cell Adhesion Molecule
- VEGF:
-
Vascular Endothelial Growth Factor
- VEGFR:
-
Vascular Endothelial Growth Factor Receptor
References
Bahou WF, Nierman WC, Durkin AS, Potter CL, Demetrick DJ. Chromosomal assignment of the human thrombin receptor gene: localization to region q13 of chromosome 5. Blood. 1993;82:1532–7.
Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci U S A. 1994;91:9208–12.
Schmidt VA, Nierman WC, Maglott DR, Cupit LD, Moskowitz KA, Wainer JA, Bahou WF. The human proteinase-activated receptor-3 (PAR-3) gene. Identification within a par gene cluster and characterization in vascular endothelial cells and platelets. J Biol Chem. 1998;273:15061–8.
Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, Farese RV Jr, Tam C, Coughlin SR. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–4.
Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–68.
Vu TK, Wheaton VI, Hung DT, Charo I, Coughlin SR. Domains specifying thrombin-receptor interaction. Nature. 1991;353:674–7.
Nystedt S, Larsson AK, Aberg H, Sundelin J. The mouse proteinase-activated receptor-2 cDNA and gene. Molecular cloning and functional expression. J Biol Chem. 1995;270:5950–5.
Nystedt S, Emilsson K, Larsson AK, Strombeck B, Sundelin J. Molecular cloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur J Biochem. 1995;232:84–9.
Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, Tram T, Coughlin SR. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature. 1997;386:502–6.
Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, Davie EW, Foster DC. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A. 1998;95:6642–6.
Gerszten RE, Chen J, Ishii M, Ishii K, Wang L, Nanevicz T, Turck CW, Vu TK, Coughlin SR. Specificity of the thrombin receptor for agonist peptide is defined by its extracellular surface. Nature. 1994;368:648–51.
Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–82.
Russo A, Soh UJ, Paing MM, Arora P, Trejo J. Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc Natl Acad Sci U S A. 2009;106:6393–7.
Zhao P, Metcalf M, Bunnett NW. Biased signaling of protease-activated receptors. Front Endocrinol (Lausanne). 2014;5:67.
Renesto P, Si-Tahar M, Moniatte M, Balloy V, Van Dorsselaer A, Pidard D, Chignard M. Specific inhibition of thrombin-induced cell activation by the neutrophil proteinases elastase, cathepsin G, and proteinase 3: evidence for distinct cleavage sites within the aminoterminal domain of the thrombin receptor. Blood. 1997;89:1944–53.
Ludeman MJ, Kataoka H, Srinivasan Y, Esmon NL, Esmon CT, Coughlin SR. PAR1 cleavage and signaling in response to activated protein C and thrombin. J Biol Chem. 2005;280:13122–8.
Chen J, Ishii M, Wang L, Ishii K, Coughlin SR. Thrombin receptor activation. Confirmation of the intramolecular tethered liganding hypothesis and discovery of an alternative intermolecular liganding mode. J Biol Chem. 1994;269:16041–5.
Scarborough RM, Naughton MA, Teng W, Hung DT, Rose J, Vu TK, Wheaton VI, Turck CW, Coughlin SR. Tethered ligand agonist peptides. Structural requirements for thrombin receptor activation reveal mechanism of proteolytic unmasking of agonist function. J Biol Chem. 1992;267:13146–9.
Chang MC, Lan WH, Chan CP, Lin CP, Hsieh CC, Jeng JH. Serine protease activity is essential for thrombin-induced protein synthesis in cultured human dental pulp cells: modulation roles of prostaglandin E2. J Oral Pathol Med. 1998;27:23–9.
Kohli M, Williams K, Yao JL, Dennis RA, Huang J, Reeder J, Ricke WA. Thrombin expression in prostate: a novel finding. Cancer Investig. 2011;29:62–7.
Burnier L, Mosnier LO. Novel mechanisms for activated protein C cytoprotective activities involving noncanonical activation of protease-activated receptor 3. Blood. 2013;122:807–16.
Kaufmann R, Schulze B, Krause G, Mayr LM, Settmacher U, Henklein P. Proteinase-activated receptors (PARs)--the PAR3 neo-N-terminal peptide TFRGAP interacts with PAR1. Regul Pept. 2005;125:61–6.
McLaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci U S A. 2007;104:5662–7.
Huang CY, Lin HJ, Chen HS, Cheng SY, Hsu HC, Tang CH. Thrombin promotes matrix metalloproteinase-13 expression through the PKCdelta c-Src/EGFR/PI3K/Akt/AP-1 signaling pathway in human chondrocytes. Mediat Inflamm. 2013;2013:326041.
Coughlin SR. How the protease thrombin talks to cells. Proc Natl Acad Sci U S A. 1999;96:11023–7.
Loew D, Perrault C, Morales M, Moog S, Ravanat C, Schuhler S, Arcone R, Pietropaolo C, Cazenave JP, van Dorsselaer A, Lanza F. Proteolysis of the exodomain of recombinant protease-activated receptors: prediction of receptor activation or inactivation by MALDI mass spectrometry. Biochemistry. 2000;39:10812–22.
Mihara K, Ramachandran R, Saifeddine M, Hansen KK, Renaux B, Polley D, Gibson S, Vanderboor C, Hollenberg MD. Thrombin-mediated direct activation of proteinase-activated receptor-2 (PAR2): another target for thrombin signaling. Mol Pharmacol. 2016;89(5):606–14.
Schuepbach RA, Madon J, Ender M, Galli P, Riewald M. Protease activated receptor-1 cleaved at R46 mediates cytoprotective effects. J Thromb Haemost. 2012;10(8):1675–84.
Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–2.
Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9:338–42.
Domotor E, Benzakour O, Griffin JH, Yule D, Fukudome K, Zlokovic BV. Activated protein C alters cytosolic calcium flux in human brain endothelium via binding to endothelial protein C receptor and activation of protease activated receptor-1. Blood. 2003;101:4797–801.
Mosnier LO, Griffin JH. Inhibition of staurosporine-induced apoptosis of endothelial cells by activated protein C requires protease-activated receptor-1 and endothelial cell protein C receptor. Biochem J. 2003;373:65–70.
Ruf W, Riewald M. Tissue factor-dependent coagulation protease signaling in acute lung injury. Crit Care Med. 2003;31:S231–7.
Ranjan S, Goihl A, Kohli S, Gadi I, Pierau M, Shahzad K, Gupta D, Bock F, Wang HJ, Shaikh H, et al. Activated protein C protects from GvHD via PAR2/PAR3 signalling in regulatory T-cells. Nat Commun. 2017;8(1):311.
Madhusudhan T, Wang H, Straub BK, Grone E, Zhou Q, Shahzad K, Muller-Krebs S, Schwenger V, Gerlitz B, Grinnell BW, et al. Cytoprotective signaling by activated protein C requires protease-activated receptor-3 in podocytes. Blood. 2012;119:874–83.
Riewald M, Kravchenko VV, Petrovan RJ, O'Brien PJ, Brass LF, Ulevitch RJ, Ruf W. Gene induction by coagulation factor Xa is mediated by activation of protease-activated receptor 1. Blood. 2001;97:3109–16.
Guo H, Liu D, Gelbard H, Cheng T, Insalaco R, Fernandez JA, Griffin JH, Zlokovic BV. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron. 2004;41:563–72.
Schuepbach RA, Riewald M. Coagulation factor Xa cleaves protease-activated receptor-1 and mediates signaling dependent on binding to the endothelial protein C receptor. J Thromb Haemost. 2010;8:379–88.
Stavenuiter F, Mosnier LO. Noncanonical PAR3 activation by factor Xa identifies a novel pathway for Tie2 activation and stabilization of vascular integrity. Blood. 2014;124:3480–9.
Camerer E, Gjernes E, Wiiger M, Pringle S, Prydz H. Binding of factor VIIa to tissue factor on keratinocytes induces gene expression. J Biol Chem. 2000;275:6580–5.
Morris DR, Ding Y, Ricks TK, Gullapalli A, Wolfe BL, Trejo J. Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res. 2006;66:307–14.
Rothmeier AS, Liu E, Chakrabarty S, Disse J, Mueller BM, Ostergaard H, Ruf W. Identification of the integrin-binding site on coagulation factor VIIa required for proangiogenic PAR2 signaling. Blood. 2018;131:674–85.
Rothmeier AS, Disse J, Mueller BM, Liu EB, Ostergaard H, Ruf W. Proangiogenic TF-FVIIa-PAR2 signaling requires Matriptase-independent integrin interaction. Blood. 2016;128:3756.
Le Gall SM, Szabo R, Lee M, Kirchhofer D, Craik CS, Bugge TH, Camerer E. Matriptase activation connects tissue factor-dependent coagulation initiation to epithelial proteolysis and signaling. Blood. 2016;127:3260–9.
Sen P, Gopalakrishnan R, Kothari H, Keshava S, Clark CA, Esmon CT, Pendurthi UR, Rao LV. Factor VIIa bound to endothelial cell protein C receptor activates protease activated receptor-1 and mediates cell signaling and barrier protection. Blood. 2011;117:3199–208.
Kondreddy V, Wang J, Keshava S, Esmon CT, Rao LVM, Pendurthi UR. Factor VIIa induces anti-inflammatory signaling via EPCR and PAR1. Blood. 2018;131:2379–92.
Riewald M, Ruf W. Mechanistic coupling of protease signaling and initiation of coagulation by tissue factor. Proc Natl Acad Sci U S A. 2001;98:7742–7.
Rinderknecht H. Activation of pancreatic zymogens. Normal activation, premature intrapancreatic activation, protective mechanisms against inappropriate activation. Dig Dis Sci. 1986;31:314–21.
Koshikawa N, Hasegawa S, Nagashima Y, Mitsuhashi K, Tsubota Y, Miyata S, Miyagi Y, Yasumitsu H, Miyazaki K. Expression of trypsin by epithelial cells of various tissues, leukocytes, and neurons in human and mouse. Am J Pathol. 1998;153:937–44.
Koivunen E, Huhtala ML, Stenman UH. Human ovarian tumor-associated trypsin. Its purification and characterization from mucinous cyst fluid and identification as an activator of pro-urokinase. J Biol Chem. 1989;264:14095–9.
Koivunen E, Ristimaki A, Itkonen O, Osman S, Vuento M, Stenman UH. Tumor-associated trypsin participates in cancer cell-mediated degradation of extracellular matrix. Cancer Res. 1991;51:2107–12.
