
Abstract
Background
In mammals, normal fertilization depends on the structural and functional integrity of the zona pellucida (ZP), which is an extracellular matrix surrounding oocytes. Mutations in ZP may affect oogenesis, fertilization and early embryonic development, which may cause female infertility.
Methods
A PubMed literature search using the keywords ‘zona pellucida’, ‘mutation’ and ‘variant’ limited to humans was performed, with the last research on June 30, 2022. The mutation types, clinical phenotypes and pregnancy outcomes were summarized and analyzed. The naive Bayes classifier was used to predict clinical pregnancy outcomes for patients with ZP mutations.
Results
A total of 29 publications were included in the final analysis. Sixty-nine mutations of the ZP genes were reported in 87 patients with different clinical phenotypes, including empty follicle syndrome (EFS), ZP-free oocytes (ZFO), ZP-thin oocytes (ZTO), degenerated and immature oocytes. The phenotypes of patients were influenced by the types and location of the mutations. The most common effects of ZP mutations are protein truncation and dysfunction. Three patients with ZP1 mutations, two with ZP2 mutations, and three with ZP4 mutations had successful pregnancies through Intracytoplasmic sperm injection (ICSI) from ZFO or ZTO. A prediction model of pregnancy outcome in patients with ZP mutation was constructed to assess the chance of pregnancy with the area under the curve (AUC) of 0.898. The normalized confusion matrix showed the true positive rate was 1.00 and the true negative rate was 0.38.
Conclusion
Phenotypes in patients with ZP mutations might be associated with mutation sites or the degree of protein dysfunction. Successful pregnancy outcomes could be achieved in some patients with identified ZP mutations. Clinical pregnancy prediction model based on ZP mutations and clinical characteristics will be helpful to precisely evaluate pregnancy chance and provide references and guidance for the clinical treatment of relevant patients.
Similar content being viewed by others


Introduction
Female infertility has become an increasingly prominent problem affecting approximately 50 million women worldwide [1]. Regular ovulation and healthy oocytes are the significant determinants of female fertility. Defects in this process usually lead to fertilization failure, as well as zygotic or embryonic arrest [2]. The anomalies of the oocyte are challenging to discover via routine examinations. Still, now with the development of in vitro fertilization (IVF) as a treatment for infertility, we have been able to retrieve oocytes and discover their defects in vitro [3].
In mammals, regular fertilization depends on the structural and functional integrity of the zona pellucida (ZP), an extracellular matrix surrounding oocytes [4]. The human ZP contains four glycoproteins (hZP1–4), only three of which (mZP1–3) are found in the mouse due to the pseudogenisation of ZP4 [5]. ZP glycoproteins contain multiple functional domains and are synthesized, processed, secreted, and assembled into long, cross-linked fibrils by growing oocytes [6, 7]. It’s reported that ZP glycoproteins play a critical role in oogenesis and ensure specific recognition of sperm-egg binding, inducing sperm exocytosis and the acrosome reaction [4, 8,9,10]. ZP glycoproteins also have a role in preventing polyspermy and protecting the growing embryo till implantation [4, 11].
Based on the crucial role of ZP, morphological assessment of ZP has become a meaningful way to determine the quality of oocytes in assisted reproductive technology (ART) [12]. ZP dysmorphology can include extracellular abnormalities such as a dark ZP, an irregularly shaped ZP, or the absence of a ZP, which contains an incidence of 2–5% of all oocytes [13, 14]. Previous reports suggested that abnormalities in the ZP of oocytes may result in poor pregnancy outcomes in vitro, but the mechanism of ZP dysmorphology is rarely reported in the literature [15]. In this context, some early evidence suggested that there might be a causal relationship between gene sequence variations (GSV) in hZP genes and female fertility [16,17,18]. Therefore, understanding the genetic mechanism behind abnormal ZP formation and maintenance is significant for the success rate of IVF.
In order to fully understand the effect of ZP mutations on female infertility, we collected all the case studies related to GSV in hZP1–4 up to June 30, 2022. In addition, we discussed the relationship between mutation types and clinical phenotypes, and established a model to predict pregnancy outcomes of patients with ZP mutations. The aim of our study was to assess the genetic mechanism of ZP defects, and to provide more reference and guidance for the clinical ART treatment and clinical outcome prediction of relevant patients.
Methods
Search strategy
Cases of patients with ZP mutations were identified through a literature review according to the Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) guideline [19]. The search was completed from January 1, 2014 to June 30, 2022 from the PubMed database using the following keywords: (“Zona Pellucida”[Mesh] OR “Zona pellucida”[All Fields]) AND (“Mutation”[Mesh] OR “Mutation”[All Fields] OR “Variant”[All Fields]). The search was restricted to publications with English titles and abstracts. Case reports and case series were included if they contained: (1) The reported new mutations need to meet the needs of previously unreported public databases, such as low allele frequencies in ExAC and gnomAD databases, mutations in exons or splice sites, and computer predictions of pathogenicity or possible pathogenicity. (2) The information of the probands is complete, including a full genetic pedigree, detailed medical records of the patients receiving ART, as well as fertilization and pregnancy outcomes. The references of publications were reviewed to identify additional cases. Publications and cases were excluded if ZP mutations were identified in other diseases instead of infertility or existed clear controversy. For each publication the following data were extracted: patient age and duration of infertility, ART treatment process, ZP mutations information, phenotypes and pregnancy outcomes.
