
Abstract
Endometriosis is major gynecological disease that affects over 10% of women worldwide and 30%-50% of these women have pelvic pain, abnormal uterine bleeding and infertility. The cause of endometriosis is unknown and there is no definite cure mainly because of our limited knowledge about its pathophysiology at the cellular and molecular levels. Therefore, demystifying the molecular mechanisms that underlie endometriosis is essential to develop advanced therapies for this disease. In this regard, HOX genes are remarkable because of their critical role in endometrial development and receptivity during implantation, which is attributed to their ability to mediate some of the sex steroid functions during the reproductive period. Access to the expression profiles of these genes would provide the necessary information to uncover new genes for endometriosis and assist with disease diagnosis and treatment. In this study we demonstrate an altered expression pattern for the HOX clusters (A-D) and their cofactors in both eutopic and ectopic conditions compared to control tissue biopsies. Remarkably, most of the intensive changes occurred in eutopic samples from endometriosis patients compared to control tissue biopsies. Pathway analysis revealed the involvement of differentially expressed genes in cancer that correlate with an association between endometriosis and cancer. Our results suggest critical roles for the HOX cluster and their cofactors in endometriosis pathophysiology.
Similar content being viewed by others

Background
Endometriosis is an estrogen-dependent gynecological disorder that affects 10%-15% of reproductive age women [1, 2]. It is characterized by the presence of endometrial tissues located outside of the uterus. The symptoms include dysmenorrhea, pelvic pain, pelvic masses, and infertility [3,4,5,6]. Social relationships, sexuality, and mental health are affected by endometriosis [7]. There is no definitive cure for all patients who suffer from endometriosis. Therefore, developing advanced efficient therapies in addition to prevention of endometriosis development and recurrence are of utmost importance [8].
It is essential to demystify the molecular mechanisms that underlie endometriosis in order to develop advanced therapies for this disease. In this regard, HOX genes are remarkable because they regulate endometrial development and its receptivity during implantation [9]. These genes are essential for endometrial growth, differentiation, and receptivity because they mediate some functions of the sex steroids during each reproductive period [10]. Dysregulation in HOXD3 [10], HOXA11 [11], and HOXB3 [10] have been reported in endometriosis patients. However, the changes in the expression levels of other HOX genes or HOX-cofactor genes in patients with endometriosis have not been elucidated and interactions between these genes remain to be determined.
Access to expression profiles of these genes would provide the information needed to uncover new genes that could assist with endometriosis diagnosis and treatment. In this study, we report about the expression profiles of HOX genes, their cofactors, and the interactions between these genes.
Materials and methods
Patients and tissue collection
The Ethics Committee of the Clinical and Research Centers for Infertility, Yazd, Iran and the Research Ethics Committee of Royan Institute, Tehran, Iran (IR.ACECR.ROYAN.REC.1396.238) approved this cross-sectional study. All methods were carried out in accordance with the approved guidelines [12]. Participants signed an informed consent in accordance with the guidelines of the Declaration of Helsinki (2000 revision) and tissue samples were collected after receipt of participants’ consent.
Study participants comprised 15 women with stages III or IV endometriosis who underwent laparoscopic surgery. The participants ranged in age from 24 to 38 years. Ectopic endometrial samples were collected from ectopic sites in the abdomen and eutopic endometrium samples were obtained by pipelle sampling. None of patients had evidence of any endometrial hyperplasia, visible endometrial hyperplasia or neoplasia, or any inflammatory diseases.
The control group consisted of 15 patients (22 to 36 years old) who underwent diagnostic laparoscopy for secondary infertility. Control group participants had no evidence of endometriosis, polyps, myoma, or reproductive inflammatory diseases and had at least one child conceived by natural pregnancy. Normal endometrial samples were taken via pipelle sampling from these women during diagnostic laparoscopy procedures. Confounding factors for samples are listed in Table S1.
All study participants did not have hormone therapy or an intrauterine device for three months before the surgical procedures, and all were in the follicular phase of their menstrual cycles. The obtained endometrial samples were placed immediately in an RNA protection reagent, RNAlater (Ambion, Austin, TX), frozen in liquid nitrogen, and stored at -80 °C.