Koshikawa N, Nagashima Y, Miyagi Y, Mizushima H, Yanoma S, Yasumitsu H, Miyazaki K. Expression of trypsin in vascular endothelial cells. FEBS Lett. 1997;409:442–8.
Nakayama T, Hirano K, Shintani Y, Nishimura J, Nakatsuka A, Kuga H, Takahashi S, Kanaide H. Unproductive cleavage and the inactivation of protease-activated receptor-1 by trypsin in vascular endothelial cells. Br J Pharmacol. 2003;138:121–30.
Molino M, Woolkalis MJ, Reavey-Cantwell J, Pratico D, Andrade-Gordon P, Barnathan ES, Brass LF. Endothelial cell thrombin receptors and PAR-2. Two protease-activated receptors located in a single cellular environment. J Biol Chem. 1997;272:11133–41.
Berger P, Tunon-De-Lara JM, Savineau JP, Marthan R. Selected contribution: tryptase-induced PAR-2-mediated Ca(2+) signaling in human airway smooth muscle cells. J Appl Physiol (1985). 2001;91:995–1003.
Berger P, Perng DW, Thabrew H, Compton SJ, Cairns JA, McEuen AR, Marthan R, Tunon De Lara JM, Walls AF. Tryptase and agonists of PAR-2 induce the proliferation of human airway smooth muscle cells. J Appl Physiol (1985). 2001;91:1372–9.
Akers IA, Parsons M, Hill MR, Hollenberg MD, Sanjar S, Laurent GJ, McAnulty RJ. Mast cell tryptase stimulates human lung fibroblast proliferation via protease-activated receptor-2. Am J Physiol Lung Cell Mol Physiol. 2000;278:L193–201.
Compton SJ, Renaux B, Wijesuriya SJ, Hollenberg MD. Glycosylation and the activation of proteinase-activated receptor 2 (PAR(2)) by human mast cell tryptase. Br J Pharmacol. 2001;134:705–18.
Schechter NM, Brass LF, Lavker RM, Jensen PJ. Reaction of mast cell proteases tryptase and chymase with protease activated receptors (PARs) on keratinocytes and fibroblasts. J Cell Physiol. 1998;176:365–73.
Seitz I, Hess S, Schulz H, Eckl R, Busch G, Montens HP, Brandl R, Seidl S, Schomig A, Ott I. Membrane-type serine protease-1/matriptase induces interleukin-6 and -8 in endothelial cells by activation of protease-activated receptor-2: potential implications in atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:769–75.
Bocheva G, Rattenholl A, Kempkes C, Goerge T, Lin CY, D'Andrea MR, Stander S, Steinhoff M. Role of matriptase and proteinase-activated receptor-2 in nonmelanoma skin cancer. J Invest Dermatol. 2009;129:1816–23.
Camerer E, Barker A, Duong DN, Ganesan R, Kataoka H, Cornelissen I, Darragh MR, Hussain A, Zheng YW, Srinivasan Y, et al. Local protease signaling contributes to neural tube closure in the mouse embryo. Dev Cell. 2010;18:25–38.
Sales KU, Friis S, Konkel JE, Godiksen S, Hatakeyama M, Hansen KK, Rogatto SR, Szabo R, Vogel LK, Chen W, et al. Non-hematopoietic PAR-2 is essential for matriptase-driven pre-malignant progression and potentiation of ras-mediated squamous cell carcinogenesis. Oncogene. 2015;34:346–56.
Caliendo G, Santagada V, Perissutti E, Severino B, Fiorino F, Frecentese F, Juliano L. Kallikrein protease activated receptor (PAR) axis: an attractive target for drug development. J Med Chem. 2012;55:6669–86.
Ramsay AJ, Dong Y, Hunt ML, Linn M, Samaratunga H, Clements JA, Hooper JD. Kallikrein-related peptidase 4 (KLK4) initiates intracellular signaling via protease-activated receptors (PARs). KLK4 and PAR-2 are co-expressed during prostate cancer progression. J Biol Chem. 2008;283:12293–304.
Oikonomopoulou K, Hansen KK, Saifeddine M, Vergnolle N, Tea I, Blaber M, Blaber SI, Scarisbrick I, Diamandis EP, Hollenberg MD. Kallikrein-mediated cell signalling: targeting proteinase-activated receptors (PARs). Biol Chem. 2006;387:817–24.
Yoon H, Radulovic M, Wu J, Blaber SI, Blaber M, Fehlings MG, Scarisbrick IA. Kallikrein 6 signals through PAR1 and PAR2 to promote neuron injury and exacerbate glutamate neurotoxicity. J Neurochem. 2013;127:283–98.
Molino M, Blanchard N, Belmonte E, Tarver AP, Abrams C, Hoxie JA, Cerletti C, Brass LF. Proteolysis of the human platelet and endothelial-cell thrombin receptor by neutrophil-derived Cathepsin-G. J Biol Chem. 1995;270:11168–75.
Molino M, Blanchard N, Belmonte E, Tarver AP, Abrams C, Hoxie JA, Cerletti C, Brass LF. Cathepsin-G cleaves the human thrombin receptor. Thromb Haemost. 1995;73:923.
Wilson TJ, Nannuru KC, Singh RK. Cathepsin G recruits osteoclast precursors via proteolytic activation of protease-activated Receptor-1. Cancer Res. 2009;69:3188–95.
Ramachandran R, Sadofsky LR, Xiao YP, Botham A, Cowen M, Morice AH, Compton SJ. Inflammatory mediators modulate thrombin and cathepsin-G signaling in human bronchial fibroblasts by inducing expression of proteinase-activated receptor-4. Am J Phys Lung Cell Mol Phys. 2007;292:L788–98.
Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA, DeFea KA, Bouvier M, Hollenberg MD. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J Biol Chem. 2011;286:24638–48.
Cumashi A, Ansuini H, Celli N, De Blasi A, O'Brien PJ, Brass LF, Molino M. Neutrophil proteases can inactivate human PAR3 and abolish the co-receptor function of PAR3 on murine platelets. Thromb Haemost. 2001;85:533–8.
Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem. 2000;275:6819–23.
Vergnolle N, Derian CK, D'Andrea MR, Steinhoff M, Andrade-Gordon P. Characterization of thrombin-induced leukocyte rolling and adherence: a potential proinflammatory role for proteinase-activated receptor-4. J Immunol. 2002;169:1467–73.
Walsh SW, Nugent WH, Solotskaya AV, Anderson CD, Grider JR, Strauss JF 3rd. Matrix metalloprotease-1 and elastase are novel uterotonic agents acting through protease-activated receptor 1. Reprod Sci. 2017. https://doi.org/10.1177/1933719117732162.
Kumar VRS, Darisipudi MN, Steiger S, Devarapu SK, Tato M, Kukarni OP, Mulay SR, Thomasova D, Popper B, Demleitner J, et al. Cathepsin S cleavage of protease-activated Receptor-2 on endothelial cells promotes microvascular diabetes complications. J Am Soc Nephrol. 2016;27:1635–49.
Elmariah SB, Reddy VB, Lerner EA. Cathepsin S signals via PAR2 and generates a novel tethered ligand receptor agonist. PLoS One. 2014;9(6):e99702.
Kuliopulos A, Covic L, Seeley SK, Sheridan PJ, Helin J, Costello CE. Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry. 1999;38:4572–85.
Domotor E, Bartha K, Machovich R, Adam-Vizi V. Protease-activated receptor-2 (PAR-2) in brain microvascular endothelium and its regulation by plasmin and elastase. J Neurochem. 2002;80:746–54.
Wang T, Lee MH, Choi E, Pardo-Villamizar CA, Lee SB, Yang IH, Calabresi PA, Nath A. Granzyme B-induced neurotoxicity is mediated via activation of PAR-1 receptor and Kv1.3 channel. PLoS One. 2012;7:e43950.
Lee PR, Johnson TP, Gnanapavan S, Giovannoni G, Wang T, Steiner JP, Medynets M, Vaal MJ, Gartner V, Nath A. Protease-activated receptor-1 activation by granzyme B causes neurotoxicity that is augmented by interleukin-1beta. J Neuroinflammation. 2017;14:131.
Cooper DM, Pechkovsky DV, Hackett TL, Knight DA, Granville DJ. Granzyme K activates protease-activated receptor-1. PLoS One. 2011;6:e21484.
Sharma M, Merkulova Y, Raithatha S, Parkinson LG, Shen Y, Cooper D, Granville DJ. Extracellular granzyme K mediates endothelial activation through the cleavage of protease-activated receptor-1. FEBS J. 2016;283:1734–47.
Alvarez-Arce A, Lee-Rivera I, Lopez E, Hernandez-Cruz A, Lopez-Colome AM. Thrombin-induced Calpain activation promotes protease-activated receptor 1 internalization. Int J Cell Biol. 2017;2017:1908310.
Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O'Callaghan K, Covic L, Kuliopulos A. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell. 2009;137:332–43.
Austin KM, Covic L, Kuliopulos A. Matrix metalloproteases and PAR1 activation. Blood. 2013;121:431–9.
Austin KM, Nguyen N, Javid G, Covic L, Kuliopulos A. Noncanonical matrix metalloprotease-1-protease-activated receptor-1 signaling triggers vascular smooth muscle cell dedifferentiation and arterial stenosis. J Biol Chem. 2013;288:23105–15.
Allen M, Ghosh S, Ahern GP, Villapol S, Maguire-Zeiss KA, Conant K. Protease induced plasticity: matrix metalloproteinase-1 promotes neurostructural changes through activation of protease activated receptor 1. Sci Rep. 2016;6:35497.
Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120:303–13.
Sebastiano M, Momi S, Falcinelli E, Bury L, Hoylaerts MF, Gresele P. A novel mechanism regulating human platelet activation by MMP-2-mediated PAR1 biased signaling. Blood. 2017;129:883–95.
Lee SE, Kim JM, Jeong SK, Jeon JE, Yoon HJ, Jeong MK, Lee SH. Protease-activated receptor-2 mediates the expression of inflammatory cytokines, antimicrobial peptides, and matrix metalloproteinases in keratinocytes in response to Propionibacterium acnes. Arch Dermatol Res. 2010;302:745–56.
Vliagoftis H, Schwingshackl A, Milne CD, Duszyk M, Hollenberg MD, Wallace JL, Befus AD, Moqbel R. Proteinase-activated receptor-2-mediated matrix metalloproteinase-9 release from airway epithelial cells. J Allergy Clin Immunol. 2000;106:537–45.
