
Abstract
Over a third of women of childbearing age in the United States are obese, and during pregnancy they are at increased risk for delayed labor onset and slow labor progress that often results in unplanned cesarean delivery. The biology behind this dysfunctional parturition is not well understood. Studies of obesity-induced changes in parturition physiology may facilitate approaches to optimize labor in obese women. In this review, we summarize known and proposed biologic effects of obesity on labor preparation, contraction/synchronization, and endurance, drawing on both clinical observation and experimental data. We present evidence from human and animal studies of interactions between obesity and parturition signaling in all elements of the birth process, including: delayed cervical ripening, prostaglandin insensitivity, amniotic membrane strengthening, decreased myometrial oxytocin receptor expression, decreased myocyte action potential initiation and contractility, decreased myocyte gap junction formation, and impaired myocyte neutralization of reactive oxygen species. We found convincing clinical data on the effect of obesity on labor initiation and successful delivery, but few studies on the underlying pathobiology. We suggest research opportunities and therapeutic interventions based on plausible biologic mechanisms.
Similar content being viewed by others

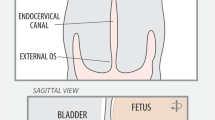

Background: The clinical phenotype of labor in obese women
Over 30% of childbearing age women in the United States are obese (body mass index [BMI] ≥ 30 kg/m2), with higher rates among racial and ethnic minority groups (31.8 % overall, 35.8 % among Hispanic and 55.8 % among non-Hispanic Black women) [1]. Obesity is associated with a number of pregnancy complications including increased risk of gestational diabetes (OR 2.83), gestational hypertension/pre-eclampsia (OR 2.68) [2], and maternal depression (OR 1.43) [3]. Maternal obesity also increases fetal risks for congenital anomalies [4] and macrosomia (birth weight > 4,500 g) [5], and for lifetime risks of heart disease [6], diabetes, and obesity [4] as an adult.
The onset of parturition in obese women is frequently delayed. Without induction, obese women are nearly twice as likely as normal-weight women to have prolonged pregnancy (≥41 weeks gestation), particularly with BMI of 35 kg/m2 or higher [7–9]. In contrast, underweight women (BMI < 17 kg/m2) are more than twice as likely to deliver preterm in spontaneous labor [10]. Prolonged pregnancy is concerning because there is a two-fold increased risk of third-trimester stillbirth in obese women [11]. Interestingly, obese women are also more likely than normal-weight women to deliver preterm [12, 13], although 60% of those early births are medically indicated [13] so the majority of these early deliveries are likely related to obesity-associated pathology.
During labor, the progress of cervical dilation in obese women is slower than in normal-weight women [14–17], a complication known as labor dystocia [18]. In two large prospective cohorts, increasing maternal BMI had a clinically relevant dose relationship with protracted labor (Table 1) [19, 20]. The time to full cervical dilation in morbidly obese (BMI ≥ 40 kg/m2) mothers was significantly longer than normal weight women, regardless of parity. Even in healthy obese women (without diabetes, chronic hypertension, or cardiovascular disease) the increased risk for slow cervical dilation (OR 3.9) and cesarean section (OR 3.2) persists [21]. We found no studies comparing labor outcomes directly in obese women with or without obesity-related metabolic dysfunction.
Obese women are more likely to be admitted to the hospital at earlier cervical dilation, and to undergo additional intervention to facilitate labor [22, 23], including administration of synthetic oxytocin (Pitocin; Table 2). Currently, a standard Pitocin regimen is typically used regardless of BMI [15] though evidence suggests that higher median dose and longer duration may be necessary for labor progress in obese women [24]. Despite increased clinical intervention, obese women are two to three times more likely than normal-weight women to have unplanned cesarean section (Table 2) [17, 25], a risk that persists after controlling for obesity-associated co-morbidities [16, 17]. Post-operative complications are also more common among obese women, including infection (18.8 % BMI > 45 kg/m2 vs. 7.5 % normal-weight) [26, 27], postpartum hemorrhage (32.6 % morbidly obese vs. 4.9 % normal-weight), and prolonged hospitalization (34.9 % morbidly obese vs. 2.3 % normal-weight) [28].
Reduced gestational weight gain in obese women improves maternal and fetal outcomes in pregnancy, and the Institute of Medicine has recommended revised prenatal care guidelines [29, 30]. Lower weight gain decreases the risk of preeclampsia, cesarean delivery, large for gestational age neonates, and small for gestational age neonates [29]. There are no data on the safety and outcomes of intentional weight loss for obese pregnant women, and that approach is controversial due to concerns for fetal growth and maternal ketosis [31]. In studies using obese mice, maternal weight loss alters epigenetic signatures in the offspring [32], emphasizing the need to understand possible influences of altered prenatal goals.
We need new approaches to improve birth outcomes in obese women, who have a unique parturition phenotype [24, 33, 34]. Biologic mechanisms linking obesity to dysfunctional labor, independent of obesity-associated comorbidities, are largely unknown. In this review, we first outline normal parturition physiology and relevant obesity pathophysiology. We then summarize studies from human and animal models identifying the possible molecular basis for differences in labor preparation, uterine contraction/synchronization, and labor endurance in obese women. We suggest opportunities for future investigation, and possible therapeutic targets and approaches to improve birth outcomes.
Overview of normal parturition physiology relevant to obesity
Human parturition can be described by myometrial phases: quiescence, preparation (activation), labor (contractions), and recovery (involution) [35]. Ninety-five percent of human gestation is spent in quiescence, when the myometrium is not contracting and the cervix is closed. In the preparation phase, the myometrium expresses contractile-associated proteins (CAPs) including oxytocin receptor (OTR), prostaglandin F receptor, and connexin-43, and the cervix softens and shortens [36]. In active labor, synchronized uterine contractions dilate the cervix for delivery of the fetus. In recovery, the uterus and cervix involute and remodel for future pregnancy. Specific signals regulate each phase of parturition.
Preparation for parturition
Preparation for parturition is initiated during the final 12 weeks of pregnancy when placental corticotropin-releasing hormone (pCRH) increases dramatically, leading some investigators to characterize pCRH as a gestational clock [37]. Simultaneously, maternal plasma CRH binding protein decreases, allowing increased free pCRH to activate myometrial CRH-receptors that switch from an isoform that enforces quiescence to an isoform that activates CAP expression [38]. pCRH also stimulates fetal pituitary adrenocorticotropic hormone (ACTH) release, increasing fetal adrenal cortisol production and maternal myometrial expression of cyclooxgenase-2 (COX-2) [35]. COX-2 synthesizes prostaglandins (PG) E2 and PGF2alpha to promote myometrial activation. Fetal cortisol increases placental pCRH production in a feed-forward loop that stimulates fetal production of dehydroepiandrosterone (DHEA) which is converted to estriol by placental enzymes [39]. Estriol, the dominant placental estrogen, increases myometrial sensitivity to oxytocin and PGs, and stimulates oxytocin release from choriodecidual tissues [40].