Data analysis
All tests were performed using the statistical software R (http://www.r-project.org/). P < 0.05 was considered statistically significant. The idea of Bayes classifier is to calculate the joint probability distribution based on the assumption of feature independence. For a given input feature, the Bayes theorem is used to calculate the maximum posterior probability output category. In the Bayes classifier, instances of known classes are learned, a probabilistic model of attributes is established, and the model is used to predict the classification of new instances. In the small sample data set, the naive Bayes classifier still has a good performance, and the result calculation depends on the prior probability. Considering that the data in this study was less and the prior probability was easy to calculate, the naive Bayes classification method was chosen. In addition, in our previous study, the naive Bayes classifier has been applied to predict clinical pregnancy outcomes for transferred blastocysts with good prediction effect [20]. Therefore, based on mutation and clinical characteristics of patients, the naive Bayes classifier was used to predict clinical pregnancy outcomes for patients with ZP mutations, and the code editing was conducted in python (https://www.python.org/).
Supplementary information collection
To acquire detailed and long-term pregnancy outcomes, we emailed all authors of the included literature. The emails contained whether there were new IVF attempts or successful pregnancies achieved in the identified patients after the publication of the literature.
Results
Literature review
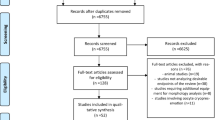
A PRISMA flowchart detailing the selection of publications is shown in Fig. 1. A total of 68 studies were identified through the keyword search. After duplicates were removed, 66 studies were screened via title and abstract for inclusion in the full-text review. Thirty-two articles underwent full-text review. Three studies were excluded because ZP mutations were identified in other diseases instead of infertility or existed clear controversy [21]. A total of 29 publications were included in the final analysis. Table 1 shows the relevant information of the included literature.

PRISMA flowchart detailing the selection of publications for this literature review of identified ZP mutations
Identified ZP mutations
A total of 47 ZP1 mutations have been reported in 19 pieces of literature, most of which are exon mutations, and only seven are intron splicing mutations. Mutations are distributed in all 12 exons of ZP1 gene. Corresponding protein variants are distributed in all seven functional domains of ZP1 protein, including 13 nonsense mutations, 16 missense mutations, seven splicing mutations and 11 frameshift mutations (Fig. 2A). Their genotypes are homozygous, heterozygous, or compound heterozygous, and are inherited in autosomal dominant (AD) or autosomal recessive (AR) manners, which cause different phenotypes like empty follicle syndrome (EFS), ZP-free oocytes (ZFO), degenerated or immature oocytes (Supplement Table 1).

Mutations in ZP genes and ZP proteins
A total of 14 ZP2 mutations have been reported in 10 pieces of literature, and only 1 is an intron splicing mutation, all the others are exon mutations. Mutations are distributed in exons 10,12,15 and 17. Corresponding protein variants are distributed in additional N-terminal ZP domain 3 (ZP-N3), ZP domain (ZPD), and consensus furin cleavage-site (CFCS), including one nonsense mutation, nine missense mutations, one splicing mutation and three frameshift mutations (Fig. 2B). Their genotypes are homozygous, heterozygous or compound heterozygous, and are inherited in AD or AR manners, which cause different phenotypes like EFS, ZFO, ZP-thin oocytes (ZTO), degenerated or immature oocytes (Supplement Table 2).
A total of 6 ZP3 gene mutations have been reported in 9 pieces of literature, all of them are exon mutations, distributed in exon 2 to exon 5, and corresponding protein variants are all distributed in ZPD, including three missense mutations and two frameshift mutations (Fig. 2C). Their genotypes are homozygous, heterozygous, or compound heterozygous, and are all inherited in AD manner, which causes EFS or ZFO (Supplement Table 3).
A total of 2 ZP4 mutations have been reported in 1 piece of literature, all of them are exon mutations, distributed in exon 3 and 10, and corresponding protein variants are distributed in additional N-terminal ZP domain 1 (ZP-N1) and ZPD, both of them are missense mutations (Fig. 2D). They are both heterozygous and inherited in an AD manner, resulting in ZTO (Supplement Table 4).
Clinical characteristics, phenotypes, and pregnancy outcomes of patients
Forty-eight primary infertile women with ZP1 mutations were reported in the literature, aged from 23 to 38 years old, with a history of infertility between 2 and 11 years. Of these patients, 37 showed EFS, degenerated or immature oocytes during different ART cycles, and all canceled the cycles (Table 2). ZFO was collected from 11 patients with different ART cycles; four were successfully fertilized and three were successfully conceived, all by intracytoplasmic sperm injection (ICSI) (Supplement Table 5). One patient carried AD heterozygous mutation c.326G > A; in her only ART cycle, eight cumulus-oocyte complexes (COCs) were collected by gonadotropin-releasing hormone agonist protocol, and eight oocytes were isolated, of which five degenerated and the remaining three were ZP-free mature oocytes. Two mature oocytes were successfully fertilized and developed to blastocyst state, while one oocyte degraded after ICSI. The patient finally successfully delivered an Apgarscore10 baby [12]. Another patient carried compound heterozygous mutation c.1100A > G and c.1215delG. The first two IVF cycles failed and the third cycle was changed to ICSI. After 2 cycles of oocyte extraction, a ZFO was obtained and fertilized successfully by ICSI. A 6 BC embryo was developed and cryopreserved. After another failure in egg retrieval, the frozen-thawed embryo transfer was performed and the patient delivered a healthy baby by planned cesarean section at 38 weeks of pregnancy [30]. The third patient carried homozygous missense mutation c.706 T > C; after the first failed ART, diagnostic intracytoplasmic sperm injection (D-ICSI) was used in the normal menstrual cycle to explore the feasibility of embryo development, then a new ART cycle was started to obtain 8 ZFO, 6 of them were fertilized successfully via ICSI, and finally delivered a healthy baby at 38 weeks of gestation by cesarean section. Additionally, we get the information from the author’s email that she is currently pregnant with twins through ICSI with the modified protocol [30, 34].