RNA extraction and cDNA synthesis
We pooled five tissue samples in the ectopic, eutopic, and normal groups as three biological repeats (each combined five tissue samples served as one biological repeat) and the following analyses were performed on the nine samples.
RNA extraction of collected tissues was performed by using an RNeasy Microarray Tissue Mini Kit (Qiagen, cat. no: 73304) according to the manufacturer’s instructions. RNA quality and concentration were measured by using a Nanodrop 2000 spectrophotometer (Thermo Scientific). Then, cDNA synthesis was performed with an RT2 First Strand Kit (Qiagen, cat. no: 330404).
PCR array
The PCR array was conducted for the HOX family genes by using an RT2 Profiler PCR Array Human Homeobox Gene Kit (Qiagen, cat. no: PAHS-083Z) and an RT2 SYBR Green ROX qPCR Master Mix (Qiagen, cat. no: 330502) [13]. There were primer sets for 84 tests and the housekeeping genes on mentioned HOX PCR array kit, according to the manufacturer’s instructions. Cycling conditions for the StepOnePlus Real-time PCR system [14] were: an initial denaturation at 95 °C for 10 min followed by 40 cycles at 95 °C for 15 s, and 60 °C for one minute. Results were expressed as values of the cycle threshold [15] and then normalized to GAPDH, as the housekeeping gene (control gene). The 2−∆∆Ct method was used to calculate fold changes in mRNA abundance.
Statistical analysis
The statistical significance of difference in expression levels between groups was analyzed using the ANOVA test. P-values < 0.05 were considered to be statistically significant. Principal component analysis (PCA) [16] was carried out using Minitab 16 statistical software to simplify the large amount of data. PCA was applied on log2 of differentially expressed genes (DEGs) and non-DEGs data separately, to underlying cluster structures of endometrial samples.
Hierarchical clustering [17] was implemented by using the correlation coefficient and complete linkage in Minitab 16 statistical software [18]. The methods were performed twice with normalized gene expression data from the DEGs and non-DEGs among the sample groups, in three clusters.
Construction of the gene co-expression network
The gene co-expression networks were constructed based on the normalized gene expression data. MATLAB was used to compute the Pearson correlation between each pair of genes, then the significant correlation pairs (p < 0.05) were imported into Cytoscape software (version 3.6.1) [19] for visualization and the circle algorithms were used [20]. The co-expression networks of the normal tissues were constructed.
Coding gene functional analysis
Gene function was annotated based on Gene Ontology (GO), the intervention in other diseases, and expression in other tissues by using DAVID (https://david.ncifcrf.gov) [21] to clarify the function of the DEGs and the mechanism of endometriosis.
Results
Differentially expressed genes (DEGs)
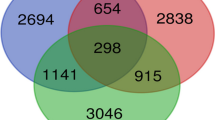
We evaluated 84 genes from the HOX clusters A-D and their cofactors in the control, eutopic, and ectopic tissue biopsies. Figure 1A-D and supplementary figures S1-S3 show the changes in expression patterns of HOX clusters A-D and their cofactors, respectively. Table S2 lists the genes that showed significant differential expression (DEGs) (P < 0.05) in the ectopic and eutopic groups compared to the control group in addition to DEGs in the ectopic compared to the eutopic group. Among the genes we evaluated in this study, 38.1% (No: 32/84) were non-DEG and about 61.9% (52/84) were differentially expressed between the ectopic, eutopic, and control groups. There were 18 genes from the DEG that were common among the studied groups (eutopic, ectopic and control tissues) (Table S2, Fig. 2 A,B). Table S2 lists the various cluster of genes that had different expression patterns in the studied groups.