In humans, progesterone does not decline dramatically at term, but serum progesterone increases at a slower rate while estradiol or estriol production accelerates, leading to an elevated estrogen to progesterone ratio. These changes are sometimes characterized as functional progesterone withdrawal [35]. Further, and perhaps more important, progesterone receptor (PR) expression changes from a predominance of PR-B to more PR-A receptors [35], thereby activating a different set of genes [41]. PR-B mediates the primary pro-gestational (i.e., quiescence) function of progesterone while the truncated N-terminal of PR-A prevents recruitment of coactivators and alters myometrial progesterone gene activation. Thus, the PR isoform switch reduces progesterone’s pregnancy maintenance function and facilitates the molecular program leading to myometrial contractility. Other factors suggested to modulate the myometrial labor program include epigenetic regulation of pCRH gene expression [42], NFkappaB inflammatory blockade of PR responsive genes [43], recruitment of inflammatory macrophages to the myometrium [44], and perhaps increased cell-free fetal DNA in the maternal circulation [45].
The cervix shortens and ripens for several weeks before labor in response to PGE2 and PGF2alpha, released by fetal membranes [35]. Cervical extracellular collagen disperses, water content increases, and fibrous stroma degrades. Matrix metalloproteinases (MMPs), released by cervical stromal fibroblasts and smooth muscle cells, break down cervical stromal proteoglycans [46, 47]. In addition, cervical epithelial and stromal cells undergo apoptosis, decreasing the cellular content of the laboring cervix [48]. The amnion and chorion overlying the internal cervical os degrade before labor, with increased apoptosis leading to rupture of membranes [49]. Disruption of fetal membranes releases amniotic fluid that exposes decidua and myometrium to PGE2 and oxytocin, enhancing labor activation [35].
Contraction & synchronization in parturition
Phasic uterine contractions are a hallmark of active labor, accompanied by progressive cervical dilation. Individual myocytes contract when actin and phosphorylated myosin interact via ATP-consuming cross-bridge cycling [50] triggered by increased intracellular calcium ion (Ca2+). Prior to parturition, Ca2+ efflux and intracellular sequestration lead to decreased intracellular Ca2+ and myocyte relaxation. Low resting Ca2+, along with potassium (K+) channel opening and plasma membrane repolarization promote the quiescent state. During labor, myosin phosphorylation is initiated by myocyte depolarization leading to calcium influx, primarily via L-type Ca2+ channels. Oxytocin signaling via the oxytocin receptor (OTR) promotes more frequent and forceful uterine contractions and release of PGs from the decidua [35, 51]. The posterior pituitary releases pulsatile oxytocin, and decidual and placental tissues produce oxytocin continuously [35, 52]. During late gestation, OTRs increase sharply in the myometrium and decidua as the primary mediator of oxytocin response in labor. The basic biology of myocyte contractility has been reviewed in detail elsewhere [35, 50, 53, 54].
During labor, myocyte contraction is coordinated, starting in the uterine “pacemaker” at the fundus and spreading toward the cervix [55, 56]. Neighboring myocytes become connected via plasma membrane gap junctions that assemble immediately before labor [57]. These low-resistance pores formed by connexins (e.g., connexin-43), allow action potentials to travel freely from cell to cell, transforming the uterus into a functional syncytium [58]. Stronger and longer originating action potentials result in organized uterine contractions of increasing force [59, 60].
Uterine endurance in parturition
During labor, the uterus must maintain sufficient strength and duration of contraction, or endurance, to expel the fetus and maintain postpartum hemostasis. During contractions, ATP hydrolysis releases protons, producing transient acidification [50]. Forceful contractions occlude blood vessels in the uterine wall, causing repetitive ischemia and reperfusion that leads to periodic acidification due to anaerobic lactic acid production [50, 61]. Cyclic hypoxia/reperfusion also produces reactive oxygen species (ROS) [62]. Myocyte regulatory mechanisms counteract the lower pH (e.g., hypoxia-resistant lactate dehydrogenase isoform) and elevated ROS (e.g., Glutathione Peroxidase [GSHPx]), maintaining labor endurance [50, 63].
Overview of obesity physiology relevant to pregnancy
Nearly 70 % of obese patients exhibit metabolic dysregulation with changes in circulating hormones [64–66] and free fatty acids (FFA). For this reason, obesity results in altered physiology that may influence normal parturition pathways.
Circulating molecules in obesity
Increasing BMI alters the secretion of a range of hormones from adipose tissue. Highly relevant to pregnancy is leptin, an adipokine secreted primarily by white adipose tissue. Leptin suppresses appetite, stimulates adipocyte hypertrophy, and increases FFA oxidation [67, 68]. Free leptin rises with BMI and increased adiposity, but obese individuals also exhibit impaired satiety feedback. This “leptin resistance” may be the result of chronic inflammatory mediators that disrupt normal hypothalamic homeostasis [67, 69, 70]. The placenta also secretes leptin [67, 71], peaking in the second trimester. Leptin is significantly increased in obese compared to normal-weight pregnant women [72]. In contrast, soluble leptin receptor decreases linearly with increasing BMI in pregnancy, leading to high serum free leptin in obese pregnant women [67].
Four other adipokines may be of importance. Apelin, secreted by adipose tissue and placenta [73, 74], is elevated in obese pregnant women and associated with insulin resistance [75]. Physiologically, apelin causes hypotension, regulates fluid homeostasis intake, and is produced in response to insulin release [73]. Visfatin increases in pregnant women near labor [76]. Visfatin is highly expressed by visceral adipose cells, and activates insulin receptors. Ghrelin, in contrast, is inversely related to maternal BMI. It stimulates insulin secretion, and regulates food intake and fat utilization during pregnancy [77, 78]. The most abundant adipokine is adiponectin, which regulates energy metabolism by increasing glucose uptake, lipid catabolism, and insulin sensitivity [79]. Adiponectin is decreased among pregnant women with higher BMI and those with more pronounced dyslipidemia [80].
Plasma cholesterol increases with obesity and also in pregnancy. Plasma lipids and lipoproteins increase throughout pregnancy, supplying nutrients to the fetus and supporting fetal cholesterol biosynthesis [80]. In reproductive age females, hypercholesterolemia increases with BMI beyond that associated with normal pregnancy [81]. Perhaps more importantly, obese insulin resistant women (i.e., more metabolically dysfunctional) show a shift to an atherogenic lipoprotein phenotype, including increased plasma triglycerides and small low density lipoproteins (LDL) [80, 82], that is associated with gestational diabetes [83], ectopic fat deposition [84], and atherosclerosis [85].
Obesity also alters vitamins, minerals, and cofactors. For example, low vitamin D levels are found more often among obese and overweight individuals, and associated with chronic disorders like cardiovascular disease and diabetes [86]. Low folic acid levels are similarly linked to obesity, resulting in increased neural tube defects in offspring of obese women [87].
FFA storage and meta-inflammation
In pregnancy, obese women have higher triglyceride and FFA levels than normal-weight pregnant women. When FFA levels overwhelm adipocyte storage capacity, excess FFA is sequestered as triglyceride in new adipocytes or within fat droplets in non-adipose tissues (ectopic fat) [65]. As FFA levels increase further, they may circulate and be used instead of glucose for cellular metabolism [88]. Mitochondrial lipid metabolism produces ROS that can damage cellular structures and impair insulin responses, a process called lipotoxicity. Normally, cells neutralize ROS with cellular antioxidants, but with increased FFA, that capacity is overwhelmed, and ROS cause RNA/DNA damage, protein carbonylation, and eventual apoptosis [63, 89, 90]. ROS-mediated cell damage releases inflammatory mediators (e.g., interleukin-6, interleukin-1ß, tumor necrosis factor-α), creating chronic low-grade inflammation throughout the body known as meta-inflammation [91, 92], which further increases ROS production [90].