Twenty primary infertile women with ZP2 mutations were reported in pieces of literature, aged from 25 to 38 years old, with a history of infertility of 3 to 13 years. In different ART cycles, nine patients showed EFS, degenerated or immature oocytes, and the other 11 collected ZTO or ZFO (Table 2). Twelve patients experienced successful fertilization among them, but up to the time of publication of the literature, only two patients had successful pregnancies (Supplement Table 6). Patient with c.1115G > C mutation obtained only one poor quality embryo after four IVF cycles, but this embryo led to a successful pregnancy (specific clinical information was unknown) [29]. In the first cycle of the patient with c.1695-2A > G mutation, all the mature oocytes were divided into the IVF group and the ICSI group. None of the IVF group was fertilized. In the ICSI group, all 8 ZTO were fertilized, and two fresh 8-cell embryos were transferred, but the transfer failed. The remaining embryos formed two blastocysts and were cryopreserved. In the following frozen embryo transfer cycle, two vitrified blastocysts were thawed and transferred (1–4 BC, 1-5 BC), resulting in a singleton pregnancy with an estimated gestational age of 39 weeks [26].
Sixteen primary infertile women with ZP3 mutations were reported in the literature, aged from 26 to 38 years old, with a history of infertility of 2–11 years. Eleven patients showed EFS; the other five collected ZFO or ZTO in different ART cycles (Table 2). However, until now, no cases of successful pregnancy have been reported (Supplement Table 7).
Three primary infertile women with ZP4 mutations were reported in the literature, aged from 33 to 34 years old, with a history of infertility of 3 to 5 years (Supplement Table 8). All patients collected ZTO (Table 2). The first patient carried heterozygous mutation c.298G > A; after two failed cycles of IVF, ICSI was performed for the third time, three ZTO were obtained and all of them were fertilized. A six-cell cleavage embryo was adopted to transfer and finally achieved clinical pregnancy. Based on this successful experience, the other two patients carrying c.298G > A or c.1330G > C also achieved clinical pregnancy through ICSI treatment [43].
In general, phenotypes of ZP mutations reported so far include EFS, ZTO, ZFO degenerated and immature oocytes (Fig. 3). The phenotypes of patients are influenced by the types and location of the mutations. Protein truncation is the most common condition caused by nonsense and frameshift. The earlier the truncated sites appear, the less the protein functional domains are retained, which leads to different degrees of ZP defects. Successful fertilization and pregnancy have been reported in ZTO and ZFO phenotypes, which are less affected by mutations. In addition, successful pregnancy cases have been reported in ZP1, ZP2, and ZP4 mutations, while there has been no report in ZP3 mutations.

Different phenotypes of patients with ZP mutations. Different mutations of ZP can cause different clinical phenotypes, including empty follicle syndrome, ZP-thin oocytes, ZP-free oocytes, degenerated oocytes, and immature oocytes. COC, cumulus-oocyte complexes; EFS, empty follicle syndrome
Correlation analysis between the truncation sites and phenotypic severity
In order to explore whether the difference in phenotypic severity is affected by different truncation sites, we performed a correlation analysis with cases of ZP1 truncation mutations, which had a relatively large amount of data. Of the 41 ZP1 truncation mutations (including mutation site truncation and frameshift truncation), 33 cases were phenotyped as EFS/degenerated/immature and eight cases were phenotyped as ZFO. The Levene variance equality test in Rstudio4.1.1 showed F = 5.536, P = 0.024 < 0.05, therefore independent sample test without assuming equal variance was calculated as P = 0.065 > 0.05, indicating no significant difference between the truncation sites of the two phenotypes in the available data.
Correlation analysis of truncation mutations and EFS
The EFS was found to occur in many cases carrying truncation mutations in the study, so the correlation between mutation types and phenotypes was analyzed in Rstudio4.1.1 for 47 ZP1 mutation samples, where the mutation types were divided into truncation and substitution, and the phenotypes were divided into EFS and non-EFS. The four-compartment table is shown in Supplement Table 9, where N > 40 and T < 5, which needs to be corrected for chi-square values, and the corrected χ2 = 2.713, P = 0.10 > 0.05, indicating that the difference in the proportion of truncation mutation in EFS and non-EFS samples is not statistically significant in the available data.
Prediction model of pregnancy outcome
Patients with different mutation types may have different phenotypes and pregnancy outcomes. Therefore, we propose that analyses of information on related mutations and phenotypes of patients may help predict pregnancy outcomes. In this study, a naive Bayes model was constructed based on the analysis of 86 samples to predict clinical pregnancy outcomes by using four characteristics: the mutated ZP gene, the protein domain where the mutation is located, the type of mutation, and the clinical phenotype of the patient. The study used one-hot coding, coding non-pregnancy as 1 and pregnancy as 0.
The study used Python3.8.8 to construct the prediction model, and the performance of the constructed model is shown in Supplement Table 10. As shown in the table, the accuracy, precision, recall and F1 of the model are high. The receiver operating characteristic (ROC) curves illustrates the efficiency of mutation-related features in predicting pregnancy outcome. Figure 4A shows an area under the curve (AUC) of 0.898, indicating the model has good classification effectiveness. The normalized confusion matrix shows the true positive rate is 1.00 and the true negative rate is 0.38 (Fig. 4B).

Prediction model of pregnancy outcome. A. The receiver operating characteristic (ROC) curves of a mixture of four features, including the mutated ZP gene, the protein domain where the mutation is located, the type of mutation, and the clinical phenotype of the patient. The area under the curve (AUC) is 0.898. B. The normalized confusion matrix shows the true positive rate is 1.00 and the true negative rate is 0.38. The study used one-hot coding, coding non-pregnancy as 1 and pregnancy as 0
Discussion
ZP is an extracellular glycoproteinaceous coat surrounding oocytes and plays a vital role during oogenesis, fertilization, and preimplantation development [4, 5]. Failure to assemble a normal ZP around growing oocytes during oogenesis results in female infertility. According to the collected literature, gene sequence variations in hZP due to missense, nonsense, splicing or frameshift have been reported as the crucial cause of ZP formation defects and female fertility.