Hou HH, Wang HC, Cheng SL, Chen YF, Lu KZ, Yu CJ. MMP-12 activates protease activated receptor (PAR)-1, upregulates placenta growth factor and leads to pulmonary emphysema. Am J Physiol Lung Cell Mol Physiol. 2018;315(3):L432–42.
Raza SL, Nehring LC, Shapiro SD, Cornelius LA. Proteinase-activated receptor-1 regulation of macrophage elastase (MMP-12) secretion by serine proteinases. J Biol Chem. 2000;275:41243–50.
Zang N, Zhuang JG, Deng Y, Yang ZM, Ye ZX, Xie XH, Ren L, Fu Z, Luo ZX, Xu FD, Liu EM. Pulmonary C fibers modulate MMP-12 production via PAR2 and are involved in the long-term airway inflammation and airway Hyperresponsiveness induced by respiratory syncytial virus infection. J Virol. 2016;90:2536–43.
Hooper JD, Nicol DL, Dickinson JL, Eyre HJ, Scarman AL, Normyle JF, Stuttgen MA, Douglas ML, Loveland KA, Sutherland GR, Antalis TM. Testisin, a new human serine proteinase expressed by premeiotic testicular germ cells and lost in testicular germ cell tumors. Cancer Res. 1999;59:3199–205.
Driesbaugh KH, Buzza MS, Martin EW, Conway GD, Kao JP, Antalis TM. Proteolytic activation of the protease-activated receptor (PAR)-2 by the glycosylphosphatidylinositol-anchored serine protease testisin. J Biol Chem. 2015;290:3529–41.
Potempa J, Pike RN. Corruption of innate immunity by bacterial proteases. J Innate Immun. 2009;1:70–87.
Kida Y, Higashimoto Y, Inoue H, Shimizu T, Kuwano K. A novel secreted protease from Pseudomonas aeruginosa activates NF-kappaB through protease-activated receptors. Cell Microbiol. 2008;10:1491–504.
Dulon S, Leduc D, Cottrell GS, D'Alayer J, Hansen KK, Bunnett NW, Hollenberg MD, Pidard D, Chignard M. Pseudomonas aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. Am J Respir Cell Mol Biol. 2005;32:411–9.
Ender M, Andreoni F, Zinkernagel AS, Schuepbach RA. Streptococcal SpeB cleaved PAR-1 suppresses ERK phosphorylation and blunts thrombin-induced platelet aggregation. PLoS One. 2013;8:e81298.
van den Boogaard FE, Brands X, Duitman J, de Stoppelaar SF, Borensztajn KS, Roelofs J, Hollenberg MD, Spek CA, Schultz MJ, van ‘t Veer C, van der Poll T. Protease-activated receptor 2 facilitates bacterial dissemination in pneumococcal pneumonia. J Infect Dis. 2018;217:1462–71.
Schouten M, Van't Veer C, Roelofs JJ, Levi M, van der Poll T. Protease-activated receptor-1 impairs host defense in murine pneumococcal pneumonia: a controlled laboratory study. Crit Care. 2012;16:R238.
Asehnoune K, Moine P. Protease-activated receptor-1: key player in the sepsis coagulation-inflammation crosstalk. Crit Care. 2013;17:119.
de Stoppelaar SF, Van't Veer C, van den Boogaard FE, Nieuwland R, Hoogendijk AJ, de Boer OJ, Roelofs JJ, van der Poll T. Protease activated receptor 4 limits bacterial growth and lung pathology during late stage Streptococcus pneumoniae induced pneumonia in mice. Thromb Haemost. 2013;110:582–92.
Holzhausen M, Spolidorio LC, Ellen RP, Jobin MC, Steinhoff M, Andrade-Gordon P, Vergnolle N. Protease-activated receptor-2 activation: a major role in the pathogenesis of Porphyromonas gingivalis infection. Am J Pathol. 2006;168:1189–99.
Francis N, Ayodele BA, O'Brien-Simpson NM, Birchmeier W, Pike RN, Pagel CN, Mackie EJ. Keratinocyte-specific ablation of protease-activated receptor-2 prevents gingival inflammation and bone loss in a mouse model of periodontal disease. Cell Microbiol. 2018;20(11):e12891.
Lourbakos A, Chinni C, Thompson P, Potempa J, Travis J, Mackie EJ, Pike RN. Cleavage and activation of proteinase-activated receptor-2 on human neutrophils by gingipain-R from Porphyromonas gingivalis. FEBS Lett. 1998;435:45–8.
Lourbakos A, Potempa J, Travis J, D'Andrea MR, Andrade-Gordon P, Santulli R, Mackie EJ, Pike RN. Arginine-specific protease from Porphyromonas gingivalis activates protease-activated receptors on human oral epithelial cells and induces interleukin-6 secretion. Infect Immun. 2001;69:5121–30.
Lourbakos A, Yuan YP, Jenkins AL, Travis J, Andrade-Gordon P, Santulli R, Potempa J, Pike RN. Activation of protease-activated receptors by gingipains from Porphyromonas gingivalis leads to platelet aggregation: a new trait in microbial pathogenicity. Blood. 2001;97:3790–7.
Papapanagiotou D, Nicu EA, Bizzarro S, Gerdes VE, Meijers JC, Nieuwland R, van der Velden U, Loos BG. Periodontitis is associated with platelet activation. Atherosclerosis. 2009;202:605–11.
Ikawa K, Nishioka T, Yu Z, Sugawara Y, Kawagoe J, Takizawa T, Primo V, Nikolic B, Kuroishi T, Sasano T, et al. Involvement of neutrophil recruitment and protease-activated receptor 2 activation in the induction of IL-18 in mice. J Leukoc Biol. 2005;78:1118–26.
Kida Y, Inoue H, Shimizu T, Kuwano K. Serratia marcescens serralysin induces inflammatory responses through protease-activated receptor 2. Infect Immun. 2007;75:164–74.
Chen X, Earley K, Luo W, Lin SH, Schilling WP. Functional expression of a human thrombin receptor in Sf9 insect cells: evidence for an active tethered ligand. Biochem J. 1996;314(Pt 2):603–11.
Ubl JJ, Sergeeva M, Reiser G. Desensitisation of protease-activated receptor-1 (PAR-1) in rat astrocytes: evidence for a novel mechanism for terminating Ca2+ signalling evoked by the tethered ligand. J Physiol. 2000;525(Pt 2):319–30.
Tripathi T, Abdi M, Alizadeh H. Protease-activated receptor 2 (PAR2) is upregulated by Acanthamoeba plasminogen activator (aPA) and induces proinflammatory cytokine in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2014;55:3912–21.
Laing GD, Compton SJ, Ramachandran R, Fuller GL, Wilkinson MC, Wagstaff SC, Watson SP, Kamiguti AS, Theakston RD, Senis YA. Characterization of a novel protein from Proatheris superciliaris venom: proatherocytin, a 34-kDa platelet receptor PAR1 agonist. Toxicon. 2005;46:490–9.
Santos BF, Serrano SM, Kuliopulos A, Niewiarowski S. Interaction of viper venom serine peptidases with thrombin receptors on human platelets. FEBS Lett. 2000;477:199–202.
Sun G, Stacey MA, Schmidt M, Mori L, Mattoli S. Interaction of mite allergens Der p3 and Der p9 with protease-activated receptor-2 expressed by lung epithelial cells. J Immunol. 2001;167:1014–21.
Asokananthan N, Graham PT, Fink J, Knight DA, Bakker AJ, McWilliam AS, Thompson PJ, Stewart GA. Activation of protease-activated receptor (PAR)-1, PAR-2, and PAR-4 stimulates IL-6, IL-8, and prostaglandin E2 release from human respiratory epithelial cells. J Immunol. 2002;168:3577–85.
Lin YP, Nelson C, Kramer H, Parekh AB. The allergen Der p3 from house dust mite stimulates store-operated ca(2+) channels and mast cell migration through PAR4 receptors. Mol Cell. 2018;70:228–241.e225.
Kondo S, Helin H, Shichijo M, Bacon KB. Cockroach allergen extract stimulates protease-activated receptor-2 (PAR-2) expressed in mouse lung fibroblast. Inflamm Res. 2004;53:489–96.
Asaduzzaman M, Nadeem A, Arizmendi N, Davidson C, Nichols HL, Abel M, Ionescu LI, Puttagunta L, Thebaud B, Gordon J, et al. Functional inhibition of PAR2 alleviates allergen-induced airway hyperresponsiveness and inflammation. Clin Exp Allergy. 2015;45:1844–55.
Polley DJ, Mihara K, Ramachandran R, Vliagoftis H, Renaux B, Saifeddine M, Daines MO, Boitano S, Hollenberg MD. Cockroach allergen serine proteinases: isolation, sequencing and signalling via proteinase-activated receptor-2. Clin Exp Allergy. 2017;47:946–60.
Chiu LL, Perng DW, Yu CH, Su SN, Chow LP. Mold allergen, pen C 13, induces IL-8 expression in human airway epithelial cells by activating protease-activated receptor 1 and 2. J Immunol. 2007;178:5237–44.
Kauffman HF, Tomee JF, van de Riet MA, Timmerman AJ, Borger P. Protease-dependent activation of epithelial cells by fungal allergens leads to morphologic changes and cytokine production. J Allergy Clin Immunol. 2000;105:1185–93.
Boitano S, Flynn AN, Schulz SM, Hoffman J, Price TJ, Vagner J. Potent agonists of the protease activated receptor 2 (PAR2). J Med Chem. 2011;54:1308–13.
Boitano S, Flynn AN, Sherwood CL, Schulz SM, Hoffman J, Gruzinova I, Daines MO. Alternaria alternata serine proteases induce lung inflammation and airway epithelial cell activation via PAR2. Am J Physiol Lung Cell Mol Physiol. 2011;300:L605–14.
Snelgrove RJ, Gregory LG, Peiro T, Akthar S, Campbell GA, Walker SA, Lloyd CM. Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J Allergy Clin Immunol. 2014;134:583–592.e586.
Rathnavelu V, Alitheen NB, Sohila S, Kanagesan S, Ramesh R. Potential role of bromelain in clinical and therapeutic applications. Biomed Rep. 2016;5:283–8.
Borrelli F, Capasso R, Severino B, Fiorino F, Aviello G, De Rosa G, Mazzella M, Romano B, Capasso F, Fasolino I, Izzo AA. Inhibitory effects of bromelain, a cysteine protease derived from pineapple stem (Ananas comosus), on intestinal motility in mice. Neurogastroenterol Motil. 2011;23:745–e331.