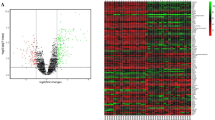
Gene expression array analysis for HOX genes. There were three biological repeats in each group. A, B, C and D in each graph show the nine data that were analyzed with SPSS software. P < 0.05 was considered to be significant

A Venn diagram for DEGs (P < 0.05). DEGs in the ectopic samples compared to normal samples (black ellipse), eutopic samples in comparison to normal samples (green ellipse), and ectopic samples compared to eutopic samples (red ellipse). Genes that were significantly upregulated are enclosed in black, green and red rectangles according to the above mentioned legend. In the multi-colored rectangles, each color represents upregulation in the relevant analysis in the corresponding agreement. B Heat map for all genes. Right column is logFC of ectopic samples compared to normal samples and the left column is the logFC of eutopic samples in comparison to normal samples. logFC: Logarithm fold change; UP: Significant upregulation; Down: Significant downregulation; None: Non-significant changes. C,D PCA analysis of gene expression in the nine sample groups. Triangle, circle, and square represent normal endometrial tissues of the control women, and ectopic endometrial tissues and eutopic endometrial tissues from patients, respectively. C: PCA of DEGs. D: PCA of non-DEGs
For further investigation, we performed PCA on the nine studied samples from the ectopic, eutopic, and control groups by separate analysis of the DEGs and non-DEGs (Fig. 2 C,D). Each point in a PCA graph represented one sample and all samples were properly classified into separate clusters based on the DEGs; however, the non-DEGs could not correctly classify the same samples. Then, we performed hierarchical clustering in the DEGs (Fig. S4-A) and non-DEGs (Fig. S4-B) in the eutopic, ectopic, and control samples. Hierarchical clustering analysis for the DEGs showed that control tissues were clustered together, and the eutopic and ectopic samples were classified in separate clusters (Supplementary figure S4).
Co-expression networks
We identified three co-expression networks based on pairwise correlation of the gene expression data and visualized them using Cytoscape (Fig. 3A). Network A was mainly occupied by genes that had significant expression changes in eutopic tissue compared to control tissue (78.6%); however, most genes in networks B and C showed significant expression changes in ectopic tissues. A large number of genes from all of the networks had significant expression changes in both ectopic and eutopic tissues compared to control tissues (Fig. 3B).

A Co-expression gene network. Blue, yellow, and green colors represent genes whose expressions significantly changed in the ectopic, eutopic and both tissues, respectively. B The number of genes with significant different expressions between sample tissues. All genes: The total number of nodes, Eutopic: Number of nodes with significant changes in gene expressions of the eutopic to control tissues, Ectopic: Number of nodes with significant changes in gene expression of the ectopic to control tissues, All DEGs: Number of nodes with significant changes in gene expressions of the patient to control tissues
Pathway analysis
The KEGG and Reactome databases were searched to determine the signaling pathways with involvement of networks A, B, and C. The number of genes grouped in networks A, B, and C were involved in “activation of anterior HOX genes in hindbrain development during early embryogenesis”. Also, the number of genes grouped in network A were involved in “signaling pathways regulating pluripotency of stem cells”. Some genes from network B were involved in “transcriptional misregulation in cancer” (Table 1).
Functional analysis
DEGs and the genes in networks A, B, and C were mapped to the DAVID database to investigate GO, interference with other diseases, and expressions in other tissues (Tables 2, 3, S2 and S4).
Analysis of interference with other diseases
Many DEGs in ectopic tissues play a role in developmental, neurological, and metabolic diseases. Numerous DEGs in eutopic tissues are also involved in developmental and neurological disease classes such as Cleft lip, Cleft palate, and Parkinson's disease. As shown in Table 2, the genes in networks A, B, and C were mostly involved in developmental diseases, while the genes in network A were involved in metabolic diseases.
Expressions in other tissues
Many DEGs in the eutopic and ectopic tissues were also expressed in different tissues, such as the cerebellar tissue, craniofacial tissue, salivary glands, and the adrenal cortex. Moreover, DEGs in the eutopic tissues were also identified in Burkitt lymphoma Raji and BM-CD33 + Myeloid (Table 3).
Network A genes were expressed in the cerebellar, craniofacial, salivary gland, lymphoma Burkitts Raji and BM-CD33 + Myeloid tissues. Genes in network B were expressed in uterine tumor tissue and embryo development. Network C genes were identified in embryo development (Table 3).
