
Abstract
Background
Polycystic ovary syndrome (PCOS) is a complex reproductive disorder, that affects approximately 5–10% of women of reproductive age. The disease is complex because its evolution may be impacted by genetic, lifestyle and environmental factors. Previous studies have emphasized the important roles of estrogen receptors in the pathogenesis of PCOS.
Objective
To use whole exome sequencing (WES) to assess possible pathogenic factors in a PCOS patient who exhibited estrogen insensitivity during hormone replacement therapy (HRT) treatment.
Methods
Genome sequencing and variant filtering via WES were performed in a patient with PCOS. DNA extraction from 364 unrelated female controls without PCOS was followed by PCR amplification, Sanger sequencing and sequence alignment. Evolutionary conservation analysis, protein structural modelling and in silico prediction were applied to analyse the potential pathogenicity of the novel ESR1 mutation.
Result(s)
During the controlled ovarian hyperstimulation (COH) period of an IVF cycle, the patient experienced markedly prolonged ovarian stimulation due to a poor response to gonadotropins (Gn) and elevated serum FSH. A novel heterozygous ESR1 mutation, c.619G > A/p.A207T, leading to the replacement of a highly conserved alanine with a threonine, was identified in this patient, via WES analysis. This novel variant was not identified in 364 unrelated female controls without PCOS, or in the Exome Aggregation Consortium (ExAC) or 1000 Genome Project.
Conclusion(s)
We identified a novel heterozygous ESR1 mutation in a Han Chinese PCOS woman exhibiting clinical signs of estrogen insensitivity. This study may provide new strategies for IVF therapy, especially for patients who exhibit estrogen insensitivity during IVF cycle.
Similar content being viewed by others

Introduction
Polycystic ovary syndrome (PCOS) is a complex reproductive disorder, that affects approximately 5–10% of women of reproductive age [1]. It is characterized by hyperandrogenemia, chronic anovulation, oligomenorrhea/amenorrhea, and multiple cysts in the ovaries as observed via ultrasound examination and thus is one of the major causes of female subfertility [2]. The disease is complex as genetic, lifestyle and environmental factors all contribute to its aetiology [3,4,5].
Estrogen regulates diverse physiological functions in vertebrates, especially in the reproductive system [6]. The physiological functions of estrogen are mediated mainly by estrogen receptor alpha (ERα, ESR1) and estrogen receptor beta (ERβ, ESR2), both of which thought to be ligand inducible transcription factors that can dimerize and regulate the transcription of multiple downstream target genes [7, 8]. Previous studies showed that ERα knockout led to the development of PCOS in female mice, with multiple cystic follicles in the ovary and elevated levels of luteinizing hormone (LH) [9], while ERβ disturbance caused partially arrested follicular development and compromised fertility in female mice [10]. In addition, other studies have suggested that the expression of ERα was lower in proliferative endometria of PCOS patients than in control women [11], and that ERα and ERβ expression was significantly lower in PCOS granulosa cells than control granulosa cells [12]. These studies emphasized the important roles of the estrogen receptors in the pathogenesis of PCOS.
To date, germline mutations in the ESR1 and ESR2 genes have been reported in samples from only a few sporadic and hereditary PCOS cases [13,14,15,16]. A germline mutation in ESR2 was identified in three members with hereditary medullary thyroid carcinoma and this mutation was shown to promote cell proliferation in MCF-7 cells [16]. While only 5 females [14, 15, 17] and 2 males [13, 15] were reported to harbor ESR1 germline mutations, all these mutations were homozygous and associated with multicystic ovaries and delayed pubertal mammary gland development in females; furthermore, the ESR1-mutated women presented with complete estrogen insensitivity, and elevated levels of serum estrogen, follicle-stimulating hormone (FSH) and luteinizing hormone (LH) [14, 15, 17].