Reddy VB, Lerner EA. Plant cysteine proteases that evoke itch activate protease-activated receptors. Br J Dermatol. 2010;163:532–5.
Lerner DJ, Chen M, Tram T, Coughlin SR. Agonist recognition by proteinase-activated receptor 2 and thrombin receptor. Importance of extracellular loop interactions for receptor function. J Biol Chem. 1996;271:13943–7.
Hollenberg MD, Yang SG, Laniyonu AA, Moore GJ, Saifeddine M. Action of thrombin receptor polypeptide in gastric smooth muscle: identification of a core pentapeptide retaining full thrombin-mimetic intrinsic activity. Mol Pharmacol. 1992;42:186–91.
Blackhart BD, Emilsson K, Nguyen D, Teng W, Martelli AJ, Nystedt S, Sundelin J, Scarborough RM. Ligand cross-reactivity within the protease-activated receptor family. J Biol Chem. 1996;271:16466–71.
Hollenberg MD, Saifeddine M, al-Ani B, Kawabata A. Proteinase-activated receptors: structural requirements for activity, receptor cross-reactivity, and receptor selectivity of receptor-activating peptides. Can J Physiol Pharmacol. 1997;75:832–41.
Vassallo RR Jr, Kieber-Emmons T, Cichowski K, Brass LF. Structure-function relationships in the activation of platelet thrombin receptors by receptor-derived peptides. J Biol Chem. 1992;267:6081–5.
al-Ani B, Saifeddine M, Hollenberg MD. Detection of functional receptors for the proteinase-activated-receptor-2-activating polypeptide, SLIGRL-NH2, in rat vascular and gastric smooth muscle. Can J Physiol Pharmacol. 1995;73:1203–7.
McGuire JJ, Saifeddine M, Triggle CR, Sun K, Hollenberg MD. 2-furoyl-LIGRLO-amide: a potent and selective proteinase-activated receptor 2 agonist. J Pharmacol Exp Ther. 2004;309:1124–31.
Al-Ani B, Saifeddine M, Kawabata A, Hollenberg MD. Proteinase activated receptor 2: role of extracellular loop 2 for ligand-mediated activation. Br J Pharmacol. 1999;128:1105–13.
Al-Ani B, Saifeddine M, Kawabata A, Renaux B, Mokashi S, Hollenberg MD. Proteinase-activated receptor 2 (PAR(2)): development of a ligand-binding assay correlating with activation of PAR(2) by PAR(1)- and PAR(2)-derived peptide ligands. J Pharmacol Exp Ther. 1999;290:753–60.
Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, Coughlin SR. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404:609–13.
Hollenberg MD, Saifeddine M, Al-Ani B, Gui Y. Proteinase-activated receptor 4 (PAR4): action of PAR4-activating peptides in vascular and gastric tissue and lack of cross-reactivity with PAR1 and PAR2. Can J Physiol Pharmacol. 1999;77:458–64.
Faruqi TR, Weiss EJ, Shapiro MJ, Huang W, Coughlin SR. Structure-function analysis of protease-activated receptor 4 tethered ligand peptides. Determinants of specificity and utility in assays of receptor function [In process citation]. J Biol Chem. 2000;275:19728–34.
Lin H, Trejo J. Transactivation of the PAR1-PAR2 heterodimer by thrombin elicits beta-arrestin-mediated endosomal signaling. J Biol Chem. 2013;288:11203–15.
O'Brien PJ, Prevost N, Molino M, Hollinger MK, Woolkalis MJ, Woulfe DS, Brass LF. Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. J Biol Chem. 2000;275:13502–9.
Shi X, Gangadharan B, Brass LF, Ruf W, Mueller BM. Protease-activated receptors (PAR1 and PAR2) contribute to tumor cell motility and metastasis. Mol Cancer Res. 2004;2:395–402.
Jaber M, Maoz M, Kancharla A, Agranovich D, Peretz T, Grisaru-Granovsky S, Uziely B, Bar-Shavit R. Protease-activated-receptor-2 affects protease-activated-receptor-1-driven breast cancer. Cell Mol Life Sci. 2014;71:2517–33.
Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, Andrade-Gordon P, Covic L, Kuliopulos A. Blocking the protease-activated receptor 1-4 heterodimer in platelet-mediated thrombosis. Circulation. 2006;113:1244–54.
Sveshnikova AN, Balatskiy AV, Demianova AS, Shepelyuk TO, Shakhidzhanov SS, Balatskaya MN, Pichugin AV, Ataullakhanov FI, Panteleev MA. Systems biology insights into the meaning of the platelet's dual-receptor thrombin signaling. J Thromb Haemost. 2016;14:2045–57.
Hansen KK, Saifeddine M, Hollenberg MD. Tethered ligand-derived peptides of proteinase-activated receptor 3 (PAR3) activate PAR1 and PAR2 in Jurkat T cells. Immunology. 2004;112:183–90.
Cunningham MR, McIntosh KA, Pediani JD, Robben J, Cooke AE, Nilsson M, Gould GW, Mundell S, Milligan G, Plevin R. Novel role for proteinase-activated receptor 2 (PAR2) in membrane trafficking of proteinase-activated receptor 4 (PAR4). J Biol Chem. 2012;287:16656–69.
Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, Kaufmann R. Proteinase-activated receptors (PARs) - focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun Signal. 2013;11:86.
Amadesi S, Cottrell GS, Divino L, Chapman K, Grady EF, Bautista F, Karanjia R, Barajas-Lopez C, Vanner S, Vergnolle N, Bunnett NW. Protease-activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent mechanisms in rats and mice. J Physiol. 2006;575:555–71.
Grant AD, Cottrell GS, Amadesi S, Trevisani M, Nicoletti P, Materazzi S, Altier C, Cenac N, Zamponi GW, Bautista-Cruz F, et al. Protease-activated receptor 2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause mechanical hyperalgesia in mice. J Physiol. 2007;578:715–33.
Zhao P, Lieu T, Barlow N, Sostegni S, Haerteis S, Korbmacher C, Liedtke W, Jimenez-Vargas NN, Vanner SJ, Bunnett NW. Neutrophil elastase activates protease-activated Receptor-2 (PAR2) and transient receptor potential Vanilloid 4 (TRPV4) to cause inflammation and pain. J Biol Chem. 2015;290:13875–87.
Saifeddine M, El-Daly M, Mihara K, Bunnett NW, McIntyre P, Altier C, Hollenberg MD, Ramachandran R. GPCR-mediated EGF receptor transactivation regulates TRPV4 action in the vasculature. Br J Pharmacol. 2015;172:2493–506.
Tsubota M, Ozaki T, Hayashi Y, Okawa Y, Fujimura A, Sekiguchi F, Nishikawa H, Kawabata A. Prostanoid-dependent bladder pain caused by proteinase-activated receptor-2 activation in mice: involvement of TRPV1 and T-type ca(2+) channels. J Pharmacol Sci. 2018;136:46–9.
Chung H, Ramachandran R, Hollenberg MD, Muruve DA. Proteinase-activated receptor-2 transactivation of epidermal growth factor receptor and transforming growth factor-beta receptor signaling pathways contributes to renal fibrosis. J Biol Chem. 2013;288:37319–31.
Sabri A, Guo J, Elouardighi H, Darrow AL, Andrade-Gordon P, Steinberg SF. Mechanisms of protease-activated receptor-4 actions in cardiomyocytes. Role of Src tyrosine kinase. J Biol Chem. 2003;278:11714–20.
Fasano A. Zonulin, regulation of tight junctions, and autoimmune diseases. Ann N Y Acad Sci. 2012;1258:25–33.
Al-Ani B, Hewett PW, Cudmore MJ, Fujisawa T, Saifeddine M, Williams H, Ramma W, Sissaoui S, Jayaraman PS, Ohba M, et al. Activation of proteinase-activated receptor 2 stimulates soluble vascular endothelial growth factor receptor 1 release via epidermal growth factor receptor transactivation in endothelial cells. Hypertension. 2010;55:689–97.
Palm E, Demirel I, Bengtsson T, Khalaf H. The role of toll-like and protease-activated receptors in the expression of cytokines by gingival fibroblasts stimulated with the periodontal pathogen Porphyromonas gingivalis. Cytokine. 2015;76:424–32.
Damien P, Cognasse F, Payrastre B, Spinelli SL, Blumberg N, Arthaud CA, Eyraud MA, Phipps RP, McNicol A, Pozzetto B, et al. NF-kappaB links TLR2 and PAR1 to soluble Immunomodulator factor secretion in human platelets. Front Immunol. 2017;8:85.
Rallabhandi P, Nhu QM, Toshchakov VY, Piao W, Medvedev AE, Hollenberg MD, Fasano A, Vogel SN. Analysis of proteinase-activated receptor 2 and TLR4 signal transduction: a novel paradigm for receptor cooperativity. J Biol Chem. 2008;283:24314–25.
Nhu QM, Shirey K, Teijaro JR, Farber DL, Netzel-Arnett S, Antalis TM, Fasano A, Vogel SN. Novel signaling interactions between proteinase-activated receptor 2 and toll-like receptors in vitro and in vivo. Mucosal Immunol. 2010;3:29–39.
Williams JC, Lee RD, Doerschuk CM, Mackman N. Effect of PAR-2 deficiency in mice on KC expression after Intratracheal LPS administration. J Signal Transduct. 2011;2011:415195.
Zhou B, Zhou H, Ling S, Guo D, Yan Y, Zhou F, Wu Y. Activation of PAR2 or/and TLR4 promotes SW620 cell proliferation and migration via phosphorylation of ERK1/2. Oncol Rep. 2011;25:503–11.
Bucci M, Vellecco V, Harrington L, Brancaleone V, Roviezzo F, Mattace Raso G, Ianaro A, Lungarella G, De Palma R, Meli R, Cirino G. Cross-talk between toll-like receptor 4 (TLR4) and proteinase-activated receptor 2 (PAR(2) ) is involved in vascular function. Br J Pharmacol. 2013;168:411–20.
Weithauser A, Bobbert P, Antoniak S, Bohm A, Rauch BH, Klingel K, Savvatis K, Kroemer HK, Tschope C, Stroux A, et al. Protease-activated receptor-2 regulates the innate immune response to viral infection in a coxsackievirus B3-induced myocarditis. J Am Coll Cardiol. 2013;62:1737–45.