The ZP genes have previously been studied in mice. ZP1-null mice showed oocytes with loosely organized ZP; the mice were less fecund but fertile [50]. ZP2-null or ZP3-null mice produced eggs without a ZP, as well as infertility [51, 52]. But human patients with ZP mutations have a variety of phenotypes, ranging from a thin or absent ZP to the most severe manifestations of EFS (Fig. 3). The phenotypic differences between mice and humans may be due to the differences in the number and transport mechanism of human and mouse ZP glycoproteins, and the differences in sensibility to ZP defects [53]. Another crucial cause of phenotypic variability may be attributed to the different positions of the variants and the associated structural changes of the protein.
In terms of the types of mutation, missense results in the substitution of corresponding amino acid residues, which may affect the function of related ZP domains (Fig. 5). The N-terminal hydrophobic signal sequence (SS) can target ZP to the secretory pathway through co-translational import into the endoplasmic reticulum and which ultimately gets cleaved from the mature proteins by signal peptidase present in the oocytes [5]. Mutations in this domain may damage the expression of ZP [31]. ZPD plays an essential role in the secretion of ZP and the polymerization of ZP into filaments; mutations mainly occur in this region [3, 54]. Dysfunction of ZPD caused by mutation will not only influence the secretion of ZP but also primarily affect the interaction between ZP proteins [12, 29, 37]. Immediately after ZPD, all four human ZP glycoproteins have CFCS. Proteolytic cleavage at CFCS by proprotein convertase enzyme is critical for the secretion and assembly of ZP proteins [55]. Mutations in this domain show secretion defects and altered protein modification [41]. Downstream of CFCS, a hydrophobic transmembrane domain (TMD) and a short cytoplasmic tail (CT) are present in all four human ZP glycoproteins [3]. TMD can prevent ZP proteins’ premature intracellular interaction and thus plays a vital role in incorporating ZP proteins into the ZP matrix [56]. Dysfunction in this domain may cause intracellular sequestration and aggregation of ZP proteins [26, 40]. Besides ZPD, additional N-terminal ZP1 domain 1 (ZP1-N1) is another domain where mutations mainly occurred. Recent research suggests that ZP1-N1 cross-link formation is essential for the assembly or maintenance of the hZP and fertility, which shows that mutations in this domain may affect the cross-linking function of ZP1 and then influence the formation of the ZP matrix [12, 57].

Three dimensional models of ZP proteins with missense mutations. The structures of ZP proteins are predicted by AlphaFold from AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/). Alpha helixes are shown in red, beta folds are shown in yellow, loops are shown in green. Amino acid residues with missense mutations are shown in blue
As for nonsense and frameshift mutations, which usually cause the premature emergence of stop codons, the severity of clinical manifestations may depend on the residual function in the mutant ZP protein. We hypothesize that the earlier the truncation site appears, the less protein function remains, which may cause a more severe phenotype, with EFS/degeneration as the most severe phenotype reported in patients with ZP mutations. However, correlation analysis shows no significant difference in truncation sites across phenotypes (P = 0.065 > 0.05) and no significant difference in the proportion of truncation mutations in EFS and non-EFS (P = 0.10 > 0.05), which may be limited by the sample size.
EFS is a condition in which the ovarian response to stimulation and follicular development seems normal but no oocytes are retrieved for fertilization [58]. EFS can be classified as either false EFS (FEFS) or genuine EFS (GEFS). FEFS is mainly caused by pharmacological or iatrogenic problems [59, 60]. However, the estimated prevalence of GEFS is 0.016% among patients who underwent IVF [61, 62]. While some previous studies have suggested that dysfunctional folliculogenesis and ovarian aging are involved in GEFS, more recent efforts implicated substantial genetic factors contributing to GEFS, including mutations in luteinizing hormone/choriogonadotropin receptor (LHCGR) [63, 64], the pericentric inversion of chromosome 2, and mutations in ZP1, ZP2, and ZP3 as shown above. The phenotypes between mutations in the LHCGR and ZP genes present differently. Neither COCs nor oocytes could be identified in patients with LHCGR mutations, while COCs could be obtained but only degenerated oocytes or no oocytes could be identified in patients with ZP mutations (Fig. 3). Thus, some articles propose standardizing the definition of ZP-related EFS as a subtype of EFS characterized by degeneration [39]. In addition, the recovery of a few mature or immature oocytes from mature follicles has been described in some literature as a borderline form of EFS [65,66,67], which can also appear in ZP mutations (Fig. 3).
According to the statistics, mutations in ZP1 and ZP3 resulted in the loss of the ZP, and EFS occurred at a high rate (Table 2). This is consistent with the function of the relevant ZP proteins; that is, ZP1 serves to cross-link ZP filaments made up of ZP2–ZP3 dimers [50, 68], and ZP3 is indispensable for the formation of ZP and is also necessary for ovulation and producing the cleaved embryos [52, 69]. Lack of ZP might interfere with the bidirectional communication between oocytes and cumulus cells, then impedes nutritional supplements from cumulus cells leading to oocyte degeneration or increases in the fragility of oocytes during follicular puncture, ultimately resulting in EFS [8, 53, 70]. In addition, patients with ZP3 mutations have been no successful pregnancies so far (Table 2), which suggests a crucial role of ZP3 in ZP assembly and supports the opinion that the absence of ZP3 cannot be overcome [71]. Mutations in ZP2 were mainly manifested as ZTO, and the proportion of successfully fertilized oocytes was relatively high via ICSI (Table 2, Supplement Table 6). This may be because ZP2 is mainly involved in sperm recognition and binding, as well as the prevention of polyspermy [72]. Oocytes obtained from patients with ZP4 mutations were free of ZP and all showed successful fertilization and pregnancy via ICSI, which suggested that ZP4 variants perhaps do not dramatically affect embryonic development [43].