In the present study, we report a Han Chinese woman with PCOS exhibiting clinical signs of estrogen insensitivity in adjuvant therapy during the IVF cycle. Via whole exome sequencing (WES) analysis, we identified a novel heterozygous ESR1 mutation, c.619G > A/p.A207T, leading to the replacement of a highly conserved alanine with threonine at the 207th residue in this patient.
Materials and methods
Patient and hormone tests
Peripheral blood was collected from the PCOS sample with estrogen insensitivity in the Reproductive Medicine Center, Jiangxi Provincial Maternal and Child Health Hospital (Nanchang, China). Additionally, 364 unrelated female controls without PCOS were recruited from Jiangxi Maternal and Child Health Hospital. All participants provided written informed consent. This study complies with the Declaration of Helsinki and was approved by the Institutional Review Board of Jiangxi Provincial Maternal and Child Health Hospital. Before the initiation of therapy, the basal serum levels of estradiol (E2), FSH, LH, thyroid stimulating hormone (TSH), progesterone (PRGO), prolactin (PRL), testosterone (T), thyrotropin (TSH), free triiodothyronine (FT3), free thyroxine (FT4) and cancer antigen 125 (CA125), were measured by commercial kits as described previously [18]. Anti-Mullerian hormone (AMH) was measured by an automated Roche Elecsys instrument (Roche; Elecsys).
WES
Genomic DNA (gDNA) was isolated from peripheral blood leukocytes with a Blood DNA kit (OMEGA Bio-tek Inc., Doraville, GA, USA) according to the manufacturer’s instructions. The gDNA sample was quantified with a NanoDrop 2000 fluorospectrometer (Thermo Fisher Scientific, MA, USA). A total of 3 μg of DNA sample was used for high-throughput sequencing. Subsequent exome capture, library construction and high throughput sequencing were carried out at the Beijing Genomics Institute (BGI, Shenzhen, China). Prior to high-throughput sequencing with a BGISEQ-500 sequencer, exome capture was performed with a BGI Exome V4 Kit (59 Mb target region) and the sequencing libraries were constructed by the MGIEasy™ DNA Library Prep Kit V1 (BGI, Shenzhen, China, Cat No. 85–05533-00). The sample had an average coverage depth of ~ 100 x. The sequenced data were aligned to the human reference genome (GRCh37/hg19). The common variants (minor allele frequency > 0.01) found in the 1000 Genomes Project (1000G, http://www.1000genomes.org), dbSNP147 (https://www.ncbi.nlm.nih.gov/projects/SNP/) or ExAC (http://exac.broadinstitute.org/) were excluded for further analysis.
Candidate gene selection
Three online bioinformatics programs, MutationTaster (www.mutationtaster.org) [19], SIFT (http://sift.jcvi.org/) [20] and PolyPhen-2 (genetics.bwh.harvard.edu/pph2) [21], were applied to predict the potential pathogenicity of the rare variants/mutations. These programs automatically predict whether rare variants/mutations are likely pathogenic or benign.
Sanger sequencing
Sanger sequencing was used to confirm the presence of the variant. Meanwhile, gDNA was isolated from the peripheral blood samples of 364 unrelated female controls without PCOS with the Blood DNA kit (OMEGA Bio-Tek Inc., Doraville, GA, USA) according to the manufacturer’s instructions. A 302 bp PCR amplicon spanning exon 2 of the ESR1 gene (NM_001291230.1) was amplified with a pair of primers (ESR1_Forward: 5′-ttctaatgttaatggatt − 3′/ESR1_Reverse: 5′-ttcctcagtcgctttggctc-3′). PCR was carried out in 30 μl reactions containing 50 ng gDNA as the template, 1 × PCR buffer, 0.5 μM of each forward and reverse primer, 2.5 mM of MgCl2 (Takara Biotechnology), 2.5 mM dNTPs (Takara Biotechnology), and 1 U LA Taq (Takara Biotechnology). The amplification program for the PCR was performed in a Thermal Cycler 2720 (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA) with a three-step PCR protocol: an initial denaturation phase at 94 °C for 3 min, followed by 35 cycles of denaturation at 94 °C for 30 sec, annealing at 53 °C for 30 sec and extension at 72 °C for 30 sec, and ending with an extension phase at 72 °C for 10 minutes. The PCR products were visualized on a 1.5% agarose gel stained with ethidium bromide. The PCR products were then purified with a DNA purification kit (Tiangen, Beijing, China) and sequenced in both directions on an ABI 3730 XL Automatic Capillary DNA Sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA) with a BigDye terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, Calif, USA). The sequencing data were assembled and aligned to the corresponding genomic sequence (ESR1, NM_001291230.1) with the SeqMan II program in the LaserGene (DNAStar, Madison, WI, USA).