Gene Ontology (GO) annotation analysis
On the basis of the GO annotation, most DEGs in eutopic and ectopic tissues were involved in the transcription and development or morphogenesis of various systems. “Anterior/posterior pattern specification”, “embryonic skeletal system morphogenesis”, “dopaminergic neuron differentiation”, and “multicellular organism development” were some of the most important pathways listed for DEGs in ectopic tissues. Eutopic tissue DEGs were also involved in the “multicellular organism development”, “neuron development”, and “dopaminergic neuron differentiation” pathways (Table S3, S4).
“Anterior/posterior pattern specification”, multicellular organism development and “skeletal system morphogenesis” were listed for all of the networks (A, B, and C). Nervous system-related pathways like dopaminergic neuron differentiation and neuron development were listed for networks A and B (Tables S3, S4).
Discussion
In this study, we have observed disruptions in expression patterns of HOX genes and their cofactors in eutopic and ectopic samples from endometriosis patients compared to samples derived from women without endometriosis.
The data demonstrated that most of the evaluated genes had altered expression patterns in the eutopic and ectopic samples from endometriosis patients compared to the control samples. Genes involved in the HOXA-D clusters showed disturbed patterns of expression in tissues derived from women with endometriosis compared to the control tissues. In line with our findings, several studies have reported an association between HOX genes and endometriosis, as well as cancer [10, 22, 23]. Differential expressions of HOXB2, B3 and B4 [10, 24], and the HOXD2 and D3 genes [10], which we detected in the current study, agreed with previous reports on endometriosis. Altered expressions of HOXB2, B3, B4, B7, and B9, which we observed in the endometriosis samples, correlated with previous reports for these genes in endometriosis and in progression and development of many cancers [24,25,26]. Here we reported misregulation of HOXC cluster genes in endometriosis. Their involvement with endometriosis, cancer, and the female reproductive tract have been shown previously [10, 27, 28]. Moreover, we reported contrariwise expression patterns for HOXD12 and HOXD13 (downregulated) and HOXD1 and D3 (upregulated) in this study on endometriotic lesions. Upregulated HOXD genes have a correlation with their expression profiles in some carcinomas [29].
HOXC12, EN1, SHOX, TLX1, VAX1, VSX1, PAX3, PITX3, DLX1, and LBX1 showed intensive changes as they are upregulated in eutopic tissues of women with endometriosis compared to the control group. Accordingly, we reported that most of these genes were hypomethylated, which correlated with elevated expressions of these genes in endometriosis patients [30]. The expression level of HOXC12 is increased in breast tumor samples [31] and TLX1 shows misregulation in cancer [32]. Therefore, our finding re-enforces the previously reported association between cancer and endometriosis [33, 34]. There is PAX3 expression in cytotrophoblast cells and decidua cells during early pregnancy [35], so its disturbed expression might be involved in implantation defects in endometriosis. EN1, PITX3, and SHOX2 are involved in neuron formation in eutopic and ectopic tissues [36, 37], and the altered expressions in these genes may be related to nerve formation and pain sensation in endometriosis patients [38].
Most DEG genes in network A showed significantly altered expressions in ectopic tissues. These genes are mainly involved in development and nerve function. In contrast, most genes categorized in networks B and C showed significantly altered expressions in eutopic tissue and are involved in developmental functions (Fig. 3A).
Our analysis indicated that ectopic and eutopic DEG genes are mainly expressed in the salivary gland and Burkett's lymphoma (Table 3). We detected pathways related to stem cell pluripotency for DEGs, which was in line with previous reports for this pathway in endometriosis [39, 40]. Moreover, we identified a “transcriptional misregulation in cancer” pathway for the studied genes that correlated with a previously proven relationship between endometriosis and cancer [41, 42].
HOX gene expressions are regulated by the hormones estradiol and progesterone. Endocrine regulation of HOX gene expressions is important for reproduction [27, 43]. Because endometriosis is associated with disturbed hormonal function (estrogen hyperactivity and progesterone resistance) [44], it is possible that an altered expression profile of HOX in endometriosis tissues is a result of hormonal dysregulation.
The results of this study provide preliminary evidence for the involvement of certain pathways (neural and immune system, as well as cancer) in endometriosis because the DEG genes identified by our study are reported to play a role in the above pathways. However, the role of these pathways in endometriosis should be investigated. It remains to be determined how neurogenesis or cancer pathways induce endometriosis or promote the development of this disease.