It’s worth noting that, the severity of patients’ phenotypes may be influenced by genotypes in addition to the degree of residual protein domains. In some patients, the presence of oocytes lacking ZP but not degenerated is likely to be dependent on the residual function of the mutant ZP protein, or the complementary role of one copy of the intact allele in heterozygous patients [36, 39]. Liu et al. used gene editing mice to further study two mutations they found in hZP (despite controversy), and found that either of the heterozygous mutations showed thinner ZP as compared to the oocytes obtained from wild mice. Moreover, it was reported that the mother of the patient carrying the ZP3 mutant was fertile, while the patient carrying both the ZP2 and ZP3 mutants showed much thinner ZP and complete fertilization failure, which suggests that these mutations have a dosage-dependent effect on the formation of ZP [73]. In addition, a case report showed that a frameshift mutation (c.860_861delTG) in ZP2 was inherited from the patient’s father, and the unaffected older sister who carried the heterozygous mutation alone had a child; only the patient who carried the compound heterozygous mutation is infertile [33]. It indicates that the genotypes of patients with homozygosis, heterozygosis, or compound heterozygosis might also affect phenotypes, and further research is needed.
According to our statistics, three patients with ZP1 mutations, two patients with ZP2 mutations, and three patients with ZP4 mutations had successful pregnancies through ICSI from ZFO or ZTO (except one patient with ZP2 mutation whose specific clinical information was not available) (Table 2), which suggests patients with ZP mutations still have chances of successful pregnancies. In fact, the developmental potential of the ZFO is reported to be similar to the zona intact oocytes in terms of fertilization, culture, and pregnancy outcomes [74]. Studies also show that although the ZP partially functions as a sperm receptor and a protective sheath for developing embryos during the cleavage stage, the ZP maybe not be an essential component for oocyte maturation and embryogenesis in the IVF settings, despite its importance during oocyte assessment and handling [32]. Therefore, for infertile patients with ZP defects or EFS, the possibility of ZP gene mutations should be considered, and whole-exome sequencing screening is recommended as a beneficial detective to identify the causes of infertility. What’s more, for patients with identified ZP mutations, the treatment plan of the following IVF attempt should consider the phenotype’s severity and the specific ZP mutation mode. If oocytes with ZP defects could be obtained from the affected patients, ICSI could be preferentially chosen instead of the traditional IVF, which may improve the probability of successful pregnancy via ART. Metwalley et al. proposed a new method called D-ICSI, an ICSI cycle performed in the natural cycle, to obtain information about embryo development potential before therapeutic ICSI [34]. For patients with the most severe phenotypes of EFS caused by ZP gene mutations, the current optimal treatment choice would be egg donation. Based on the evidence reported that the oocytes existed at least progressing normally through the preantral stages of folliculogenesis in EFS [25], a further approach may involve culturing early-stage oocytes in vitro to maturity before they can degenerate, but this treatment requires further study. In addition, for patients continuing to attempt IVF, the use of the pregnancy prediction model for ZP mutations is strongly recommended to assess the chances of pregnancy (Fig. 4), so as to provide better fertility counseling and reproductive guidance for patients.
Conclusion
In general, we summarized the currently reported cases with mutations in ZP genes. We found that different phenotypes in patients with ZP mutations might be associated with differences in mutation sites or the degree of protein dysfunction. Despite the significant impact on fertility, some patients with ZP mutations still have chances of successful pregnancies. Clinical pregnancy prediction model based on ZP mutations and clinical characteristics will be helpful to precisely evaluate pregnancy chance and provide references and guidance for the clinical treatment of relevant patients.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- AD:
-
autosomal dominant
- AR:
-
autosomal recessive
- ART:
-
assisted reproductive technology
- AUC:
-
area under the curve
- CFCS:
-
consensus furin cleavage-site
- COCs:
-
cumulus-oocyte complexes
- CT:
-
cytoplasmic tail
- D-ICSI:
-
diagnostic intracytoplasmic sperm injection
- EFS:
-
empty follicle syndrome
- FEFS:
-
false empty follicle syndrome
- GEFS:
-
genuine empty follicle syndrome
- GSV:
-
gene sequence variations
- hZP:
-
human zona pellucida
- ICSI:
-
intracytoplasmic sperm injection
- IVF:
-
in vivo fertilization
- LHCGR :
-
luteinizing hormone/choriogonadotropin receptor
- mZP:
-
mouse zona pellucida
- PRISM:
-
Preferred Reporting Items for Systematic reviews and Meta-Analyses
- ROC:
-
receiver operating characteristic
- SS:
-
signal sequence
- TMD:
-
transmembrane domain
- ZFO:
-
ZP-free oocyte
- ZP:
-
zona pellucida
- ZPD:
-
ZP domain
- ZP-N1:
-
N-terminal ZP domain 1
- ZP-N3:
-
N-terminal ZP domain 3
- ZP1-N1:
-
N-terminal ZP1 domain 1
- ZTO:
-
ZP-thin oocyte
References
Mascarenhas MN, Flaxman SR, Boerma T, Vanderpoel S, Stevens GA. National, regional, and global trends in infertility prevalence since 1990: a systematic analysis of 277 health surveys. PLoS Med. 2012;9(12):e1001356.
Nikiforov D, Grøndahl ML, Hreinsson J, Andersen CY. Human oocyte morphology and outcomes of infertility treatment: a systematic review. Reprod Sci. 2022;29(10):2768–85.