Evolutionary conservation analysis
The ESR1 protein sequences from 18 different vertebrate species, including Homo sapiens (NP_000116), Pan troglodytes (XP_009450519), Mus musculus (NP_001289460), Rattus norvegicus (NP_036821), Ovis aries (NP_000116), Bos taurus (NP_001001443), Gallus gallus (NP_990514), Sus scrofa (NP_999385), Canis lupus familiaris (NP_001273887), Equus caballus (NP_001075241), Tupaia chinensis (NP_001304001), Mustela putorius furo (XP_004753629), Oryctolagus cuniculus (XP_008261925), Pongo abelii (XP_002817538), Coturnix japonica (NP_001310118), Alligator sinensis (XP_014375965), Ceratotherium simum simum (NP_001266182) and Xenopus tropicalis (NP_988866), were subjected to evolutionary conservation analysis. Multiple sequence alignment was carried out with Molecular Evolutionary Genetics Analysis (MEGA) software (version 7.0) developed by the laboratory of Dr. Kumar [22].
Protein structural modelling
The PDB file of ESR1 was generated by SWISS-MODEL in the ExPASy database (http://www.expasy.org) based on the protein sequences of human ESR1. With the generated PDB file, the protein structure of ESR1 was generated by DeepView Swiss-PdbViewer 4.0 software, by selecting “show dots surface”, “show backbone oxygen” and “sender in solid 3D”. Within the structure of the wild-type ESR1 protein, mutated ESR1 protein (p.A207T) was generated by changing alanine to threonine at the 207th residue.
Results
Clinical findings
The patient was 36 years old and was diagnosed with PCOS when she was undergoing in vitro fertilization (IVF) treatment due to secondary infertility. Her height was 158 cm and weight was 53 kg, and her body mass index (BMI) was 21.23 (within the normal rang for adult women, 18.5–24.9). She gave birth to a daughter when she was 20 years old, and then suffered from three spontaneous abortions between 2013 and 2016. Before the initiation of therapy, the laboratory tests showed that she had a mildly elevated level of basic serum FSH (14.56 IU/L, normal follicular phase, 3.5 to 12.5) and AMH (6.45 ng/mL, normal follicular phase, 0.777 to 5.24), while the levels of other associated factors, including E2, LH, TSH, PRGO, PRL, T, TSH, FT3, FT4 and CA125, were within the normal range on the third day of menstruation (Table 1).