Recently developed endometriosis organoids are suitable pre-clinical models to investigate molecular mechanisms that underlie this disease [30, 45]. We suggest knock out the upregulated genes with extensive fold changes like HOXC12 in endometriosis organoids and over-express these genes in endometrial organoids developed from non-endometriosis patients to explore the role of these genes in the development of endometriosis. Endometriosis/endometrium organoids and stromal cell cultures provide a suitable platform to investigate the role of progesterone and estrogen in the disturbed expression profiles of HOX genes.
Conclusion
Our study provides the first insight into the expression pattern of HOX clusters and their cofactors in endometriosis. The results suggest an important role for these genes in the pathophysiology of this disease. Some genes from this platform should be additionally investigated in the future because of their thousand-fold changes in endometriosis tissue biopsies compared to control tissue biopsies.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- cDNA:
-
Complementary DNA
- DAVID:
-
Database for Annotation, Visualization, and Integrated Discovery
- DEG:
-
Differentially expressed gene
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- non-DEG:
-
Non-differentially expressed gene
- PCA:
-
Principal component analysis
- CtoN:
-
Ratio of gene expression level in ectopic tissue to normal tissue
- CtoU:
-
Ratio of gene expression level in ectopic tissue to eutopic tissue
- UtoN:
-
Ratio of gene expression level in eutopic tissue to normal tissue
References
Mehedintu C, Plotogea MN, Ionescu S, Antonovici M. Endometriosis still a challenge. J Med Life. 2014;7(3):349–57.
Sheveleva T, Bejenar V, Komlichenko E, Dedul A, Malushko A. Innovative approach in assessing the role of neurogenesis, angiogenesis, and lymphangiogenesis in the pathogenesis of external genital endometriosis. Gynecol Endocrinol. 2016;32(sup2):75–9.
Wang Y, Li Y, Yang Z, Liu K, Wang D. Genome-wide microarray analysis of long non-coding rnas in eutopic secretory endometrium with endometriosis. Cell Physiol Biochem. 2015;37(6):2231–45.
Samadieh Y, Favaedi R, Ramezanali F, Afsharian P, Aflatoonian R, Shahhoseini M. Epigenetic dynamics of HOXA10 gene in infertile women with endometriosis. Reprod Sci. 2019;26(1):88–96.
Mahdian S, Aflatoonian R, Yazdi RS, Yaghmaei P, Ramazanali F, Afsharian P, et al. Macrophage migration inhibitory factor as a potential biomarker of endometriosis. Fertil Steril. 2015;103(1):153–9 e3.
Soriano D, Schonman R, Gat I, Schiff E, Seidman DS, Carp H, et al. Thoracic endometriosis syndrome is strongly associated with severe pelvic endometriosis and infertility. J Minim Invasive Gynecol. 2012;19(6):742–8.
Lagana AS, La Rosa VL, Rapisarda AMC, Valenti G, Sapia F, Chiofalo B, et al. Anxiety and depression in patients with endometriosis: impact and management challenges. Int J Womens Health. 2017;9:323–30.
Yotova IY, Quan P, Leditznig N, Beer U, Wenzl R, Tschugguel W. Abnormal activation of Ras/Raf/MAPK and RhoA/ROCKII signalling pathways in eutopic endometrial stromal cells of patients with endometriosis. Hum Reprod. 2011;26(4):885–97.
Merabet S, Mann RS. To be specific or not: The critical relationship between hox and tale proteins. Trends Genet. 2016;32(6):334–47.
Golestan Jahromi M, Aflatoonian R, Afsharian P, Aghajanpour S, Shahhoseini M, Aflatoonian A. Altered expression of 3 paralogus HOX A-D clusters in endometriosis disease: a case-control study. Int J Reprod Biomed (Yazd). 2018;16(9):549–56.
Xu X, Ding J, Ding H, Shen J, Gattuso P, Prinz RA, et al. Immunohistochemical detection of heparanase-1 expression in eutopic and ectopic endometrium from women with endometriosis. Fertil Steril. 2007;88(5):1304–10.