Wassarman PM, Litscher ES. Zona Pellucida Genes and Proteins: Essential Players in Mammalian Oogenesis and Fertility. Genes (Basel). 2021;12(8).
Litscher ES, Wassarman PM. Zona Pellucida proteins, fibrils, and matrix. Annu Rev Biochem. 2020;89:695–715.
Gupta SK. Human zona Pellucida glycoproteins: binding characteristics with human spermatozoa and induction of acrosome reaction. Front Cell Dev Biol. 2021;9:619868.
Bokhove M, Jovine L. Structure of zona Pellucida module proteins. Curr Top Dev Biol. 2018;130:413–42.
Louros NN, Chrysina ED, Baltatzis GE, Patsouris ES, Hamodrakas SJ, Iconomidou VA. A common 'aggregation-prone' interface possibly participates in the self-assembly of human zona pellucida proteins. FEBS Lett. 2016;590(5):619–30.
Matzuk MM, Burns KH, Viveiros MM, Eppig JJ. Intercellular communication in the mammalian ovary: oocytes carry the conversation. Science. 2002;296(5576):2178–80.
Familiari G, Relucenti M, Heyn R, Micara G, Correr S. Three-dimensional structure of the zona pellucida at ovulation. Microsc Res Tech. 2006;69(6):415–26.
Pang P-C, Chiu PCN, Lee C-L, Chang L-Y, Panico M, Morris HR, et al. Human sperm binding is mediated by the sialyl-Lewis(x) oligosaccharide on the zona pellucida. Science. 2011;333(6050):1761–4.
Wassarman PM. Zona pellucida glycoproteins. J Biol Chem. 2008;283(36):24285–9.
Cao Q, Zhao C, Zhang X, Zhang H, Lu Q, Wang C, et al. Heterozygous mutations in ZP1 and ZP3 cause formation disorder of ZP and female infertility in human. J Cell Mol Med. 2020;24(15):8557–66.
Rienzi L, Vajta G, Ubaldi F. Predictive value of oocyte morphology in human IVF: a systematic review of the literature. Hum Reprod Update. 2011;17(1):34–45.
Balaban B, Urman B, Sertac A, Alatas C, Aksoy S, Mercan R. Oocyte morphology does not affect fertilization rate, embryo quality and implantation rate after intracytoplasmic sperm injection. Hum Reprod. 1998;13(12):3431–3.
Sousa M. Teixeira da Silva J, Silva J, Cunha M, Viana P, Oliveira E, et al. embryological, clinical and ultrastructural study of human oocytes presenting indented zona pellucida. Zygote. 2015;23(1):145–57.
Männikkö M, Törmälä RM, Tuuri T, Haltia A, Martikainen H, Ala-Kokko L, et al. Association between sequence variations in genes encoding human zona pellucida glycoproteins and fertilization failure in IVF. Hum Reprod. 2005;20(6):1578–85.
Pökkylä R-M, Lakkakorpi JT, Nuojua-Huttunen SH, Tapanainen JS. Sequence variations in human ZP genes as potential modifiers of zona pellucida architecture. Fertil Steril. 2011;95(8):2669–72.
Margalit M, Paz G, Yavetz H, Yogev L, Amit A, Hevlin-Schwartz T, et al. Genetic and physiological study of morphologically abnormal human zona pellucida. Eur J Obstet Gynecol Reprod Biol. 2012;165(1):70–6.
Moher D, Shamseer L, Clarke M, Ghersi D, Liberati A, Petticrew M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst. Rev. 2015;4:1.
Zhu L, Li J, Wang M, Fang Z, Zheng F, Li Z, et al. Normalized mitochondrial DNA copy number can optimize pregnancy outcome prediction in IVF. Reprod Sci. 2021;28(5):1439–46.
Barbaux S, El Khattabi L, Ziyyat A. ZP2 heterozygous mutation in an infertile woman. Hum Genet. 2017;136(11–12):1489–91.
Huang H-L, Lv C, Zhao Y-C, Li W, He X-M, Li P, et al. Mutant ZP1 in familial infertility. N Engl J Med. 2014;370(13):1220–6.
Chen T, Bian Y, Liu X, Zhao S, Wu K, Yan L, et al. A recurrent missense mutation in ZP3 causes empty follicle syndrome and female infertility. Am J Hum Genet. 2017;101(3):459–65.
Yang P, Luan X, Peng Y, Chen T, Su S, Zhang C, et al. Novel zona pellucida gene variants identified in patients with oocyte anomalies. Fertil Steril. 2017;107(6):1364–9.
Dai C, Chen Y, Hu L, Du J, Gong F, Dai J, et al. ZP1 mutations are associated with empty follicle syndrome: evidence for the existence of an intact oocyte and a zona pellucida in follicles up to the early antral stage. A case report. Hum Reprod. 2019;34(11):2201–7.
Dai C, Hu L, Gong F, Tan Y, Cai S, Zhang S, et al. ZP2 pathogenic variants cause in vitro fertilization failure and female infertility. Genet Med. 2019;21(2):431–40.
Sun L, Fang X, Chen Z, Zhang H, Zhang Z, Zhou P, et al. Compound heterozygous ZP1 mutations cause empty follicle syndrome in infertile sisters. Hum Mutat. 2019;40(11):2001–6.
Yuan P, Li R, Li D, Zheng L, Ou S, Zhao H, et al. Novel mutation in the ZP1 gene and clinical implications. J Assist Reprod Genet. 2019;36(4):741–7.
Zhou Z, Ni C, Wu L, Chen B, Xu Y, Zhang Z, et al. Novel mutations in ZP1, ZP2, and ZP3 cause female infertility due to abnormal zona pellucida formation. Hum Genet. 2019;138(4):327–37.