A “long-term” GnRH-a regimen protocol was selected, Pituitary downregulation with GnRH-a (3.75 mg, long acting Diphereline, Beaufour Ipsen, France) on Day 3 of the menstrual cycle, and 28 days later, endocrine examination showed that the serum FSH of the patient was still high (9.18 IU/L, reference value < 5 IU/L). The patient was injected with recombinant human FSH (r-FSH, 112.5 IU/d, Gonal-F, Merck Serono, Switzerland) for 6 days, and the follicles did not grow significantly. Then, human menopausal gonadotropin (HMG, 75 IU/d, H20045720, China) was injected for 4 days, but the follicles continued to develop slowly. The dose of HMG was increased (400 IU/d) and treatment continued for 24 days, only one dominant follicle (follicular diameter > 1.8 cm) developed, while other follicles were less than 1.4 cm in diameter. Afterwards, Gn was adjusted to HMG (375 IU/d) and recombinant human lutropin (r-LH, 75 IU/d, Luveris, Merck Serono, Switzerland) due to the low level of serum LH (0.45 IU/L). At day 31 of ovulation induction, transvaginal ultrasound monitors showed that bilateral follicles of the patient grow poorly, while E2 levels continued to decline (from 4763 pg/mL to 4132 pg/mL). Generally, during the controlled ovarian hyperstimulation (COH) regime in the IVF cycle, the patient had a markedly prolonged ovarian stimulation period due to poor response to Gn and elevated serum FSH, who experienced a 31-day-Gn administration after pituitary down-regulation with GnRH-a and exhibited slowly increased and fluctuant serum level of estrogen in the cycle (Fig. 1). These clinical features suggest an estrogen insensitivity condition.

Fluctuating serum estrogen (E2), FSH and LH levels of the patient with PCOS during a controlled ovarian hyperstimulation (COH) regime in the IVF cycle. A prolong- acting agonist protocol was selected. Before the initiation of therapy, the baseline serum E2, FSH and LH levels were tested on the third day of menstruation (0). Twenty-eight days after pituitary downregulation with GnRHa (3.75 mg), the patient received 31-day Gn administration and exhibited slowly increasing and fluctuating levels of serum E2 in the cycle
Exome sequencing and sanger sequencing validation
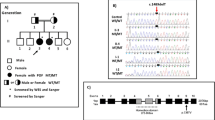
A total of 26.99 million raw reads were produced by sequencing, and over 96% of the sequenced bases possessed a quality score of Q20. The raw sequencing data generated more than 5000 Mb of effective data and the average sequencing depth on target areas was over 90×. In total, 1419 SNPs and 362 indels, including 1398 SNPs and 274 indels in the exonic regions, and 93 SNPs and 88 indels in splicing sites, were obtained. For the identified SNPs and indels, the synonymous variants and common variants (MAF > 0.01) deposited in the dbSNP147, 1000 Genomes Project and ExAC databases, were excluded. For the remaining nonsynonymous variants, we evaluated their potential pathogenicities with a combination of the SIFT, PolyPhen-2 and MutationTaster prediction programs. Based on these filtering criteria, a novel heterozygous missense variant in the exon 4 in the ESR1 gene (NM_001291230.1), c.619G > A (p.A207T), was identified. The identified ESR1 variant was validated by direct Sanger sequencing of the patient sample (Fig. 2A). This novel variant was not identified in 364 unrelated female controls without PCOS, or in the ExAC database and 1000 Genomes Project databases.

Evolutionary conservation analysis and protein structural modelling of the ESR1 (p.A207T) mutatiom. A Representative sequencing electropherogram of the ESR1 (p.A207T) mutation. The arrow indicates the location of the mutation. B Evolutionary conservation analysis of the ESR1 (p.A207T) mutation. Sequence alignment with other species indicates that the affected Ala (A) 207 residue is highly conserved in all 18 vertebrate species. C Structural difference between wild type ESR1 (p.A207) and mutated ESR1 (p.T207) proteins. The protein structures of wild type and mutated ESR1 proteins were modelled based on the crystal model of the human ESR1 protein. D Location of ESR1 missense mutations found in clinical samples. The A207 residue (red text) shows the novel mutation identified in our study. AF1, activation function 1; DBD, DNA binding domain; AF2, activation function 2; LBD, ligand binding domain
Evolutionary conservation analysis and protein structural modelling
Evolutionary conservation analysis based on 18 vertebrate species, from Homo sapiens to Xenopus tropicalis, showed that the ESR1 A207T mutation changed a highly conserved alanine to threonine at the 207th residue (Fig. 2B). The results of protein structural prediction showed that the ESR1 A207T mutation caused marked structural change when compared with its wild-type counterpart (Fig. 2C). The location of all of the reported ESR1 missense germline mutations in prior studies [13,14,15] and somatic mutations in clinical samples from the Catalogue of Somatic Mutations in Cancer (COSMIC) database (https://cancer.sanger.ac.uk/cosmic); among these mutations, A207T is located in the DNA binding domain (DBD) (Fig. 2D).