Abbas Suleiman A, Nader A, Winzenborg I, Beck D, Polepally AR, Ng J, et al. Exposure-Safety Analyses Identify Predictors of Change in Bone Mineral Density and Support Elagolix Labeling for Endometriosis-Associated Pain. CPT Pharmacometrics Syst Pharmacol. 2020;9(11):639–48.
Dickel H, Kuss O, Kamphowe J, Altmeyer P, Hoxtermann S. Association of CD69 up-regulation on CD4+ Cla+ T cells versus patch test, strip patch test and clinical history in nickel sensitization. Eur J Med Res. 2010;15:303–8.
Delevich K, Abi-Dargham A, Hall J, Sawa A. Studying schizophrenia in the post-genomic era: perspectives from the 2016 summer Banbury workshop at cold spring harbor laboratory. Mol Psychiatry. 2017;22(1):2–3.
Wahl KJ, Yong PJ, Bridge-Cook P, Allaire C, EndoAct C. Endometriosis in Canada: It Is Time for Collaboration to Advance Patient-Oriented, Evidence-Based Policy, Care, and Research. J Obstet Gynaecol Can. 2021;43(1):88–90.
Yeung KY, Ruzzo WL. Principal component analysis for clustering gene expression data. Bioinformatics. 2001;17(9):763–74.
Cameron DA, Middleton FA, Chenn A, Olson EC. Hierarchical clustering of gene expression patterns in the Eomes + lineage of excitatory neurons during early neocortical development. BMC Neurosci. 2012;13:90.
Osman MI. Exploring a mixture of distributions using Minitab. Comput Biol Med. 1997;27(3):223–32.
Prieto C, Risueno A, De las Rivas J. Human gene coexpression landscape: confident network derived from tissue transcriptomic profiles. PLoS One. 2008;3(12):e3911.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
Makiyama K, Hamada J, Takada M, Murakawa K, Takahashi Y, Tada M, et al. Aberrant expression of HOX genes in human invasive breast carcinoma. Oncol Rep. 2005;13(4):673–9.
Kishibuchi R, Kondo K, Soejima S, Tsuboi M, Kajiura K, Kawakami Y, et al. DNA methylation of GHSR, GNG4, HOXD9 and SALL3 is a common epigenetic alteration in thymic carcinoma. Int J Oncol. 2020;56(1):315–26.
AlKusayer GM, Pon JR, Peng B, Klausen C, Lisonkova S, Kinloch M, et al. HOXB4 immunoreactivity in endometrial tissues from women with or without endometriosis. Reprod Sci. 2018;25(6):950–7.
Lopez-Romero R, Marrero-Rodriguez D, Romero-Morelos P, Villegas V, Valdivia A, Arreola H, et al. The role of developmental HOX genes in cervical cancer. Rev Med Inst Mex Seguro Soc. 2015;53(Suppl 2):S188–93.
Kar SP, Tyrer JP, Li Q, Lawrenson K, Aben KK, Anton-Culver H, et al. Network-based integration of GWAS and gene expression identifies a hox-centric network associated with serous ovarian cancer risk. Cancer Epidemiol Biomarkers Prev. 2015;24(10):1574–84.
Du H, Taylor HS. The Role of Hox Genes in Female Reproductive Tract Development, Adult Function, and Fertility. Cold Spring Harb Perspect Med. 2015;6(1):a023002.
Bhatlekar S, Fields JZ, Boman BM. HOX genes and their role in the development of human cancers. J Mol Med (Berl). 2014;92(8):811–23.
Li L, Wang Y, Zhang X, Huang Q, Diao Y, Yin H, et al. Long non-coding RNA HOXD-AS1 in cancer. Clin Chim Acta. 2018;487:197–201.
Esfandiari F, Favaedi R, Heidari-Khoei H, Chitsazian F, Yari S, Piryaei A, et al. Insight into epigenetics of human endometriosis organoids: DNA methylation analysis of HOX genes and their cofactors. Fertil Steril. 2021;115(1):125–37.
Luo Z, Rhie SK, Farnham PJ. The Enigmatic HOX Genes: Can We Crack Their Code? Cancers (Basel). 2019;11(3):323.