Insulin is crucial for glucose uptake, glycogen synthesis, lipogenesis, and cell growth, and stimulates adipose uptake and storage of FFA [93]. Insulin resistance is normal in human pregnancy, a necessary adaptation to ensure appropriate maternal nutrient shunting to the fetus [94]. As insulin resistance exacerbates the FFA storage problem, pregnancy may worsen obesity-related ROS damage and meta-inflammation [93].
Methods: a literature search for interactions between obesity and parturition signaling
Considering the known physiology described above, we explored the experimental evidence for interactions between the physiology of obesity and normal parturition signaling. Our primary question was: What biologic mechanisms could be responsible for parturition dysfunction in obesity? In June 2015, we performed a comprehensive literature review using PubMed, Google Scholar, Web of Science, and MEDLINE databases with the primary medical subject heading (MeSH) search terms: obesity, labor, parturition, and pregnancy. Additional search terms included: BMI, uterus, myometrium, dystocia, mechanism, and dysfunction. We considered all original research in the English language from any year. We also considered relevant articles from citations in these publications. After initial screening, we obtained full-text articles for evaluation of content, quality, and relevance and included all studies relevant to potential interactions between physiologic changes of obesity and dysfunctional labor (Additional file 1: Table S1).
Review of evidence: biologic mechanisms of labor dysfunction in obesity
We identified 31 studies for review (Additional file 1: Table S1) and have grouped them into three categories: labor onset, contraction/synchronization, and endurance. They include human, animal, and cell culture studies. Figure 1 summarizes interactions between obesity and parturition. Some mechanisms were corroborated by multiple investigators, while others involve only early work and suggested hypotheses.

Overview of Obesity-Related Biologic Dysfunction of Labor. PGE2 = prostaglandin E2 (dinoprostone; naturally occurring prostaglandin). Blue italicized script = Proposed mechanism or mechanism with limited evidence
Changes in labor preparation due to obesity
Placental function
Obesity increases placental weight and hypertrophy, but reduces the fetal/placental weight ratio (sometimes referred to as “placental efficiency”) [95]. A high fat diet decreases uterine blood flow, potentially leading to placental relative hypoxia and trophoblast dysfunction [96]. Placental amino acid transport is reduced in women with normal birthweight infants [97], but increased in obese mice [98] and in women with large babies [99]. Altered placental steroid hormone biosynthesis in obese pregnant women has not been established, but obesity is associated with lower mid- and late-pregnancy pCRH [100, 101]. Decreased pCRH together with changes in placental structure/function could alter estrogen/progesterone production, metabolism, or ratio resulting in delayed onset of parturition [102]. We found no studies on the changes in placental parturition steroid hormone signaling with obesity that might account for altered parturition timing.
Elevated leptin
Leptin stimulates PGE2 release from placental and adipose tissue via inflammatory signaling pathways [103]. Chronically elevated PGE2 in late pregnancy among obese women with meta-inflammation could decrease the sensitivity of maternal tissues to PGE2 during labor activation, a finding that has been documented clinically [104] and is supported by the known elevated PGE2 in obesity [105, 106]. Insensitivity to PGE2 during labor induction is linked to failed functional progesterone withdrawal [107]. Thus, the chronic inflammatory state of obese women could impede functional progesterone withdrawal and PGE2 activation. There is no current evidence examining changes in myometrial PRB/PRA switching with obesity.
Leptin also disrupts in vitro collagen degradation by MMPs, as well as cervical cell apoptosis [108, 109], two effects that may inhibit cervical ripening in obese women. Further, leptin stimulates cervical collagen synthesis in late pregnancy [108, 151], possibly explaining the decreased cervical ripening at term in obese women that we see clinically [110]. High circulating leptin in the second trimester may also inhibit fetal membrane weakening through decreased membrane apoptosis [109], thereby inhibiting spontaneous rupture of membranes in obese women [49]. The clinical finding that there is an increased requirement for artificial rupture of membranes with increasing BMI supports this hypothesis (Table 2) [111].
Changes in labor contraction/synchronization due to obesity
Adipokines
Leptin exerts an inhibitory effect on spontaneous and oxytocin-stimulated myometrial contractions in vitro [112] however, the second trimester serum leptin level does not explain the risk of failed first-stage labor in an adjusted model [113]. To our knowledge, there are no published reports on labor outcome related to third-trimester leptin levels. Visfatin also decreases both spontaneous and oxytocin-induced contractions [76]. There are no data on whether adiponectin influences myometrial contractility [114]. Apelin also inhibits spontaneous and oxytocin-induced myometrial contractions in organ bath, but has not been evaluated clinically [73]. Ghrelin may stimulate myometrial contractions [114], although there are conflicting reports [78].
Cholesterol
Cholesterol supports plasma membrane channel function in cholesterol-rich “lipid rafts” [115]. Smooth muscle cells, including myocytes, are rich in a particular type of lipid raft, the caveolae [116], on which potassium channels such as Maxi-K and human ether-a-go-go–related gene (hERG) K+ channels cluster [60, 117]. These hyperpolarizing, and therefore pro-relaxant channels, are more active with the hypercholesterolemia that is more common among obese than normal-weight women. Both estrogen receptors [118] and oxytocin receptors [119] associate with myocyte membrane caveolae, but it is unknown whether maternal hypercholesterolemia alters the stability or function of these receptors. Cholesterol and LDL reduced contraction force and frequency in human and rodent myometrial strips, effects that were not alleviated by exogenous oxytocin [120, 121]. Mice with dysfunctional hepatic cholesterol clearance exhibited reduced oxytocin responses and abnormal labor without pup expulsion [122]. In humans, elevated maternal cholesterol at 14–16 weeks gestation is not a risk factor for first stage labor dystocia leading to cesarean section [123]. We found no studies that evaluated labor progress or outcomes by maternal cholesterol or maternal triglyceride levels near the time of birth.
Oxytocin receptors (OTR)
Obese women require more Pitocin for labor induction, even controlling for neonate size, parity, and epidural anesthesia [33, 124, 125]. Existing reports on obesity-dependent changes in OTR are contradictory [152]. In one study, OTR mRNA was decreased in myometrial biopsies from term pregnancies in women with higher BMI at delivery [126], but another group found no change in OTR gene or protein expression with increasing BMI [127], though greater than 30% of those subjects were laboring. Biopsy location, timing in pregnancy, and stage of labor could all influence OTR results [51], and women with protracted labor and delayed transition to active labor have different OTR gene polymorphisms than those with efficient labor [128]. Thus, labor progress is demonstrably sensitive to OTR expression, suggesting the important question of whether altered OTR is in part responsible for delayed labor in obese women. Genome-wide association studies have not evaluated interactions between obesity, OTR, and labor outcomes.
Gap junctions
We found no human studies investigating uterine connexin-43 expression in obesity or with labor dystocia. Uterine biopsies from a mixed-weight sample of women with prolonged labor have decreased connexin-43 mRNA compared to normal labor [58]. Rats fed a high-fat, high-cholesterol diet during pregnancy showed significantly lower connexin-43 expression compared to animals eating regular chow, although contractility was not examined [129].