In silico prediction of ESR1 mutation
Three online bioinformatic programs, SIFT, PolyPhen-2 and MutationTaster, were applied to predict the potential pathogenicity of the ESR1 mutation. The ESR1 p.A207T mutation was predicted to be “damaging” and to affect protein function by SIFT, predicted to be “disease causing” by MutationTaster, and to be “probably damaging” with PolyPhen-2 (Table 2).
Discussion
Prior studies revealed that ERα-knockout mice presented with clinical characteristics of PCOS, including disturbance of late maturation process of follicle development and failure of ovulation and the presence of large cysts, with few effects on development at the primordial, primary and antral follicle stages [9, 23]. Furthermore, ERα-knockout mice exhibited reduced ovulatory capacity, impaired mammary gland development and increased levels of serum estradiol and LH, compared with age-matched wild-type mice [23,24,25,26]. In addition, a prior study also revealed that two knock-in mutations in the AF-2 domain of ESR1, p.L543A and p.L544A, led to PCOS and elevated serum levels of estradiol and LH in mice [27]. To data, in humans, germline mutations in the ESR1 gene have been reported in only three females [14, 15] and two males in human [13, 15]. All these patients exhibited clinical signs of estrogen insensitivity, namely, markedly elevated serum estrogen levels and mildly elevated gonadotropins (FSH and LH) [13,14,15]. Additionally, all ESR1-mutated women presented with bilateral multicystic ovaries and the absence of breast development [14, 15].
Here, we report a PCOS patient harboring a novel missense ESR1 mutation, who had normal basal serum estradiol and LH levels and elevated FSH and AMH, levels and underwent a markedly prolonged ovarian stimulation period and poor response to this stimulus after pituitary downregulation with GnRH-a, exhibiting characteristics of estrogen insensitivity during the COH phase of an IVF cycle. In contrast, all previously reported females with ESR1 germline mutations presented with markedly elevated levels of serum estrogen, FSH and LH, and complete estrogen insensitivity [14, 15]. Furthermore, breast development in this patient was unaffected, in contrast to the absence of breast development in previously described ESR1-mutated women [14, 15]. We speculate that the differences in clinical features and estrogen insensitivity severity between the present and prior studies [14, 15] could be due to the homozygous vs. heterozygous status of the ESR1 mutation, as the wild type ESR1 allele might confer some normal ERα function in this patient with heterozygous mutations.
The patient reported in the present study was diagnosed with PCOS and had multiple small follicles in the ovary, consistent with prior observations that ESR1-knockout and ESR1-mutated mice presented with characteristic features of PCOS [27]. The ovarian phenotypes of this patient seemed to be different from those observed previously, where bilateral enlarged multicystic ovaries, rather than PCOS phenotypes, were present [14, 15]. To date, all reported ESR1-mutated women, including those in the present and prior studies [14, 15], exhibited ovarian phenotypes; furthermore, PCOS is a multiple-factor complex disorder and a large proportion of women are diagnosed with PCOS at 30 years of age or older. Considering the young age of the previously reported female patients with ESR1 mutations (18, 21 and 25 years old, respectively), it is uncertain whether these patients will develop PCOS in the future.