Wu X, Miao J, Jiang J, Liu F. Analysis of methylation profiling data of hyperplasia and primary and metastatic endometrial cancers. Eur J Obstet Gynecol Reprod Biol. 2017;217:161–6.
Dawson A, Fernandez ML, Anglesio M, Yong PJ, Carey MS. Endometriosis and endometriosis-associated cancers: new insights into the molecular mechanisms of ovarian cancer development. Ecancermedicalscience. 2018;12:803.
Anglesio MS, Papadopoulos N, Ayhan A, Nazeran TM, Noe M, Horlings HM, et al. Cancer-associated mutations in endometriosis without cancer. N Engl J Med. 2017;376(19):1835–48.
Jinping Zhang LZ, Xuehong Liu, Fuqun Zhou. Expression of Cx43 and Pax3 proteins in the human placental villi and decidua during early pregnancy. Bio Med Mater Eng. 2014;24(6):3841–7.
Kouwenhoven WM, von Oerthel L, Smidt MP. Pitx3 and En1 determine the size and molecular programming of the dopaminergic neuronal pool. PLoS One. 2017;12(8):e0182421.
Rosin JM, Kurrasch DM, Cobb J. Shox2 is required for the proper development of the facial motor nucleus and the establishment of the facial nerves. BMC Neurosci. 2015;16:39.
Morotti M, Vincent K, Becker CM. Mechanisms of pain in endometriosis. Eur J Obstet Gynecol Reprod Biol. 2017;209:8–13.
Adammek M, Greve B, Kassens N, Schneider C, Bruggemann K, Schuring AN, et al. MicroRNA miR-145 inhibits proliferation, invasiveness, and stem cell phenotype of an in vitro endometriosis model by targeting multiple cytoskeletal elements and pluripotency factors. Fertil Steril. 2013;99(5):1346–55 e5.
Othman ER, Meligy FY, Sayed AA, El-Mokhtar MA, Refaiy AM. Stem cell markers describe a transition from somatic to pluripotent cell states in a rat model of endometriosis. Reprod Sci. 2018;25:873–81.
Bas-Esteve E, Perez-Arguedas M, Guarda-Muratori GA, Acien M, Acien P. Endometriosis and ovarian cancer: Their association and relationship. Eur J Obstet Gynecol Reprod Biol. 2019;3.
Farland LV, Lorrain S, Missmer SA, Dartois L, Cervenka I, Savoye I, et al. Endometriosis and the risk of skin cancer: a prospective cohort study. Cancer Causes Control. 2017;28(10):1011–9.
Daftary GS, Taylor HS. Endocrine regulation of HOX genes. Endocr Rev. 2006;27(4):331–55.
Marquardt RM, Kim TH, Shin JH, Jeong JW. Progesterone and estrogen signaling in the endometrium: what goes wrong in endometriosis? Int J Med Sci. 2019;20(15):3822.
Esfandiari F, Heidari-Kkoei H, Saber M, Favaedi R, Piryaei A, Moini A, Shahhoseini M, Ramezanaali F, Ghafari F, Baharvand H. Disturbed. progesterone signaling in an advanced preclinical model of endometriosis. Reprod Biomed Online. 2021;43(1):139–47.
Acknowledgements
The authors thank the participants who consented to the use of their tissue samples for research.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
F.C.: Conducted statistical and bioinformatics analysis, and data interpretation, and wrote the manuscript. F.E.: Data interpretation and wrote the manuscript. R.F.: Methodological design and performed the experiments. M.G.J.: Performed the experiments and collected the samples. M.B.: Provided expertise. R.A.: Provided expertise. P.A.: Provided expertise. A.A.: Provided expertise, supervision, and administrative support for collecting the samples. M.S.: Provided expertise, supervision, and revised the content of the manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the Ethical Committee of Royan Institute for Reproductive Biomedicine (IR.ACECR.ROYAN.REC.1396.238). Informed consent was obtained from all the participants prior to their participation in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Esfandiari, F., Chitsazian, F., Jahromi, M.G. et al. HOX cluster and their cofactors showed an altered expression pattern in eutopic and ectopic endometriosis tissues. Reprod Biol Endocrinol 19, 132 (2021). https://doi.org/10.1186/s12958-021-00816-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12958-021-00816-y