Changes in uterine endurance in labor due to obesity
Women with slow labor have lower oxygen saturation, increased myometrial lactate, and more acidic capillary pH compared to normal controls (pH 7.35 in dysfunctional labor vs. 7.48 in normal labor) [130]. One labor dystocia theory posits that lower pH and increased ROS cause unorganized and ineffective contractions, and an individual’s capacity to buffer pH and neutralize ROS predicts labor dystocia [130]. It is possible that obesity-related lipotoxicity when combined with normal myometrial ROS production during labor leads to excess ROS that might cause labor dystocia. Increased pre-pregnancy BMI is associated with excessive placental ROS and decreased ATP production [131]. However, high-fat/cholesterol diet did not change term non-laboring rat myometrial mitochondrial function [132]. We found no evidence for changes in myometrial cellular respiration, ROS with obesity, or effects of either on labor endurance.
Discussion
In a theoretical model of successful human parturition, labor occurs as a result of “integrative and synergistic coordination” of separate biological processes or modules, occurring across many tissues [133]. With obesity, the delayed labor and labor dystocia phenotypes could result from multiple accumulated parturition malfunctions (Fig. 2) in the placenta, cervix, amnion, and myometrium. Obesity may also decrease labor endurance, with meta-inflammation and excess FFA causing excess myometrial ROS accumulation. Changes in biologic signals or responses with obesity could alter the onset, synchronization, and endurance of labor. Our literature search identified several questions that require additional basic and translational investigation, and as the effects of obesity are better understood, additional hypotheses will likely be generated. Considering the evidence for the pathobiology of labor dystocia in obesity, we suggest several investigative and therapeutic opportunities to improve vaginal delivery rates:

Comparison of normal and obesity-associated mechanisms of labor. MMP = matrix metalloproteinase, hERG K + =human ether-a-go-go-related gene potassium channel, pCRH = placental corticotropin releasing hormone, FFA = free fatty acid, ROS = reactive oxygen species, GSHPX = glutathione peroxidase, OTR = oxytocin receptor, PGE2 = prostaglandin E2, connexin-43 = myometrial gap junction. Blue italicized script = proposed mechanism or mechanism with limited evidence
-
Basic science investigation: We have summarized the existing evidence that obesity produces important changes in parturition signaling and the molecular program of labor in placenta, myometrium, and cervix. It is clear that each known step in the parturition process could be altered by hormones or other unique regulation produced by adipose tissue or associated meta-inflammation. However, we need to identify specific mechanisms.
-
o It will be important to define the type of estrogens (estriol or estradiol) produced in obese parturients and the effect of obesity on the ratio of estrogen to progesterone at term. In addition, the effect of obesity on placental hormone production and myometrial progesterone receptor expression must be quantified in both animal models and women if we are to develop new approaches to modifying the deleterious effects of obesity.
-
o Preclinical animal models of obesity management in pregnancy, for example limited weight gain and intentional weight loss, may reveal important parturition and fetal effects.
-
o Basic investigation of whether increased fat mass alone or associated metabolic syndrome and meta-inflammation drive parturition dysfunction is warranted. Metabolomic characterization of obese women with labor dystocia or Pitocin resistance could further our understanding of the signals linking obesity and labor outcome versus the separate influence of metabolic syndrome.
-
-
Clinical management: Allowing additional time for obese women to complete cervical ripening and the first stage of labor before proceeding to cesarean delivery for slow labor progress could immediately improve outcomes [134, 135]. Clinical tools to identify aberrant labor progress with obesity, such as BMI-determined labor partograms, are needed. Further, investigating parturition changes with decreased weight gain or weight loss during obese pregnancy may be informative. The safety and outcome of altered prenatal and obstetric management requires thorough clinical evaluation and prospective trials [136].
-
Medication management: Obese women may benefit from optimized protocols for Pitocin (and other induction/augmentation agents), rather than the universal protocols currently in use for women of any BMI. Higher doses and/or longer infusion times may be necessary for induction of labor in an obese woman. The differences in OTR expression and function and myometrial contractility between obese and normal-weight women are not yet defined. All induction agents may need to be examined to find the most effective regimen for obese cohorts.
-
Therapeutic investigation:
-
o Cholesterol-lowering therapies (e.g., statins, niacin, bile acid sequestrants) might prevent hypercholesterolemia-linked suppression of contractions in obese women. Although statins are rated Category X in pregnancy, the teratogenic potential appears to be limited to extraordinarily high doses in animal models, and in select cases the potential benefits may outweigh the risks, even during pregnancy [137]. Randomized controlled trials (RCT) are needed.
-
o Thiazolidinedione (TZD) activates peroxisome proliferator-activated receptors (PPARs), allowing for safe storage of FFA, improved insulin sensitivity, and decreased cellular ROS damage [138]. Activation of the PPAR system is known to reduce recruitment of immune cells and inhibit inflammation, allowing better utilization of glucose as an energy source for cellular processes [65]. TZD therapy could improve labor endurance by decreasing myometrial ROS damage from FFA metabolism, although the known association of TZD with increased weight gain [139] could carry other risks. A RCT of TZD therapy to improve labor endurance is needed.
-
-
Nutrition intervention:
-
o High-fat diet apparently decreases uterine blood flow [96] and lowers connexin-43 expression [129] in animal models. Prospective investigation of the effect of dietary fat in normal and obese women on labor length, induction response, and dystocia incidence is needed.
-
o Serum vitamin D is inversely related to both visceral and subcutaneous fat and insulin resistance [140], and significantly decreases biomarkers of oxidative stress [141]. A RCT investigating nutritional supplementation of anti-oxidants (e.g., vitamins D, C, E, and DHA/EPA) to modulate lipotoxicity-linked ROS elevation and improve labor outcomes is needed.
-
Conclusion
Obesity is associated with changes in the placenta, cervix, amnion, and myometrium that could alter labor preparation, contraction/synchronization, and endurance. Studies investigating the biologic mechanisms of labor dystocia and failed induction due to obesity, both basic and translational, are needed. With a better understanding of the unique biology of obese parturients, we may develop better techniques and treatments to optimize labor outcomes, increase the rate of vaginal deliveries, and reduce the risk of obstetric complications. Even without new medications, changing our clinical management of obese women offers an immediate opportunity to decrease unplanned cesareans. Simply offering additional time in labor for obese women could result in increased vaginal deliveries. Therefore, while initial efforts to address obesity-related labor dysfunction could be directed to clinical management, future interventions can be designed to correct or overcome the altered parturition physiology in obese women. Moreover, as additional molecular details of human parturition are discovered, there may be substantial benefit to exploring the interactions with obesity. As obesity increases in the population, addressing the interactions between obesity and normal physiology is a women’s health priority, and may lead to important improvements in obstetric care and the well-being of mothers and their newborn children. For now, we suggest giving more time clinically to circumvent the parturition dysfunction in obesity, while we invest additional time and research in understanding better the pathobiological effects of obesity on normal parturition signaling.