It will be interesting to see whether there is any difference among the female individuals harboring either homozygous or heterozygous ESR1 mutation. For the unaffected mother and sister of the proband harboring heterozygous ESR1 mutation (p.R394H), they had normal puberty and underwent menarche at 14 and 15 years of age, respectively; furthermore, the proband’s mother has given birth to seven children and thus should be fertile [15], contrary to the secondary infertility of the patient in the present study. Notably, a newly published research reported two sisters from a consanguineous Jordanian family, displayed endocrine and ovarian defects of different severities despite they harbored the same homozygous ESR1 mutation (p.E385V); while we failed to get more information regarding the mutation status of ESR1 and the potential puberty and menarche condition for their mother [17]. For the homozygous ESR1 p.R157X men [13] and ESR1 p.Q375H women [14], we failed to know whether their mother and sister would exhibit similar phenotype with our sample [13]. Taken together, the clinical phenotype/symptoms of these females with either homozygous or heterozygous mutation in the ESR1 gene might be affected by age, genetic and environmental factors, it might be more complex than we thought.
A limitation of the present study was that we failed to recruit the family members of ESR1-mutated patients to determine whether other females in the family also harbor ESR1 mutation, and whether they might exhibit estrogen insensitivity, in certain circumstances. In addition, functional assays should be performed to clarify the potential mechanism of estrogen insensitivity conferred by the novel ESR1 mutation.
In summary, we identified a novel heterozygous ESR1 mutation in a Han Chinese PCOS patient who exhibited clinical signs of estrogen insensitivity. This study may provide new strategies for IVF therapy, especially for patients who exhibit estrogen insensitivity in the IVF cycle. Furthermore, WES is a cost-effective method to identify gene mutations that might be associated with clinically heterogeneous disorders and thus facilitate improvements in clinical therapy.
Availability of data and materials
The datasets used and/or analysed in the current study are available from the corresponding author on reasonable request.
References
March WA, Moore VM, Willson KJ, Phillips DI, Norman RJ, Davies MJ. The prevalence of polycystic ovary syndrome in a community sample assessed under contrasting diagnostic criteria. Hum Reprod. 2010;25(2):544–51.
Braillon A. Polycystic ovary syndrome. N Engl J Med. 2005;352(26):2756–7.
Azziz R. PCOS in 2015: new insights into the genetics of polycystic ovary syndrome. Nat Rev Endocrinol. 2016;12(2):74–5.
Martorell R, Khan LK, Hughes ML, Grummer-Strawn LM. Obesity in women from developing countries. Eur J Clin Nutr. 2000;54(3):247–52.
Barkley GS. Factors influencing health behaviors in the National Health and nutritional examination survey, III (NHANES III). Soc Work Health Care. 2008;46(4):57–79.
Gustafsson JA. What pharmacologists can learn from recent advances in estrogen signalling. Trends Pharmacol Sci. 2003;24(9):479–85.
Arao Y, Korach KS. The F domain of estrogen receptor α is involved in species-specific, tamoxifen-mediated transactivation. J Biol Chem. 2018;293(22):8495–507.
Xu XL, Deng SL, Lian ZX, Yu K. Estrogen receptors in polycystic ovary syndrome. Cells. 2021;10(2):459.
Schomberg DW, Couse JF, Mukherjee A, Lubahn DB, Sar M, Mayo KE, et al. Targeted disruption of the estrogen receptor-alpha gene in female mice: characterization of ovarian responses and phenotype in the adult. Endocrinology. 1999;140(6):2733–44.
Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, et al. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci U S A. 1998;95(26):15677–82.
Hulchiy M, Nybacka Å, Sahlin L, Hirschberg AL. Endometrial expression of estrogen receptors and the androgen receptor in women with polycystic ovary syndrome: a lifestyle intervention study. J Clin Endocrinol Metab. 2016;101(2):561–71.
Artimani T, Saidijam M, Aflatoonian R, Amiri I, Ashrafi M, Shabab N, et al. Estrogen and progesterone receptor subtype expression in granulosa cells from women with polycystic ovary syndrome. Gynecol Endocrinol. 2015;31(5):379–83.
Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331(16):1056–61.