Chu K, He Y, Wang L, Ji Y, Hao M, Pang W, et al. Novel ZP1 pathogenic variants identified in an infertile patient and a successful live birth following ICSI treatment. Clin Genet. 2020;97(5):787–8.
Liu M, Shen Y, Zhang X, Wang X, Li D, Wang Y. Novel biallelic loss-of-function variants in ZP1 identified in an infertile female with empty follicle syndrome. J Assist Reprod Genet. 2020;37(9):2151–7.
Zhang Z, Shangguan T, Li Y, He W. Loss of zona pellucida in oocytes due to compound heterozygous variants of ZP1 gene. Chin. J. Med. Genet. 2020;37(7):789–91.
Luo G, Zhu L, Liu Z, Yang X, Xi Q, Li Z, et al. Novel mutations in ZP1 and ZP2 cause primary infertility due to empty follicle syndrome and abnormal zona pellucida. J Assist Reprod Genet. 2020;37(11):2853–60.
Metwalley A, Brasha N, Esteves SC, Fawzy M, Brasha H, Hellani A, et al. Role of diagnostic intracytoplasmic sperm injection (ICSI) in the management of genetically determined zona pellucida-free oocytes during fertilization: a case report. Zygote. 2020;28(6):519–23.
Okutman Ö, Demirel C, Tülek F, Pfister V, Büyük U, Muller J, et al. Homozygous Splice Site Mutation in Causes Familial Oocyte Maturation Defect. Genes (Basel). 2020;11(4).
Xu Q, Zhu X, Maqsood M, Li W, Tong X, Kong S, et al. A novel homozygous nonsense ZP1 variant causes human female infertility associated with empty follicle syndrome (EFS). Mol Genet Genomic Med. 2020;8(7):e1269.
Chen Y, Wang Z, Wu Y, He W, Du J, Cai S, et al. Case report: a novel heterozygous deletion associated with empty follicle syndrome and abnormal follicular development. Front Genet. 2021;12:690070.
Sun Y, Zeng Y, Chen H, Zhou Z, Fu J, Sang Q, et al. A novel homozygous variant in ZP2 causes abnormal zona pellucida formation and female infertility. J Assist Reprod Genet. 2021;38(5):1239–45.
Wang J, Yang X, Sun X, Ma L, Yin Y, He G, et al. A novel homozygous nonsense mutation in zona pellucida 1 (ZP1) causes human female empty follicle syndrome. J Assist Reprod Genet. 2021;38(6):1459–68.
Wu L, Li M, Yin M, Ou Y, Yan Z, Kuang Y, et al. Novel mutations in ZP1: expanding the mutational spectrum associated with empty follicle syndrome in infertile women. Clin Genet. 2021;99(4):583–7.
Yang P, Chen T, Liu Y, Hou Z, Wu K, Cao Y, et al. The critical role of ZP genes in female infertility characterized by empty follicle syndrome and oocyte degeneration. Fertil Steril. 2021;115(5):1259–69.
Zhang D, Zhu L, Liu Z, Ren X, Yang X, Li D, et al. A novel mutation in ZP3 causes empty follicle syndrome and abnormal zona pellucida formation. J Assist Reprod Genet. 2021;38(1):251–9.
Wei X, Li Y, Liu Q, Liu W, Yan X, Zhu X, et al. Mutations in ZP4 are associated with abnormal zona pellucida and female infertility. J Clin Pathol. 2022;75(3):201–4.
Hou M, Zhu L, Jiang J, Liu Z, Li Z, Jia W, et al. Novel heterozygous mutations in ZP2 cause abnormal zona Pellucida and female infertility. Reprod Sci. 2022;29(10):3047–54.
Huo M, Zhang Y, Shi S, Shi H, Liu Y, Zhang L, et al. Gene Spectrum and clinical traits of nine patients with oocyte maturation arrest. Front Genet. 2022;13:772143.
Jia W, Xi Q, Zhu L, Luo Y, Li Z, Hou M, et al. Novel mutations in ZP2 and ZP3 cause female infertility in three patients. J Assist Reprod Genet. 2022;39(5):1205–15.
Shen Y, Guo J, Zhang X, Wang X, Zhu S, Chen D, et al. Identification of a heterozygous variant of ZP2 as a novel cause of empty follicle syndrome in humans and mice. Hum Reprod. 2022;37(4):859–72.
Zhang Z, Guo Q, Jia L, Zhou C, He S, Fang C, et al. A novel gene mutation in ZP3 loop region identified in patients with empty follicle syndrome. Hum Mutat. 2022;43(2):180–8.
Zou T, Xi Q, Liu Z, Li Z, Hou M, Zhu L, et al. A novel homozygous nonsense mutation in ZP1 causes female infertility due to empty follicle syndrome. Reprod Sci. 2022.
Rankin T, Talbot P, Lee E, Dean J. Abnormal zonae pellucidae in mice lacking ZP1 result in early embryonic loss. Development. 1999;126(17):3847–55.
Rankin TL, O'Brien M, Lee E, Wigglesworth K, Eppig J, Dean J. Defective zonae pellucidae in Zp2-null mice disrupt folliculogenesis, fertility and development. Development. 2001;128(7):1119–26.
Liu C, Litscher ES, Mortillo S, Sakai Y, Kinloch RA, Stewart CL, et al. Targeted disruption of the mZP3 gene results in production of eggs lacking a zona pellucida and infertility in female mice. Proc Natl Acad Sci U S A. 1996;93(11):5431–6.
Wang Y, Lv C, Huang H-L, Zeng M-H, Yi D-J, Tan H-J, et al. Influence of mouse defective zona pellucida in folliculogenesis on apoptosis of granulosa cells and developmental competence of oocytes†. Biol Reprod. 2019;101(2):457–65.