Abbreviations
- ATP:
-
adenosine triphosphate
- ACTH:
-
adrenocorticotropin hormone
- BMI:
-
body mass index
- Ca2+ :
-
calcium ion
- CAP:
-
contractile associated protein
- COX-2:
-
cyclooxgenase-2 enzyme
- CRH:
-
corticotropin-releasing hormone
- DHA/EPA:
-
docosahexaenoic acid and eicosapentaenoic acid (omega-3 fatty acids)
- DHEA:
-
dehydroepiandrosterone
- DM:
-
diabetes mellitus
- FFA:
-
free fatty acid
- GDM:
-
gestational diabetes mellitus
- GSHPx:
-
glutathione peroxidase
- hERG:
-
human ether-a-go-go-related gene (potassium channel)
- K+ :
-
potassium ion
- LDL:
-
low-density lipoprotein
- Maxi-K channel:
-
large conductance potassium channel
- MMP:
-
matrix metalloproteinase
- OR:
-
odds ratio
- OTR:
-
oxytocin receptor
- pCRH:
-
placental corticotropin-releasing hormone
- PGE2 :
-
prostaglandin E2 (dinoprostone; naturally occurring prostaglandin)
- PGF2alpha :
-
prostaglandin F2alpha (dinoprost; naturally occurring prostaglandin)
- PR:
-
progesterone receptor
- ROS:
-
reactive oxygen species
- PPAR:
-
peroxisome proliferator-activated receptor
- Pitocin:
-
synthetic oxytocin
- TZD:
-
thiazolidinedione
References
Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA. 2014;311:806–14.
Chung JH, Melsop KA, Gilbert WM, Caughey AB, Walker CK, Main EK. Increasing pre-pregnancy body mass index is predictive of a progressive escalation in adverse pregnancy outcomes. J Matern Fetal Neonatal Med. 2012;25:1635–9.
Molyneaux E, Poston L, Ashurst-Williams S, Howard LM. Obesity and mental disorders during pregnancy and postpartum: a systematic review and meta-analysis. Obstet Gynecol. 2014;123:857–67.
O'Reilly JR, Reynolds RM. The risk of maternal obesity to the long-term health of the offspring. Clin Endocrinol (Oxf). 2013;78:9–16.
Ovesen P, Rasmussen S, Kesmodel U. Effect of prepregnancy maternal overweight and obesity on pregnancy outcome. Obstet Gynecol. 2011;118:305–12.
Forsen T, Eriksson JG, Tuomilehto J, Teramo K, Osmond C, Barker DJ. Mother’s weight in pregnancy and coronary heart disease in a cohort of Finnish men: follow up study. BMJ. 1997;315:837–40.
Arrowsmith S, Wray S, Quenby S. Maternal obesity and labor complications after induction of labor in prolonged pregnancy. Obstet Anesth Dig. 2012;32:39.
Bogaerts A, Witters I, Van den Bergh BRH, Jans G, Devlieger R. Obesity in pregnancy: altered onset and progression of labour. Midwifery. 2013;29:1303–13.
Bhattacharya S, Campbell DM, Liston WA, Bhattacharya S. Effect of body mass index on pregnancy outcomes in nulliparous women delivering singleton babies. BMC Public Health. 2007;7:168.
Lynch AM, Hart JE, Agwu OC, Fisher BM, West NA, Gibbs RS. Association of extremes of prepregnancy BMI with the clinical presentations of preterm birth. Am J Obstet Gynecol. 2014;210:428 e421–429.
Chu SY, Kim SY, Lau J, Schmid CH, Dietz PM, Callaghan WM, et al. Maternal obesity and risk of stillbirth: a metaanalysis. Am J Obstet Gynecol. 2007;197:223–8.
Suidan RS, Apuzzio JJ, Williams SF. Obesity, Comorbidities, and the Cesarean Delivery Rate. Am J Perinatol. 2012;29:623–8.
Cnattingius S, Villamor E, Johansson S, Edstedt Bonamy AK, Persson M, Wikstrom AK, et al. Maternal obesity and risk of preterm delivery. JAMA. 2013;309:2362–70.
Dietz PM, Callaghan WM, Morrow B, Cogswell ME. Population-Based Assessment of the Risk of Primary Cesarean Delivery Due to Excess Prepregnancy Weight Among Nulliparous Women Delivering Term Infants. Matern Child Health J. 2005;9:237–44.
Arrowsmith S, Wray S, Quenby S. Maternal obesity and labour complications following induction of labour in prolonged pregnancy. BJOG. 2011;118:578–88.
Fyfe EM, Anderson NH, North RA, Chan EH, Taylor RS, Dekker GA, et al. Risk of first-stage and second-stage cesarean delivery by maternal body mass index among nulliparous women in labor at term. Obstet Gynecol. 2011;117:1315–22.
Poobalan AS, Aucott LS, Gurung T, Smith WCS, Bhattacharya S. Obesity as an independent risk factor for elective and emergency caesarean delivery in nulliparous women – systematic review and meta‐analysis of cohort studies. Obes Rev. 2009;10:28–35.
ACOG. Practice Bulletin Number 49, December 2003: Dystocia and augmentation of labor. Obstet Gynecol. 2003;102:1445–54.
Kominiarek M, Zhang J, VanVeldhuisen P, Troendle J, Beaver J, Hibbard JU. Contemporary labor patterns: the impact of maternal body mass index. Am J Obstet Gynecol. 2011;205:244.e241–8.
Norman SM, Tuuli MG, Odibo AO, Caughey AB, Roehl KA, Cahill AG. The effects of obesity on the first stage of labor. Obstet Gynecol. 2012;120:130–5.
Gilead R, Yaniv Salem S, Sergienko R, Sheiner E. Maternal “isolated” obesity and obstetric complications. J Matern Fetal Neonatal Med. 2012;25:2579–82.
Buhimschi CS, Buhimschi IA, Malinow AM, Weiner CP. Intrauterine pressure during the second stage of labor in obese women. [Erratum appears in Obstet Gynecol. 2004 May;103(5 Pt 1):1019]. Obstet Gynecol. 2004;2004(103):225–30.
Vahratian A, Zhang J, Troendle JF, Savitz DA, Siega-Riz AM. Maternal prepregnancy overweight and obesity and the pattern of labor progression in term nulliparous women. Obstet Gynecol. 2004;104:943–51.
Pevzner L, Powers BL, Rayburn WF, Rumney P, Wing DA. Effects of maternal obesity on duration and outcomes of prostaglandin cervical ripening and labor induction. Obstet Gynecol. 2009;114:1315–21.
Chu SY, Kim SY, Schmid CH, Dietz PM, Callaghan WM, Lau J, et al. Maternal obesity and risk of cesarean delivery: a meta-analysis. Obes Rev. 2007;8:385–94.
Leth RA, Uldbjerg N, Norgaard M, Moller JK, Thomsen RW. Obesity, diabetes, and the risk of infections diagnosed in hospital and post-discharge infections after cesarean section: a prospective cohort study. Acta Obstet Gynecol Scand. 2011;90:501–9.
Stamilio DM, Scifres CM. Extreme obesity and postcesarean maternal complications. Obstet Gynecol. 2014;124:227–32.
Perlow JH, Morgan MA. Massive maternal obesity and perioperative cesarean morbidity. Am J Obstet Gynecol. 1994;170:560–5.
Rasmussen KM, Yaktine AL, Institute of Medicine (U.S.). Committee to Reexamine IOM Pregnancy Weight Guidelines. Weight gain during pregnancy: reexamining the guidelines. Washington, DC: National Academies Press; 2009.