Oldham WM, Hamm HE. How do receptors activate G proteins? Adv Protein Chem. 2007;74:67–93.
Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71.
Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–84.
Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, Ye SQ, Garcia JG. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem. 2005;280:17286–93.
Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood. 2012;120:5237–46.
Hung DT, Wong YH, Vu TK, Coughlin SR. The cloned platelet thrombin receptor couples to at least two distinct effectors to stimulate phosphoinositide hydrolysis and inhibit adenylyl cyclase. J Biol Chem. 1992;267:20831–4.
Hung DT, Vu TK, Wheaton VI, Ishii K, Coughlin SR. Cloned platelet thrombin receptor is necessary for thrombin-induced platelet activation. J Clin Invest. 1992;89:1350–3.
Santulli RJ, Derian CK, Darrow AL, Tomko KA, Eckardt AJ, Seiberg M, Scarborough RM, Andrade-Gordon P. Evidence for the presence of a protease-activated receptor distinct from the thrombin receptor in human keratinocytes. Proc Natl Acad Sci U S A. 1995;92:9151–5.
McLaughlin JN, Shen L, Holinstat M, Brooks JD, Dibenedetto E, Hamm HE. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem. 2005;280:25048–59.
Offermanns S, Laugwitz KL, Spicher K, Schultz G. G proteins of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. Proc Natl Acad Sci U S A. 1994;91:504–8.
Klages B, Brandt U, Simon MI, Schultz G, Offermanns S. Activation of G12/G13 results in shape change and rho/rho-kinase- mediated myosin light chain phosphorylation in mouse platelets. J Cell Biol. 1999;144:745–54.
Moers A, Nieswandt B, Massberg S, Wettschureck N, Gruner S, Konrad I, Schulte V, Aktas B, Gratacap MP, Simon MI, et al. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat Med. 2003;9:1418–22.
Klarenbach SW, Chipiuk A, Nelson RC, Hollenberg MD, Murray AG. Differential actions of PAR2 and PAR1 in stimulating human endothelial cell exocytosis and permeability: the role of Rho-GTPases. Circ Res. 2003;92:272–8.
Fortunato TM, Vara DS, Wheeler-Jones CP, Pula G. Expression of protease-activated receptor 1 and 2 and anti-tubulogenic activity of protease-activated receptor 1 in human endothelial colony-forming cells. PLoS One. 2014;9:e109375.
Kim YV, Di Cello F, Hillaire CS, Kim KS. Differential Ca2+ signaling by thrombin and protease-activated receptor-1-activating peptide in human brain microvascular endothelial cells. Am J Physiol Cell Physiol. 2004;286:C31–42.
Soh UJ, Trejo J. Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through beta-arrestin and dishevelled-2 scaffolds. Proc Natl Acad Sci U S A. 2011;108:E1372–80.
Roy RV, Ardeshirylajimi A, Dinarvand P, Yang L, Rezaie AR. Occupancy of human EPCR by protein C induces beta-arrestin-2 biased PAR1 signaling by both APC and thrombin. Blood. 2016;128:1884–93.
DeFea KA, Zalevsky J, Thoma MS, Dery O, Mullins RD, Bunnett NW. Beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol. 2000;148:1267–81.
Ge L, Shenoy SK, Lefkowitz RJ, DeFea K. Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both beta-arrestin-1 and -2. J Biol Chem. 2004;279:55419–24.
Stalheim L, Ding Y, Gullapalli A, Paing MM, Wolfe BL, Morris DR, Trejo J. Multiple independent functions of arrestins in the regulation of protease-activated receptor-2 signaling and trafficking. Mol Pharmacol. 2005;67:78–87.
Hein L, Ishii K, Coughlin SR, Kobilka BK. Intracellular targeting and trafficking of thrombin receptors. A novel mechanism for resensitization of a G protein-coupled receptor. J Biol Chem. 1994;269:27719–26.
Takafuta T, Wu G, Murphy GF, Shapiro SS. Human beta-filamin is a new protein that interacts with the cytoplasmic tail of glycoprotein Ibalpha. J Biol Chem. 1998;273:17531–8.
Vouret-Craviari V, Grall D, Chambard JC, Rasmussen UB, Pouyssegur J, Van Obberghen-Schilling E. Post-translational and activation-dependent modifications of the G protein-coupled thrombin receptor. J Biol Chem. 1995;270:8367–72.
Schuepbach RA, Feistritzer C, Brass LF, Riewald M. Activated protein C-cleaved protease activated receptor-1 is retained on the endothelial cell surface even in the presence of thrombin. Blood. 2008;111:2667–73.
Trejo J, Hammes SR, Coughlin SR. Termination of signaling by protease-activated receptor-1 is linked to lysosomal sorting. Proc Natl Acad Sci U S A. 1998;95:13698–702.
Paing MM, Johnston CA, Siderovski DP, Trejo J. Clathrin adaptor AP2 regulates thrombin receptor constitutive internalization and endothelial cell resensitization. Mol Cell Biol. 2006;26:3231–42.
Dores MR, Chen B, Lin H, Soh UJ, Paing MM, Montagne WA, Meerloo T, Trejo J. ALIX binds a YPX(3)L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J Cell Biol. 2012;197:407–19.
Grimsey NJ, Coronel LJ, Cordova IC, Trejo J. Recycling and endosomal sorting of protease-activated Receptor-1 is distinctly regulated by Rab11A and Rab11B proteins. J Biol Chem. 2016;291:2223–36.
Trejo J, Coughlin SR. The cytoplasmic tails of protease-activated receptor-1 and substance P receptor specify sorting to lysosomes versus recycling. J Biol Chem. 1999;274:2216–24.
Ishii K, Chen J, Ishii M, Koch WJ, Freedman NJ, Lefkowitz RJ, Coughlin SR. Inhibition of thrombin receptor signaling by a G-protein coupled receptor kinase. J Biol Chem. 1994;269:1125–30.
Paing MM, Stutts AB, Kohout TA, Lefkowitz RJ, Trejo J. Beta -Arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. J Biol Chem. 2002;277:1292–300.
Chen J, De S, Damron DS, Chen WS, Hay N, Byzova TV. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood. 2004;104:1703–10.
Wolfe BL, Marchese A, Trejo J. Ubiquitination differentially regulates clathrin-dependent internalization of protease-activated receptor-1. J Cell Biol. 2007;177:905–16.
Soh UJ, Dores MR, Chen B, Trejo J. Signal transduction by protease-activated receptors. Br J Pharmacol. 2010;160:191–203.
Jacob C, Cottrell GS, Gehringer D, Schmidlin F, Grady EF, Bunnett NW. c-Cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J Biol Chem. 2005;280:16076–87.
Dery O, Thoma MS, Wong H, Grady EF, Bunnett NW. Trafficking of proteinase-activated receptor-2 and beta-arrestin-1 tagged with green fluorescent protein. Beta-Arrestin-dependent endocytosis of a proteinase receptor. J Biol Chem. 1999;274:18524–35.
Ricks TK, Trejo J. Phosphorylation of protease-activated receptor-2 differentially regulates desensitization and internalization. J Biol Chem. 2009;284:34444–57.
Jimenez-Vargas NN, Pattison LA, Zhao P, Lieu T, Latorre R, Jensen DD, Castro J, Aurelio L, Le GT, Flynn B, et al. Protease-activated receptor-2 in endosomes signals persistent pain of irritable bowel syndrome. Proc Natl Acad Sci U S A. 2018;115:E7438–47.
Steinhoff M, Buddenkotte J, Shpacovitch V, Rattenholl A, Moormann C, Vergnolle N, Luger TA, Hollenberg MD. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr Rev. 2005;26:1–43.
Smith TH, Coronel LJ, Li JG, Dores MR, Nieman MT, Trejo J. Protease-activated receptor-4 signaling and trafficking is regulated by the Clathrin adaptor protein complex-2 independent of beta-Arrestins. J Biol Chem. 2016;291:18453–64.
Shapiro MJ, Weiss EJ, Faruqi TR, Coughlin SR. Protease-activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J Biol Chem. 2000;275:25216–21.
Smith TH, Li JG, Dores MR, Trejo J. Protease-activated receptor-4 and purinergic receptor P2Y12 dimerize, co-internalize, and activate Akt signaling via endosomal recruitment of beta-arrestin. J Biol Chem. 2017;292:13867–78.
Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103:879–87.
Andrade-Gordon P, Derian CK, Maryanoff BE, Zhang HC, Addo MF, Cheung W, Damiano BP, D'Andrea MR, Darrow AL, de Garavilla L, et al. Administration of a potent antagonist of protease-activated receptor-1 (PAR-1) attenuates vascular restenosis following balloon angioplasty in rats. J Pharmacol Exp Ther. 2001;298:34–42.
Bae JS, Rezaie AR. Thrombin inhibits nuclear factor kappaB and RhoA pathways in cytokine-stimulated vascular endothelial cells when EPCR is occupied by protein C. Thromb Haemost. 2009;101:513–20.
Buddenkotte J, Stroh C, Engels IH, Moormann C, Shpacovitch VM, Seeliger S, Vergnolle N, Vestweber D, Luger TA, Schulze-Osthoff K, Steinhoff M. Agonists of proteinase-activated receptor-2 stimulate upregulation of intercellular cell adhesion molecule-1 in primary human keratinocytes via activation of NF-kappa B. J Invest Dermatol. 2005;124:38–45.
Howells GL, Macey MG, Chinni C, Hou L, Fox MT, Harriott P, Stone SR. Proteinase-activated receptor-2: expression by human neutrophils. J Cell Sci. 1997;110(Pt 7):881–7.
Borbiev T, Birukova A, Liu F, Nurmukhambetova S, Gerthoffer WT, Garcia JG, Verin AD. p38 MAP kinase-dependent regulation of endothelial cell permeability. Am J Physiol Lung Cell Mol Physiol. 2004;287:L911–8.
Schuepbach RA, Feistritzer C, Fernandez JA, Griffin JH, Riewald M. Protection of vascular barrier integrity by activated protein C in murine models depends on protease-activated receptor-1. Thromb Haemost. 2009;101:724–33.
Birukova AA, Alekseeva E, Mikaelyan A, Birukov KG. HGF attenuates thrombin-induced endothelial permeability by Tiam1-mediated activation of the Rac pathway and by Tiam1/Rac-dependent inhibition of the rho pathway. FASEB J. 2007;21:2776–86.