Jovine L, Darie CC, Litscher ES, Wassarman PM. Zona pellucida domain proteins. Annu Rev Biochem. 2005;74.
Kiefer SM, Saling P. Proteolytic processing of human zona pellucida proteins. Biol Reprod. 2002;66(2):407–14.
Jimenez-Movilla M, Dean J. ZP2 and ZP3 cytoplasmic tails prevent premature interactions and ensure incorporation into the zona pellucida. J Cell Sci. 2011;124(Pt 6):940–50.
Nishimura K, Dioguardi E, Nishio S, Villa A, Han L, Matsuda T, et al. Molecular basis of egg coat cross-linking sheds light on ZP1-associated female infertility. Nat Commun. 2019;10(1):3086.
Coulam CB, Bustillo M, Schulman JD. Empty follicle syndrome. Fertil Steril. 1986;46(6):1153–5.
Stevenson TL, Lashen H. Empty follicle syndrome: the reality of a controversial syndrome, a systematic review. Fertil Steril. 2008;90(3):691–8.
Beck-Fruchter R, Weiss A, Lavee M, Geslevich Y, Shalev E. Empty follicle syndrome: successful treatment in a recurrent case and review of the literature. Hum Reprod. 2012;27(5):1357–67.
Mesen TB, Yu B, Richter KS, Widra E, DeCherney AH, Segars JH. The prevalence of genuine empty follicle syndrome. Fertil Steril. 2011;96(6):1375–7.
Revelli A, Carosso A, Grassi G, Gennarelli G, Canosa S, Benedetto C. Empty follicle syndrome revisited: definition, incidence, aetiology, early diagnosis and treatment. Reprod BioMed Online. 2017;35(2):132–8.
Yuan P, He Z, Zheng L, Wang W, Li Y, Zhao H, et al. Genetic evidence of 'genuine' empty follicle syndrome: a novel effective mutation in the LHCGR gene and review of the literature. Hum Reprod. 2017;32(4):944–53.
Chen C, Xu X, Kong L, Li P, Zhou F, Zhao S, et al. Novel homozygous nonsense mutations in LHCGR lead to empty follicle syndrome and 46, XY disorder of sex development. Hum Reprod. 2018;33(7):1364–9.
Işik AZ, Vicdan K. Borderline form of empty follicle syndrome: is it really an entity? Eur J Obstet Gynecol Reprod Biol. 2000;88(2):213–5.
Cao X-L, Sun Z-G. Borderline form of empty follicle syndrome treated with a novel dual trigger method combined with delayed oocyte retrieval: a case report. World J Clin Cases. 2020;8(4):825–30.
Song J, Sun Z. A borderline form of empty follicle syndrome treated with a double-trigger of gonadotropin-releasing hormone agonist and human chorionic gonadotropin: a case report. Medicine (Baltimore). 2019;98(27):e16213.
Wassarman PM, Qi H, Litscher ES. Mutant female mice carrying a single mZP3 allele produce eggs with a thin zona pellucida, but reproduce normally. Proc Biol Sci. 1997;264(1380):323–8.
Rankin T, Familari M, Lee E, Ginsberg A, Dwyer N, Blanchette-Mackie J, et al. Mice homozygous for an insertional mutation in the Zp3 gene lack a zona pellucida and are infertile. Development. 1996;122(9):2903–10.
Tiwari M, Prasad S, Tripathi A, Pandey AN, Ali I, Singh AK, et al. Apoptosis in mammalian oocytes: a review. Apoptosis. 2015;20(8):1019–25.
Fahrenkamp E, Algarra B, Jovine L. Mammalian egg coat modifications and the block to polyspermy. Mol Reprod Dev. 2020;87(3):326–40.
Burkart AD, Xiong B, Baibakov B, Jiménez-Movilla M, Dean J. Ovastacin, a cortical granule protease, cleaves ZP2 in the zona pellucida to prevent polyspermy. J Cell Biol. 2012;197(1):37–44.
Liu W, Li K, Bai D, Yin J, Tang Y, Chi F, et al. Dosage effects of ZP2 and ZP3 heterozygous mutations cause human infertility. Hum Genet. 2017;136(8):975–85.
Ueno S, Bodri D, Uchiyama K, Okimura T, Okuno T, Kobayashi T, et al. Developmental potential of zona pellucida-free oocytes obtained following mild in vitro fertilization. Fertil Steril. 2014;102(6):1602–7.
Acknowledgments
We appreciate Dr. Jingyu Chen’s help in the data analysis of the prediction model.
Funding
This work was supported by research grants from the Health Commission of Hubei Province scientific research project (WJ2021M110), and the National Key Research and Development Project (2018YFC1002103).
Author information
Authors and Affiliations
Contributions
Juepu Zhou: literature review, writing and revising of text and tables. Meng Wang: Lead for prediction model, literature review, writing and revising. Qiyu Yang: Literature search and review, writing and revising. Dan Li: Lead for figures and Tables. Zhou Li: Tables, figures, literature review. Juan Hu: Literature review and revising. Lei Jin: Primary supervisor, conception and design, writing and revising. Lixia Zhu: Conception and design, writing and revising. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study has been approved by the Ethics Committee of Tongji Hospital of Tongji Medical College of Huazhong University of Science and Technology. Reference number for ethics approval is TJ-IRB20220450.
Consent for publication
All data were collected from published literature.
Competing interests
We have no conflicts of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, J., Wang, M., Yang, Q. et al. Can successful pregnancy be achieved and predicted from patients with identified ZP mutations? A literature review. Reprod Biol Endocrinol 20, 166 (2022). https://doi.org/10.1186/s12958-022-01046-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12958-022-01046-6