Quaynor SD, Stradtman EW Jr, Kim HG, Shen Y, Chorich LP, Schreihofer DA, et al. Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant. N Engl J Med. 2013;369(2):164–71.
Bernard V, Kherra S, Francou B, Fagart J, Viengchareun S, Guéchot J, et al. Familial multiplicity of estrogen insensitivity associated with a loss-of-function ESR1 mutation. J Clin Endocrinol Metab. 2017;102(1):93–9.
Smith J, Read ML, Hoffman J, Brown R, Bradshaw B, Campbell C, et al. Germline ESR2 mutation predisposes to medullary thyroid carcinoma and causes up-regulation of RET expression. Hum Mol Genet. 2016;25(9):1836–45.
Delcour C, Khawaja N, Gonzalez-Duque S, Lebon S, Talbi A, Drira L, et al. Estrogen receptor α inactivation in 2 sisters: different phenotypic severities for the same pathogenic variant. J Clin Endocrinol Metab. 2022;107(6):e2553–62.
Wu J, Zou Y, Luo Y, Guo JB, Liu FY, Zhou JY, et al. Prevalence and clinical significance of mediator complex subunit 12 mutations in 362 Han Chinese samples with uterine leiomyoma. Oncol Lett. 2017;14(1):47–54.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–2.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20(3):358–417.
Couse JF, Bunch DO, Lindzey J, Schomberg DW, Korach KS. Prevention of the polycystic ovarian phenotype and characterization of ovulatory capacity in the estrogen receptor-alpha knockout mouse. Endocrinology. 1999;140(12):5855–65.
Couse JF, Yates MM, Walker VR, Korach KS. Characterization of the hypothalamic-pituitary-gonadal axis in estrogen receptor (ER) null mice reveals hypergonadism and endocrine sex reversal in females lacking ERalpha but not ERbeta. Mol Endocrinol. 2003;17(6):1039–53.
Hewitt SC, Kissling GE, Fieselman KE, Jayes FL, Gerrish KE, Korach KS. Biological and biochemical consequences of global deletion of exon 3 from the ER alpha gene. FASEB J. 2010;24(12):4660–7.
Arao Y, Hamilton KJ, Ray MK, Scott G, Mishina Y, Korach KS. Estrogen receptor α AF-2 mutation results in antagonist reversal and reveals tissue selective function of estrogen receptor modulators. Proc Natl Acad Sci U S A. 2011;108(36):14986–91.
Funding
This work was supported by the Social Development Foundation of Jiangxi Province (No. 20203BBGL73159, 20203BBGL73135 and 20203BBG73042), the Postdoctoral Science Foundation of Jiangxi Province of China (No. 2018KY21), and the Science and Technology Plan of Health Commission of Jiangxi Province (No. 202130760).
Author information
Authors and Affiliations
Contributions
Liu FY: project development, project manager, manuscript writing. Tian LF: data collection/analysis, manuscript editing. Tan J: data extraction, data analysis. Li ZM: data extraction, data analysis. Qin HY: data extraction, data analysis. Xu DF: material preparation, DNA isolation, exomic sequencing. Huang ZH: bioinformatic evaluation. Wu XW: bioinformatic evaluation. Chen G: material preparation, DNA isolation, Sanger sequencing. Wu QF: project development, manuscript editing, supervision. Zou Y: project manager, manuscript editing, supervision. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to particiapte
This study was approved by the ethics committee of Jiangxi Provincial Maternal and Child Health Hospital. All procedures in this study involving human participants were performed in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. This manuscript does not contain any studies with animals performed by any of the authors.
Consent for publication
Consent for publication was obtained from all authors. Informed consent was obtained from the participant in the study.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, F., Tian, L., Tan, J. et al. Identification of a novel ESR1 mutation in a Chinese PCOS woman with estrogen insensitivity in IVF treatment. Reprod Biol Endocrinol 20, 157 (2022). https://doi.org/10.1186/s12958-022-01029-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12958-022-01029-7