Grimsey NJ, Aguilar B, Smith TH, Le P, Soohoo AL, Puthenveedu MA, Nizet V, Trejo J. Ubiquitin plays an atypical role in GPCR-induced p38 MAP kinase activation on endosomes. J Cell Biol. 2015;210:1117–31.
Hamilton JR, Frauman AG, Cocks TM. Increased expression of protease-activated receptor-2 (PAR2) and PAR4 in human coronary artery by inflammatory stimuli unveils endothelium-dependent relaxations to PAR2 and PAR4 agonists. Circ Res. 2001;89:92–8.
Saifeddine M, al-Ani B, Cheng CH, Wang L, Hollenberg MD. Rat proteinase-activated receptor-2 (PAR-2): cDNA sequence and activity of receptor-derived peptides in gastric and vascular tissue. Br J Pharmacol. 1996;118:521–30.
Moffatt JD, Cocks TM. Endothelium-dependent and -independent responses to protease-activated receptor-2 (PAR-2) activation in mouse isolated renal arteries. Br J Pharmacol. 1998;125:591–4.
Sobey CG, Moffatt JD, Cocks TM. Evidence for selective effects of chronic hypertension on cerebral artery vasodilatation to protease-activated receptor-2 activation. Stroke. 1999;30:1933–40 discussion 1941.
McGuire JJ, Hollenberg MD, Andrade-Gordon P, Triggle CR. Multiple mechanisms of vascular smooth muscle relaxation by the activation of proteinase-activated receptor 2 in mouse mesenteric arterioles. Br J Pharmacol. 2002;135:155–69.
Villari A, Giurdanella G, Bucolo C, Drago F, Salomone S. Apixaban enhances vasodilatation mediated by protease-activated receptor 2 in isolated rat arteries. Front Pharmacol. 2017;8:480.
Roy SS, Saifeddine M, Loutzenhiser R, Triggle CR, Hollenberg MD. Dual endothelium-dependent vascular activities of proteinase-activated receptor-2-activating peptides: evidence for receptor heterogeneity. Br J Pharmacol. 1998;123:1434–40.
Arisato T, Sarker KP, Kawahara K, Nakata M, Hashiguchi T, Osame M, Kitajima I, Maruyama I. The agonist of the protease-activated receptor-1 (PAR) but not PAR3 mimics thrombin-induced vascular endothelial growth factor release in human smooth muscle cells. Cell Mol Life Sci. 2003;60:1716–24.
Seymour ML, Binion DG, Compton SJ, Hollenberg MD, MacNaughton WK. Expression of proteinase-activated receptor 2 on human primary gastrointestinal myofibroblasts and stimulation of prostaglandin synthesis. Can J Physiol Pharmacol. 2005;83:605–16.
Iablokov V, Hirota CL, Peplowski MA, Ramachandran R, Mihara K, Hollenberg MD, MacNaughton WK. Proteinase-activated receptor 2 (PAR2) decreases apoptosis in colonic epithelial cells. J Biol Chem. 2014;289:34366–77.
Rolland-Fourcade C, Denadai-Souza A, Cirillo C, Lopez C, Jaramillo JO, Desormeaux C, Cenac N, Motta JP, Larauche M, Tache Y, et al. Epithelial expression and function of trypsin-3 in irritable bowel syndrome. Gut. 2017;66:1767–78.
Vergnolle N, Macnaughton WK, Al-Ani B, Saifeddine M, Wallace JL, Hollenberg MD. Proteinase-activated receptor 2 (PAR2)-activating peptides: identification of a receptor distinct from PAR2 that regulates intestinal transport. Proc Natl Acad Sci U S A. 1998;95:7766–71.
Buresi MC, Schleihauf E, Vergnolle N, Buret A, Wallace JL, Hollenberg MD, MacNaughton WK. Protease-activated receptor-1 stimulates Ca(2+)-dependent Cl(-) secretion in human intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2001;281:G323–32.
Chin AC, Vergnolle N, MacNaughton WK, Wallace JL, Hollenberg MD, Buret AG. Proteinase-activated receptor 1 activation induces epithelial apoptosis and increases intestinal permeability. Proc Natl Acad Sci U S A. 2003;100:11104–9.
Idell S, Gonzalez K, Bradford H, Macarthur CK, Fein AM, Maunder RJ, Garcia JGN, Griffith DE, Weiland J, Martin TR, et al. Procoagulant activity in Bronchoalveolar lavage in the adult respiratory-distress syndrome - contribution of tissue factor associated with factor-vii. Am Rev Respir Dis. 1987;136:1466–74.
Jarjour NN, Calhoun WJ, Schwartz LB, Busse WW. Elevated bronchoalveolar lavage fluid histamine levels in allergic asthmatics are associated with increased airway obstruction. Am Rev Respir Dis. 1991;144:83–7.
Clark JM, Abraham WM, Fishman CE, Forteza R, Ahmed A, Cortes A, Warne RL, Moore WR, Tanaka RD. Tryptase inhibitors block allergen-induced airway and inflammatory responses in allergic sheep. Am J Respir Crit Care Med. 1995;152:2076–83.
Rice KD, Tanaka RD, Katz BA, Numerof RP, Moore WR. Inhibitors of tryptase for the treatment of mast cell-mediated diseases. Curr Pharm Des. 1998;4:381–96.
Hauck RW, Schulz C, Schomig A, Hoffman RK, Panettieri RA Jr. Alpha-thrombin stimulates contraction of human bronchial rings by activation of protease-activated receptors. Am J Phys. 1999;277:L22–9.
Cocks TM, Fong B, Chow JM, Anderson GP, Frauman AG, Goldie RG, Henry PJ, Carr MJ, Hamilton JR, Moffatt JD. A protective role for protease-activated receptors in the airways. Nature. 1999;398:156–60.
Cocks TM, Moffatt JD. Protease-activated receptor-2 (PAR2) in the airways. Pulm Pharmacol Ther. 2001;14:183–91.
Schmidlin F, Amadesi S, Vidil R, Trevisani M, Martinet N, Caughey G, Tognetto M, Cavallesco G, Mapp C, Geppetti P, Bunnett NW. Expression and function of proteinase-activated receptor 2 in human bronchial smooth muscle. Am J Respir Crit Care Med. 2001;164:1276–81.
Schmidlin F, Amadesi S, Dabbagh K, Lewis DE, Knott P, Bunnett NW, Gater PR, Geppetti P, Bertrand C, Stevens ME. Protease-activated receptor 2 mediates eosinophil infiltration and hyperreactivity in allergic inflammation of the airway. J Immunol. 2002;169:5315–21.
Knight DA, Lim S, Scaffidi AK, Roche N, Chung KF, Stewart GA, Thompson PJ. Protease-activated receptors in human airways: upregulation of PAR-2 in respiratory epithelium from patients with asthma. J Allergy Clin Immunol. 2001;108:797–803.
Miotto D, Hollenberg MD, Bunnett NW, Papi A, Braccioni F, Boschetto P, Rea F, Zuin A, Geppetti P, Saetta M, et al. Expression of protease activated receptor-2 (PAR-2) in central airways of smokers and non-smokers. Thorax. 2002;57:146–51.
Ricciardolo FL, Steinhoff M, Amadesi S, Guerrini R, Tognetto M, Trevisani M, Creminon C, Bertrand C, Bunnett NW, Fabbri LM, et al. Presence and bronchomotor activity of protease-activated receptor-2 in Guinea pig airways. Am J Respir Crit Care Med. 2000;161:1672–80.
Carr MJ, Schechter NM, Undem BJ. Trypsin-induced, neurokinin-mediated contraction of Guinea pig bronchus. Am J Respir Crit Care Med. 2000;162:1662–7.
Lin C, von der Thusen J, Daalhuisen J, ten Brink M, Crestani B, van der Poll T, Borensztajn K, Spek CA. Protease-activated receptor (PAR)-2 is required for PAR-1 signalling in pulmonary fibrosis. J Cell Mol Med. 2015;19:1346–56.
Cork MJ, Danby SG, Vasilopoulos Y, Hadgraft J, Lane ME, Moustafa M, Guy RH, Macgowan AL, Tazi-Ahnini R, Ward SJ. Epidermal barrier dysfunction in atopic dermatitis. J Invest Dermatol. 2009;129:1892–908.
Derian CK, Eckardt AJ, Andrade-Gordon P. Differential regulation of human keratinocyte growth and differentiation by a novel family of protease-activated receptors. Cell Growth Differ. 1997;8:743–9.
D'Andrea MR, Derian CK, Santulli RJ, Andrade-Gordon P. Differential expression of protease-activated receptors-1 and -2 in stromal fibroblasts of normal, benign, and malignant human tissues. Am J Pathol. 2001;158:2031–41.
Wakita H, Furukawa F, Takigawa M. Thrombin and trypsin induce granulocyte-macrophage colony-stimulating factor and interleukin-6 gene expression in cultured normal human keratinocytes. Proc Assoc Am Physicians. 1997;109:190–207.
Kanke T, Macfarlane SR, Seatter MJ, Davenport E, Paul A, McKenzie RC, Plevin R. Proteinase-activated receptor-2-mediated activation of stress-activated protein kinases and inhibitory kappa B kinases in NCTC 2544 keratinocytes. J Biol Chem. 2001;276:31657–66.
Kawagoe J, Takizawa T, Matsumoto J, Tamiya M, Meek SE, Smith AJ, Hunter GD, Plevin R, Saito N, Kanke T, et al. Effect of protease-activated receptor-2 deficiency on allergic dermatitis in the mouse ear. Jpn J Pharmacol. 2002;88:77–84.
Lee SE, Jeong SK, Lee SH. Protease and protease-activated receptor-2 signaling in the pathogenesis of atopic dermatitis. Yonsei Med J. 2010;51:808–22.
Steinhoff M, Neisius U, Ikoma A, Fartasch M, Heyer G, Skov PS, Luger TA, Schmelz M. Proteinase-activated receptor-2 mediates itch: a novel pathway for pruritus in human skin. J Neurosci. 2003;23:6176–80.
Calabresi E, Petrelli F, Bonifacio AF, Puxeddu I, Alunno A. One year in review 2018: pathogenesis of rheumatoid arthritis. Clin Exp Rheumatol. 2018;36:175–84.
Kelso EB, Lockhart JC, Hembrough T, Dunning L, Plevin R, Hollenberg MD, Sommerhoff CP, McLean JS, Ferrell WR. Therapeutic promise of proteinase-activated receptor-2 antagonism in joint inflammation. J Pharmacol Exp Ther. 2006;316:1017–24.