Rodgers AB, Yaktine AL, Institute of Medicine (U.S.). Food and Nutrition Board, National Research Council (U.S.), Committee on Implementation of the IOM Pregnancy Weight Gain Guidelines. Leveraging action to support dissemination of the pregnancy weight gain guidelines : workshop summary. Washington, D.C.: National Academies Press; 2013.
Nicklas JM, Barbour LA. Optimizing Weight for Maternal and Infant Health - Tenable, or Too Late? Expert Rev Endocrinol Metab. 2015;10:227–42.
Wei Y, Yang CR, Wei YP, Ge ZJ, Zhao ZA, Zhang B, et al. Enriched environment-induced maternal weight loss reprograms metabolic gene expression in mouse offspring. J Biol Chem. 2015;290:4604–19.
Hill M, Reed KL, Cohen WR. Oxytocin utilization for labor induction in obese and lean women. J Perinat Med. 2015;43:703–6.
Walsh J, Foley M, O'Herlihy C. Dystocia correlates with body mass index in both spontaneous and induced nulliparous labors. J Matern Fetal Neonatal Med. 2011;24:817–21.
Cunningham FG, Williams JW. Williams obstetrics. 23rd ed. New York: McGraw-Hill Medical; 2010.
Word RA, Li XH, Hnat M, Carrick K. Dynamics of cervical remodeling during pregnancy and parturition: mechanisms and current concepts. Semin Reprod Med. 2007;25:69–79.
McLean M, Bisits A, Davies J, Woods R, Lowry P, Smith R. A placental clock controlling the length of human pregnancy. Nat Med. 1995;1:460–3.
Markovic D, Vatish M, Gu M, Slater D, Newton R, Lehnert H, et al. The onset of labor alters corticotropin-releasing hormone type 1 receptor variant expression in human myometrium: putative role of interleukin-1beta. Endocrinology. 2007;148:3205–13.
Heffner LJ, Schust DJ. The reproductive system at a glance. 4th ed. Hoboken: John Wiley & Sons, Ltd.; 2014.
Chibbar R, Miller FD, Mitchell BF. Synthesis of oxytocin in amnion, chorion, and decidua may influence the timing of human parturition. J Clin Invest. 1993;91:185–92.
Jacobsen BM, Horwitz KB. Progesterone receptors, their isoforms and progesterone regulated transcription. Mol Cell Endocrinol. 2012;357:18–29.
Hogg K, Blair JD, McFadden DE, von Dadelszen P, Robinson WP. Early onset pre-eclampsia is associated with altered DNA methylation of cortisol-signalling and steroidogenic genes in the placenta. PLoS ONE. 2013;8:e62969.
Allport VC, Pieber D, Slater DM, Newton R, White JO, Bennett PR. Human labour is associated with nuclear factor-kappaB activity which mediates cyclo-oxygenase-2 expression and is involved with the 'functional progesterone withdrawal'. Mol Hum Reprod. 2001;7:581–6.
Hamilton S, Oomomian Y, Stephen G, Shynlova O, Tower CL, Garrod A, et al. Macrophages infiltrate the human and rat decidua during term and preterm labor: evidence that decidual inflammation precedes labor. Biol Reprod. 2012;86:39.
Farina A, LeShane ES, Romero R, Gomez R, Chaiworapongsa T, Rizzo N, et al. High levels of fetal cell-free DNA in maternal serum: a risk factor for spontaneous preterm delivery. Am J Obstet Gynecol. 2005;193:421–5.
Canty EG, Kadler KE. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci. 2005;118:1341–53.
Stygar D, Wang H, Vladic YS, Ekman G, Eriksson H, Sahlin L. Increased level of matrix metalloproteinases 2 and 9 in the ripening process of the human cervix. Biol Reprod. 2002;67:889–94.
Hassan SS, Romero R, Haddad R, Hendler I, Khalek N, Tromp G, et al. The transcriptome of the uterine cervix before and after spontaneous term parturition. Am J Obstet Gynecol. 2006;195:778–86.
Reti NG, Lappas M, Riley C, Wlodek ME, Permezel M, Walker S, et al. Why do membranes rupture at term? Evidence of increased cellular apoptosis in the supracervical fetal membranes. Am J Obstet Gynecol. 2007;196:484.e1–10.
Wray S. Insights into the uterus. Exp Physiol. 2007;92:621–31.
Garfield RE, Beier S. Increased myometrial responsiveness to oxytocin during term and preterm labor. Am J Obstet Gynecol. 1989;161:454–61.
Fuchs AR, Romero R, Keefe D, Parra M, Oyarzun E, Behnke E. Oxytocin secretion and human parturition: pulse frequency and duration increase during spontaneous labor in women. Am J Obstet Gynecol. 1991;165:1515–23.
Young RC. Myocytes, myometrium, and uterine contractions. Ann N Y Acad Sci. 2007;1101:72–84.
Smith R. Parturition. N Engl J Med. 2007;356:271–83.
Aslanidi O, Atia J, Benson AP, van den Berg HA, Blanks AM, Choi C, et al. Towards a computational reconstruction of the electrodynamics of premature and full term human labour. Prog Biophys Mol Biol. 2011;107:183–92.
Buhimschi CS. Spatiotemporal electromyography during human labor to monitor propagation of the uterine contraction wave and diagnose dystocia. Am J Obstet Gynecol. 2009;200:1–3.
Bru-Mercier G, Gullam JE, Thornton S, Blanks AM, Shmygol A. Characterization of the tissue-level Ca2+ signals in spontaneously contracting human myometrium. J Cell Mol Med. 2012;16:2990–3000.
Cluff A, Bystrom B, Klimaviciute A, Dahlqvist C, Cebers G, Malmstrom A, et al. Prolonged labour associated with lower expression of syndecan 3 and connexin 43 in human uterine tissue. Reprod Biol Endocrinol. 2006;4:24.
Burdyga T, Borisova L, Burdyga AT, Wray S. Temporal and spatial variations in spontaneous Ca events and mechanical activity in pregnant rat myometrium. Eur J Obstet Gynecol Reprod Biol. 2009;144 Suppl 1:S25–32.
Parkington HC, Stevenson J, Tonta MA, Paul J, Butler T, Maiti K, et al. Diminished hERG K+ channel activity facilitates strong human labour contractions but is dysregulated in obese women. Nat Commun. 2014;5:4108.
Taggart MJ, Wray S. Occurrence of intracellular pH transients during spontaneous contractions in rat uterine smooth muscle. J Physiol. 1993;472:23–31.
Khan KS, Kunz R, Kleijnen J, Antes G. Five steps to conducting a systematic review. J R Soc Med. 2003;96:118–21.
Khan RN, Matharoo-Ball B, Shaw RW. Antioxidant enzyme expression, lipid peroxidation, and protein oxidation in human myometrium with parturition. Reprod Sci. 2010;17:78–84.
Karelis AD, St-Pierre DH, Conus F, Rabasa-Lhoret R, Poehlman ET. Metabolic and body composition factors in subgroups of obesity: what do we know? J Clin Endocrinol Metab. 2004;89:2569–75.
Frayn KN. Metabolic regulation : a human perspective. 3rd ed. Chichester, U.K; Malden, MA: Wiley-Blackwell Pub.; 2010.