Ferrell WR, Lockhart JC, Kelso EB, Dunning L, Plevin R, Meek SE, Smith AJ, Hunter GD, McLean JS, McGarry F, et al. Essential role for proteinase-activated receptor-2 in arthritis. J Clin Invest. 2003;111:35–41.
McCulloch K, McGrath S, Huesa C, Dunning L, Litherland G, Crilly A, Hultin L, Ferrell WR, Lockhart JC, Goodyear CS. Rheumatic disease: protease-activated Receptor-2 in synovial joint pathobiology. Front Endocrinol (Lausanne). 2018;9:257.
Crilly A, Palmer H, Nickdel MB, Dunning L, Lockhart JC, Plevin R, McInnes IB, Ferrell WR. Immunomodulatory role of proteinase-activated receptor-2. Ann Rheum Dis. 2012;71:1559–66.
Andrade-Gordon P, Maryanoff BE, Derian CK, Zhang HC, Addo MF, Darrow AL, Eckardt AJ, Hoekstra WJ, McComsey DF, Oksenberg D, et al. Design, synthesis, and biological characterization of a peptide-mimetic antagonist for a tethered-ligand receptor. Proc Natl Acad Sci U S A. 1999;96:12257–62.
Brass LF, Vassallo RR Jr, Belmonte E, Ahuja M, Cichowski K, Hoxie JA. Structure and function of the human platelet thrombin receptor. Studies using monoclonal antibodies directed against a defined domain within the receptor N terminus. J Biol Chem. 1992;267:13795–8.
Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS, Boykow G, Hsieh Y, Palamanda J, Agans-Fantuzzi J, et al. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J Med Chem. 2008;51:3061–4.
Kogushi M, Matsuoka T, Kawata T, Kuramochi H, Kawaguchi S, Murakami K, Hiyoshi H, Suzuki S, Kawahara T, Kajiwara A, Hishinuma I. The novel and orally active thrombin receptor antagonist E5555 (Atopaxar) inhibits arterial thrombosis without affecting bleeding time in guinea pigs. Eur J Pharmacol. 2011;657:131–7.
Covic L, Gresser AL, Talavera J, Swift S, Kuliopulos A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc Natl Acad Sci U S A. 2002;99:643–8.
Aisiku O, Peters CG, De Ceunynck K, Ghosh CC, Dilks JR, Fustolo-Gunnink SF, Huang M, Dockendorff C, Parikh SM, Flaumenhaft R. Parmodulins inhibit thrombus formation without inducing endothelial injury caused by vorapaxar. Blood. 2015;125:1976–85.
Becker RC, Moliterno DJ, Jennings LK, Pieper KS, Pei J, Niederman A, Ziada KM, Berman G, Strony J, Joseph D, et al. Safety and tolerability of SCH 530348 in patients undergoing non-urgent percutaneous coronary intervention: a randomised, double-blind, placebo-controlled phase II study. Lancet. 2009;373:919–28.
Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, Fox KA, Lipka LJ, Liu X, Nicolau JC, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366:1404–13.
Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, Van de Werf F, White HD, Aylward PE, Wallentin L, Chen E, et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012;366:20–33.
Goto S, Ogawa H, Takeuchi M, Flather MD, Bhatt DL, Investigators JL. Double-blind, placebo-controlled phase II studies of the protease-activated receptor 1 antagonist E5555 (atopaxar) in Japanese patients with acute coronary syndrome or high-risk coronary artery disease. Eur Heart J. 2010;31:2601–13.
Wiviott SD, Flather MD, O'Donoghue ML, Goto S, Fitzgerald DJ, Cura F, Aylward P, Guetta V, Dudek D, Contant CF, et al. Randomized trial of atopaxar in the treatment of patients with coronary artery disease: the lessons from antagonizing the cellular effect of thrombin-coronary artery disease trial. Circulation. 2011;123:1854–63.
O'Donoghue ML, Bhatt DL, Wiviott SD, Goodman SG, Fitzgerald DJ, Angiolillo DJ, Goto S, Montalescot G, Zeymer U, Aylward PE, et al. Safety and tolerability of atopaxar in the treatment of patients with acute coronary syndromes: the lessons from antagonizing the cellular effects of thrombin-acute coronary syndromes trial. Circulation. 2011;123:1843–53.
Gurbel PA, Bliden KP, Turner SE, Tantry US, Gesheff MG, Barr TP, Covic L, Kuliopulos A. Cell-penetrating Pepducin therapy targeting PAR1 in subjects with coronary artery disease. Arterioscler Thromb Vasc Biol. 2016;36:189–97.
Wilson SJ, Ismat FA, Wang Z, Cerra M, Narayan H, Raftis J, Gray TJ, Connell S, Garonzik S, Ma X, et al. PAR4 (protease-activated receptor 4) antagonism with BMS-986120 inhibits human ex vivo Thrombus formation. Arterioscler Thromb Vasc Biol. 2018;38:448–56.
Rasmussen UB, Gachet C, Schlesinger Y, Hanau D, Ohlmann P, Van Obberghen-Schilling E, Pouyssegur J, Cazenave JP, Pavirani A. A peptide ligand of the human thrombin receptor antagonizes alpha- thrombin and partially activates platelets. J Biol Chem. 1993;268:14322–8.
Seiler SM, Peluso M, Tuttle JG, Pryor K, Klimas C, Matsueda GR, Bernatowicz MS. Thrombin receptor activation by thrombin and receptor-derived peptides in platelet and CHRF-288 cell membranes: receptor-stimulated GTPase and evaluation of agonists and partial agonists. Mol Pharmacol. 1996;49:190–7.
Cisowski J, O'Callaghan K, Kuliopulos A, Yang J, Nguyen N, Deng Q, Yang E, Fogel M, Tressel S, Foley C, et al. Targeting protease-activated receptor-1 with cell-penetrating pepducins in lung cancer. Am J Pathol. 2011;179:513–23.
Cheung WM, D'Andrea MR, Andrade-Gordon P, Damiano BP. Altered vascular injury responses in mice deficient in protease-activated receptor-1. Arterioscler Thromb Vasc Biol. 1999;19:3014–24.
Young SE, Duvernay MT, Schulte ML, Lindsley CW, Hamm HE. Synthesis of indole derived protease-activated receptor 4 antagonists and characterization in human platelets. PLoS One. 2013;8:e65528.
Zhang HC, Derian CK, Andrade-Gordon P, Hoekstra WJ, McComsey DF, White KB, Poulter BL, Addo MF, Cheung WM, Damiano BP, et al. Discovery and optimization of a novel series of thrombin receptor (par-1) antagonists: potent, selective peptide mimetics based on indole and indazole templates. J Med Chem. 2001;44:1021–4.
Xu QL, Guo XH, Liu JX, Chen B, Liu ZF, Su L. Blockage of protease-activated receptor 1 ameliorates heat-stress induced intestinal high permeability and bacterial translocation. Cell Biol Int. 2015;39:411–7.
Nawata S, Murakami A, Hirabayashi K, Sakaguchi Y, Ogata H, Suminami Y, Numa F, Nakamura K, Kato H. Identification of squamous cell carcinoma antigen-2 in tumor tissue by two-dimensional electrophoresis [in process citation]. Electrophoresis. 1999;20:614–7.
Kato Y, Kita Y, Nishio M, Hirasawa Y, Ito K, Yamanaka T, Motoyama Y, Seki J. In vitro antiplatelet profile of FR171113, a novel non-peptide thrombin receptor antagonist. Eur J Pharmacol. 1999;384:197–202.
Tanaka M, Arai H, Liu N, Nogaki F, Nomura K, Kasuno K, Oida E, Kita T, Ono T. Role of coagulation factor Xa and protease-activated receptor 2 in human mesangial cell proliferation. Kidney Int. 2005;67:2123–33.
Kogushi M, Matsuoka T, Kobayashi H, Sato N, Suzuki S, Kawahara T, Kajiwara A, Hishinuma I. Biological characterization of ER129614-06, a novel, non-peptide protease-activated receptor-1 (PAR-1) antagonist. Atheroscler Suppl. 2003;4:245–6.
Kawahara T, Suzuki S, Kogushi M, Matsuoka T, Kobayashi H, Kajiwara A, Hishinuma I. Discovery and optimization of potent orally active small molecular thrombin receptor PAR-1 antagonists. Abstr Pap Am Chem Soc. 2003;225:U215.
Perez M, Lamothe M, Maraval C, Mirabel E, Loubat C, Planty B, Horn C, Michaux J, Marrot S, Letienne R, et al. Discovery of novel protease activated receptors 1 antagonists with potent antithrombotic activity in vivo. J Med Chem. 2009;52:5826–36.
Monjotin N, Gillespie J, Farrie M, Le Grand B, Junquero D, Vergnolle N. F16357, a novel protease-activated receptor 1 antagonist, improves urodynamic parameters in a rat model of interstitial cystitis. Br J Pharmacol. 2016;173:2224–36.
Ahn HS, Foster C, Boykow G, Stamford A, Manna M, Graziano M. Inhibition of cellular action of thrombin by N3-cyclopropyl-7-[[4-(1-methylethyl)phenyl]methyl]-7H-pyrrolo[3, 2-f]quinazoline-1,3-diamine (SCH 79797), a nonpeptide thrombin receptor antagonist. Biochem Pharmacol. 2000;60:1425–34.
Gupta N, Liu R, Shin S, Sinha R, Pogliano J, Pogliano K, Griffin JH, Nizet V, Corriden R. SCH79797 improves outcomes in experimental bacterial pneumonia by boosting neutrophil killing and direct antibiotic activity. J Antimicrob Chemother. 2018;73:1586–94.
Kogushi M, Kobayashi H, Matsuoka T, Suzuki S, Kawahara T, Kajiwara A, Hishinuma L. Anti-thrombotic and bleeding time effects of E5555, an orally active protease-activated receptor-1 antagonist, in Guinea pigs. Circulation. 2003;108:280.
Asteriti S, Daniele S, Porchia F, Dell'anno MT, Fazzini A, Pugliesi I, Trincavelli ML, Taliani S, Martini C, Mazzoni MR, Gilchrist A. Modulation of PAR(1) signaling by benzimidazole compounds. Br J Pharmacol. 2012;167(1):80–94.
Deng X, Mercer PF, Scotton CJ, Gilchrist A, Chambers RC. Thrombin induces fibroblast CCL2/JE production and release via coupling of PAR1 to Galphaq and cooperation between ERK1/2 and rho kinase signaling pathways. Mol Biol Cell. 2008;19:2520–33.
Tressel SL, Kaneider NC, Kasuda S, Foley C, Koukos G, Austin K, Agarwal A, Covic L, Opal SM, Kuliopulos A. A matrix metalloprotease-PAR1 system regulates vascular integrity, systemic inflammation and death in sepsis. EMBO Mol Med. 2011;3:370–84.
Lin C, Duitman J, Daalhuisen J, Ten Brink M, von der Thusen J, van der Poll T, Borensztajn K, Spek CA. Targeting protease activated receptor-1 with P1pal-12 limits bleomycin-induced pulmonary fibrosis. Thorax. 2014;69:152–60.
Dockendorff C, Aisiku O, Verplank L, Dilks JR, Smith DA, Gunnink SF, Dowal L, Negri J, Palmer M, Macpherson L, et al. Discovery of 1,3-diaminobenzenes as selective inhibitors of platelet activation at the PAR1 receptor. ACS Med Chem Lett. 2012;3:232–7.
De Ceunynck K, Peters CG, Jain A, Higgins SJ, Aisiku O, Fitch-Tewfik JL, Chaudhry SA, Dockendorff C, Parikh SM, Ingber DE, Flaumenhaft R. PAR1 agonists stimulate APC-like endothelial cytoprotection and confer resistance to thromboinflammatory injury. Proc Natl Acad Sci U S A. 2018;115:E982–91.
Zhong W, Chen S, Zhang Q, Xiao T, Qin Y, Gu J, Sun B, Liu Y, Jing X, Hu X, et al. Doxycycline directly targets PAR1 to suppress tumor progression. Oncotarget. 2017;8:16829–42.
Zhong W, Chen S, Qin Y, Zhang H, Wang H, Meng J, Huai L, Zhang Q, Yin T, Lei Y, et al. Doxycycline inhibits breast cancer EMT and metastasis through PAR-1/NF-kappaB/miR-17/E-cadherin pathway. Oncotarget. 2017;8:104855–66.
Bendeck MP, Conte M, Zhang M, Nili N, Strauss BH, Farwell SM. Doxycycline modulates smooth muscle cell growth, migration, and matrix remodeling after arterial injury. Am J Pathol. 2002;160:1089–95.
Wei H, Wei Y, Tian F, Niu T, Yi G. Blocking proteinase-activated receptor 2 alleviated neuropathic pain evoked by spinal cord injury. Physiol Res. 2016;65:145–53.
Al-Ani B, Saifeddine M, Wijesuriya SJ, Hollenberg MD. Modified proteinase-activated receptor-1 and -2 derived peptides inhibit proteinase-activated receptor-2 activation by trypsin. J Pharmacol Exp Ther. 2002;300:702–8.
Kanke T, Kabeya M, Kubo S, Kondo S, Yasuoka K, Tagashira J, Ishiwata H, Saka M, Furuyama T, Nishiyama T, et al. Novel antagonists for proteinase-activated receptor 2: inhibition of cellular and vascular responses in vitro and in vivo. Br J Pharmacol. 2009;158:361–71.
Goh FG, Ng PY, Nilsson M, Kanke T, Plevin R. Dual effect of the novel peptide antagonist K-14585 on proteinase-activated receptor-2-mediated signalling. Br J Pharmacol. 2009;158:1695–704.
Boitano S, Hoffman J, Flynn AN, Asiedu MN, Tillu DV, Zhang Z, Sherwood CL, Rivas CM, DeFea KA, Vagner J, Price TJ. The novel PAR2 ligand C391 blocks multiple PAR2 signalling pathways in vitro and in vivo. Br J Pharmacol. 2015;172:4535–45.
Suen JY, Barry GD, Lohman RJ, Halili MA, Cotterell AJ, Le GT, Fairlie DP. Modulating human proteinase activated receptor 2 with a novel antagonist (GB88) and agonist (GB110). Br J Pharmacol. 2012;165:1413–23.
Gardell LR, Ma JN, Seitzberg JG, Knapp AE, Schiffer HH, Tabatabaei A, Davis CN, Owens M, Clemons B, Wong KK, et al. Identification and characterization of novel small-molecule protease-activated receptor 2 agonists. J Pharmacol Exp Ther. 2008;327:799–808.
Chanakira A, Westmark PR, Ong IM, Sheehan JP. Tissue factor-factor VIIa complex triggers protease activated receptor 2-dependent growth factor release and migration in ovarian cancer. Gynecol Oncol. 2017;145:167–75.
Sun Q, Wang Y, Zhang J, Lu J. ENMD-1068 inhibits liver fibrosis through attenuation of TGF-beta1/Smad2/3 signaling in mice. Sci Rep. 2017;7:5498.
Barry GD, Suen JY, Le GT, Cotterell A, Reid RC, Fairlie DP. Novel agonists and antagonists for human protease activated receptor 2. J Med Chem. 2010;53:7428–40.
Lieu T, Savage E, Zhao P, Edgington-Mitchell L, Barlow N, Bron R, Poole DP, McLean P, Lohman RJ, Fairlie DP, Bunnett NW. Antagonism of the proinflammatory and pronociceptive actions of canonical and biased agonists of protease-activated receptor-2. Br J Pharmacol. 2016;173:2752–65.
Cheng RKY, Fiez-Vandal C, Schlenker O, Edman K, Aggeler B, Brown DG, Brown GA, Cooke RM, Dumelin CE, Dore AS, et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature. 2017;545:112–5.
Sevigny LM, Austin KM, Zhang P, Kasuda S, Koukos G, Sharifi S, Covic L, Kuliopulos A. Protease-activated receptor-2 modulates protease-activated receptor-1-driven neointimal hyperplasia. Arterioscler Thromb Vasc Biol. 2011;31:e100–6.
Michael ES, Kuliopulos A, Covic L, Steer ML, Perides G. Pharmacological inhibition of PAR2 with the pepducin P2pal-18S protects mice against acute experimental biliary pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2013;304:G516–26.
Ishikawa C, Tsuda T, Konishi H, Nakagawa N, Yamanishi K. Tetracyclines modulate protease-activated receptor 2-mediated proinflammatory reactions in epidermal keratinocytes. Antimicrob Agents Chemother. 2009;53:1760–5.
Liu XJ, Mu ZL, Zhao Y, Zhang JZ. Topical tetracycline improves MC903-induced atopic dermatitis in mice through inhibition of inflammatory cytokines and Thymic stromal Lymphopoietin expression. Chin Med J. 2016;129:1483–90.
Castro ML, Franco GC, Branco-de-Almeida LS, Anbinder AL, Cogo-Muller K, Cortelli SC, Duarte S, Saxena D, Rosalen PL. Downregulation of proteinase-activated Receptor-2, Interleukin-17, and other Proinflammatory genes by subantimicrobial doxycycline dose in a rat periodontitis model. J Periodontol. 2016;87:203–10.
Hollenberg MD, Saifeddine M. Proteinase-activated receptor 4 (PAR4): activation and inhibition of rat platelet aggregation by PAR4-derived peptides. Can J Physiol Pharmacol. 2001;79:439–42.
Kim HY, Goo JH, Joo YA, Lee HY, Lee SM, Oh CT, Ahn SM, Kim NH, Hwang JS. Impact on inflammation and recovery of skin barrier by nordihydroguaiaretic acid as a protease-activated receptor 2 antagonist. Biomol Ther (Seoul). 2012;20:463–9.
Yang J, Wu J, Kowalska MA, Dalvi A, Prevost N, O'Brien PJ, Manning D, Poncz M, Lucki I, Blendy JA, Brass LF. Loss of signaling through the G protein, Gz, results in abnormal platelet activation and altered responses to psychoactive drugs. Proc Natl Acad Sci U S A. 2000;97:9984–9.
Wu CC, Huang SW, Hwang TL, Kuo SC, Lee FY, Teng CM. YD-3, a novel inhibitor of protease-induced platelet activation. Br J Pharmacol. 2000;130:1289–96.
Wu CC, Hwang TL, Liao CH, Kuo SC, Lee FY, Lee CY, Teng CM. Selective inhibition of protease-activated receptor 4-dependent platelet activation by YD-3. Thromb Haemost. 2002;87:1026–33.
Young SE, Duvernay MT, Schulte ML, Nance KD, Melancon BJ, Engers J, Wood MR, Hamm HE, Lindsley CW. A novel and selective PAR4 antagonist: ML354. In: Probe reports from the NIH molecular libraries program. Bethesda: National Center for Biotechnology Information; 2015.
Wen W, Young SE, Duvernay MT, Schulte ML, Nance KD, Melancon BJ, Engers J, Locuson CW 2nd, Wood MR, Daniels JS, et al. Substituted indoles as selective protease activated receptor 4 (PAR-4) antagonists: discovery and SAR of ML354. Bioorg Med Chem Lett. 2014;24:4708–13.
Peek GJ, Elbourne D, Mugford M, Tiruvoipati R, Wilson A, Allen E, Clemens F, Firmin R, Hardy P, Hibbert C, et al. Randomised controlled trial and parallel economic evaluation of conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR). Health Technol Assess. 2010;14:1–46.
Wong PC, Seiffert D, Bird JE, Watson CA, Bostwick JS, Giancarli M, Allegretto N, Hua J, Harden D, Guay J, et al. Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding. Sci Transl Med. 2017;9. https://doi.org/10.1126/scitranslmed.aaf5294.
Hollenberg MD, Saifeddine M, Sandhu S, Houle S, Vergnolle N. Proteinase-activated receptor-4: evaluation of tethered ligand-derived peptides as probes for receptor function and as inflammatory agonists in vivo. Br J Pharmacol. 2004;143:443–54.
Acknowledgements
We would like to thank Hermenegild K. Heuberger for the drawing of the figures.
We thank Enago (http://www.enago.com) for the english language review.
Funding
We would like to express our sincere gratitude for the support given to RAS by the Swiss National Science Foundation grant #PZ00P3_136639 for the salary of DMH.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Author information
Authors and Affiliations
Contributions
Manuscript preparation by DMH and RAS. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Heuberger, D.M., Schuepbach, R.A. Protease-activated receptors (PARs): mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thrombosis J 17, 4 (2019). https://doi.org/10.1186/s12959-019-0194-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12959-019-0194-8