Unger RH, Scherer PE. Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity. Trends Endocrinol Metab. 2010;21:345–52.
Tsai PJ, Davis J, Bryant-Greenwood G. Systemic and Placental Leptin and Its Receptors in Pregnancies Associated With Obesity. Reprod Sci. 2015;22:189–97.
Unger RH, Clark GO, Scherer PE, Orci L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim BiophysActa . 2010;1801:209–14.
Hamed EA, Zakary MM, Ahmed NS, Gamal RM. Circulating leptin and insulin in obese patients with and without type 2 diabetes mellitus: relation to ghrelin and oxidative stress. Diabetes Res Clin Pract. 2011;94:434–41.
de Git KC, Adan RA. Leptin resistance in diet-induced obesity: the role of hypothalamic inflammation. Obes Rev. 2015;16:207–24.
Tessier DR, Ferraro ZM, Gruslin A. Role of leptin in pregnancy: consequences of maternal obesity. Placenta. 2013;34:205–11.
Domali E, Messinis IE. Leptin in pregnancy. J Matern Fetal Neonatal Med. 2002;12:222–30.
Hehir MP, Morrison JJ. The adipokine apelin and human uterine contractility. Am J Obstet Gynecol. 2012;206:359.e1–5.
Hanssens S, Laura B, Laurent S, Lesage J, Deruelle P. 267: Plasma apelin variations during pregnancy in obese mice. Am J Obstet Gynecol. 2014;210:S142.
Boucher J, Masri B, Daviaud D, Gesta S, Guigne C, Mazzucotelli A, et al. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology. 2005;146:1764–71.
Mumtaz S, AlSaif S, Wray S, Noble K. Inhibitory effect of visfatin and leptin on human and rat myometrial contractility. Life Sci. 2015;125:57–62.
Broglio F, Prodam F, Me E, Riganti F, Lucatello B, Granata R, et al. Ghrelin: endocrine, metabolic and cardiovascular actions. J Endocrinol Invest. 2005;28:23–5.
Hehir MP, Glavey SV, Morrison JJ. Uterorelaxant effect of ghrelin on human myometrial contractility. Am J Obstet Gynecol. 2008;198:323.e321–325.
Axelsson J, Heimburger O, Lindholm B, Stenvinkel P. Adipose tissue and its relation to inflammation: the role of adipokines. J Ren Nutr. 2005;15:131–6.
Meyer BJ, Steward FM, Brown EA, Cooney J, Nilsson S, Olivecrona G, et al. Maternal obesity is associated with the formation of small dense LDL and hypoadiponectinemia in the third trimester. J Clin Endocrinol Metab. 2013;98:643–52.
Gostynski M, Gutzwiller F, Kuulasmaa K, Doring A, Ferrario M, Grafnetter D, et al. Analysis of the relationship between total cholesterol, age, body mass index among males and females in the WHO MONICA Project. Int J Obes Relat Metab Disords. 2004;28:1082–90.
Selby JV, Austin MA, Newman B, Zhang D, Quesenberry Jr CP, Mayer EJ, et al. LDL subclass phenotypes and the insulin resistance syndrome in women. Circulation. 1993;88:381–7.
Rizzo M, Berneis K, Altinova AE, Toruner FB, Akturk M, Ayvaz G, et al. Atherogenic lipoprotein phenotype and LDL size and subclasses in women with gestational diabetes. Diabet Med. 2008;25:1406–11.
Mackay VA, Huda SS, Stewart FM, Tham K, McKenna LA, Martin I, et al. Preeclampsia is associated with compromised maternal synthesis of long-chain polyunsaturated fatty acids, leading to offspring deficiency. Hypertension. 2012;60:1078–85.
Watson TD, Caslake MJ, Freeman DJ, Griffin BA, Hinnie J, Packard CJ, et al. Determinants of LDL subfraction distribution and concentrations in young normolipidemic subjects. Arterioscler Thromb. 1994;14:902–10.
Samuel L, Borrell LN. The effect of body mass index on optimal vitamin D status in U.S. adults: The National Health and Nutrition Examination Survey 2001–2006. Ann Epidemiol. 2013;23:409–14.
McMahon DM, Liu J, Zhang H, Torres ME, Best RG. Maternal obesity, folate intake, and neural tube defects in offspring. Birth Defects Res A Clin Mol Teratol. 2013;97:115–22.
Schrauwen P, Schrauwen-Hinderling V, Hoeks J, Hesselink MK. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 1801;2010:266–71.
Demozay D, Mas JC, Rocchi S, Van Obberghen E. FALDH reverses the deleterious action of oxidative stress induced by lipid peroxidation product 4-hydroxynonenal on insulin signaling in 3T3-L1 adipocytes. Diabetes. 2008;57:1216–26.
Curtis JM, Hahn WS, Long EK, Burrill JS, Arriaga EA, Bernlohr DA. Protein carbonylation and metabolic control systems. Trends Endocrinol Metab. 2012;23:399–406.
Segovia SA, Vickers MH, Gray C, Reynolds CM. Maternal obesity, inflammation, and developmental programming. Biomed Res Int. 2014;2014:418975.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–7.
Jung UJ, Choi MS. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci. 2014;15:6184–223.
Catalano PM. Carbohydrate metabolism and gestational diabetes. Clin Obstet Gynecol. 1994;37:25–38.
Wallace JM, Horgan GW, Bhattacharya S. Placental weight and efficiency in relation to maternal body mass index and the risk of pregnancy complications in women delivering singleton babies. Placenta. 2012;33:611–8.
Frias AE, Morgan TK, Evans AE, Rasanen J, Oh KY, Thornburg KL, et al. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology. 2011;152:2456–64.
Farley DM, Choi J, Dudley DJ, Li C, Jenkins SL, Myatt L, et al. Placental amino acid transport and placental leptin resistance in pregnancies complicated by maternal obesity. Placenta. 2010;31:718–24.
Rosario FJ, Kanai Y, Powell TL, Jansson T. Increased placental nutrient transport in a novel mouse model of maternal obesity with fetal overgrowth. Obesity (Silver Spring). 2015;23:1663–70.
Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, et al. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab. 2013;98:105–13.
Kramer MS, Lydon J, Seguin L, Goulet L, Kahn SR, McNamara H, et al. Non-stress-related factors associated with maternal corticotrophin-releasing hormone (CRH) concentration. Paediatr Perinat Epidemiol. 2010;24:390–7.
Stirrat LI, O'Reilly JR, Riley SC, Howie AF, Beckett GJ, Smith R, et al. Altered maternal hypothalamic-pituitary-adrenal axis activity in obese pregnancy is associated with macrosomia and prolonged pregnancy. Pregnancy Hypertens. 2014;4:231–9.
Denison FC, Price J, Graham C, Wild S, Liston WA. Maternal obesity, length of gestation, risk of postdates pregnancy and spontaneous onset of labour at term. BJOG. 2008;115:720–5.
Lappas M, Permezel M, Rice GE. Leptin and adiponectin stimulate the release of proinflammatory cytokines and prostaglandins from human placenta and maternal adipose tissue via nuclear factor-kappaB, peroxisomal proliferator-activated receptor-gamma and extracellularly regulated kinase 1/2. Endocrinology. 2005;146:3334–42.
Suidan RS, Rondon KC, Apuzzio JJ, Williams SF. Labor Outcomes of Obese Patients Undergoing Induction of Labor with Misoprostol compared to Dinoprostone. Am J Perinatol. 2015;30:187–92.
Subbaramaiah K, Morris PG, Zhou XK, Morrow M, Du B, Giri D, et al. Increased levels of COX-2 and prostaglandin E2 contribute to elevated aromatase expression in inflamed breast tissue of obese women. Cancer Discov. 2012;2:356–65.
Martinez ME, Heddens D, Earnest DL, Bogert CL, Roe D, Einspahr J, et al. Physical activity, body mass index, and prostaglandin E2 levels in rectal mucosa. J Natl Cancer Inst. 1999;91:950–3.
Konopka CK, Morais EN, Naidon D, Pereira AM, Rubin MA, Oliveira JF, et al. Maternal serum progesterone, estradiol and estriol levels in successful dinoprostone-induced labor. Braz J Med Biol Res. 2013;46:91–7.
Wendremaire M, Goirand F, Barrichon M, Lirussi F, Peyronel C, Dumas M, et al. Leptin prevents MMP activation in an in vitro model of myometrial inflammation. Fundam Clin Pharmacol. 2012;26:83.
Wendremaire M, Bardou M, Peyronel C, Hadi T, Sagot P, Morrison JJ, et al. Effects of leptin on lipopolysaccharide-induced myometrial apoptosis in an in vitro human model of chorioamnionitis. Am J Obstet Gynecol. 2011;205:363.e1–9.
Zelig CM, Nichols SF, Dolinsky BM, Hecht MW, Napolitano PG. Interaction between maternal obesity and Bishop score in predicting successful induction of labor in term, nulliparous patients. Am J Perinatol. 2013;30:75–80.
Carlson NS, Lowe NK. Intrapartum management associated with obesity in nulliparous women. J Midwifery Womens Health. 2014;59:43–53.
Moynihan AT, Hehir MP, Glavey SV, Smith TJ, Morrison JJ. Inhibitory effect of leptin on human uterine contractility in vitro. Am J Obstet Gynecol. 2006;195:504–9.
Zera CA, McElrath TF, Rifas-Shiman SL, Parker M, Rich-Edwards JW, Gillman MW. A prospective study of second trimester leptin level and failed first stage of labor. Reprod Sci. 2010;17:211A.
AlSaif S, Mumtaz S, Wray S. A short review of adipokines, smooth muscle and uterine contractility. Life Sci. 2015;125:2–8.
Dopico AM, Bukiya AN, Singh AK. Large conductance, calcium- and voltage-gated potassium (BK) channels: regulation by cholesterol. Pharmacol Ther. 2012;135:133–50.
Noble K, Zhang J, Wray S. Lipid rafts, the sarcoplasmic reticulum and uterine calcium signalling: an integrated approach. J Physiol. 2006;570:29–35.
Shmygol A, Noble K, Wray S. Depletion of membrane cholesterol eliminates the Ca2 + −activated component of outward potassium current and decreases membrane capacitance in rat uterine myocytes. J Physiol London. 2007;581:445–56.
Turi A, Kiss AL, Mullner N. Estrogen downregulates the number of caveolae and the level of caveolin in uterine smooth muscle. Cell Biol Int. 2001;25:785–94.
Klein U, Gimpl G, Fahrenholz F. Alteration of the myometrial plasma membrane cholesterol content with beta-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry. 1995;34:13784–93.
Smith RD, Babiychuk EB, Noble K, Draeger A, Wray S. Increased cholesterol decreases uterine activity: functional effects of cholesterol alteration in pregnant rat myometrium. Am J Physiol Cell Physiol. 2005;288:C982–8.
Zhang J, Kendrick A, Quenby S, Wray S. Contractility and calcium signaling of human myometrium are profoundly affected by cholesterol manipulation: implications for labor? Reprod Sci. 2007;14:456–66.
Mouzat K, Prod'Homme M, Volle DH, Sion B, Dechelotte P, Gauthier K, et al. Oxysterol nuclear receptor LXR beta regulates cholesterol homeostasis and contractile function in mouse uterus. J Biol Chem. 2007;282:4693–701.
Fyfe EM, Rivers KS, Thompson JM, Thiyagarajan KP, Groom KM, Dekker GA, et al. Elevated maternal lipids in early pregnancy are not associated with risk of intrapartum caesarean in overweight and obese nulliparous women. BMC Pregnancy Childbirth. 2013;13:143.
Nuthalapaty FS, Rouse DJ, Owen J. The association of maternal weight with cesarean risk, labor duration, and cervical dilation rate during labor induction. Obstet Gynecol. 2004;103:452–6.
Roloff K, Peng S, Sanchez-Ramos L, Valenzuela GJ. Cumulative oxytocin dose during induction of labor according to maternal body mass index. Int J Gynecol Obstet. 2015;131:54–8.
Garabedian MJ, Hansen WF, Manning MA, McCord LA, Curry TE. Expression of uterine oxytocin receptor mrna increases with maternal obesity. Reprod Sci. 2011;18:131A.
Grotegut CA, Gunatilake RP, Feng L, Heine RP, Murtha AP. The influence of maternal body mass index on myometrial oxytocin receptor expression in pregnancy. Reprod Sci. 2013;20:1471–7.
Terkawi AS, Jackson WM, Thiet MP, Hansoti S, Tabassum R, Flood P. Oxytocin and catechol-O-methyltransferase receptor genotype predict the length of the first stage of labor. Am J Obstet Gynecol. 2012;207:184.e1–8.
Elmes MJ, Tan DS, Cheng Z, Wathes DC, McMullen S. The effects of a high-fat, high-cholesterol diet on markers of uterine contractility during parturition in the rat. Reproduction. 2011;141:283–90.
Quenby S, Pierce SJ, Brigham S, Wray S. Dysfunctional labor and myometrial lactic acidosis. Obstet Gynecol. 2004;103:718–23.
Mele J, Muralimanoharan S, Maloyan A, Myatt L. Impaired mitochondrial function in human placenta with increased maternal adiposity. Am J Physiol Endocrinol Metab. 2014;307:E419–425.
Gam CM, Mortensen OH, Qvortrup K, Damm P, Quistorff B. Effect of high-fat diet on rat myometrium during pregnancy-isolated myometrial mitochondria are not affected. Pflugers Arch. 2015;467:1539–49.
Mitchell BF, Taggart MJ. Are animal models relevant to key aspects of human parturition? Am J Physiol Regul Integr Comp Physiol. 2009;297:R525–545.
Kaiser PS, Kirby RS. Obesity as a risk factor for cesarean in a low-risk population. Obstet Gynecol. 2001;97:39–43.
Kominiarek M, Vanveldhuisen P, Hibbard J, Landy H, Haberman S, Learman L, et al. The maternal body mass index: a strong association with delivery route. Am J Obstet Gynecol. 2010;203:264.e261–267.
Rasmussen T, Stene LC, Samuelsen SO, Cinek O, Wetlesen T, Torjesen PA, et al. Maternal BMI before pregnancy, maternal weight gain during pregnancy, and risk of persistent positivity for multiple diabetes-associated autoantibodies in children with the high-risk HLA genotype: the MIDIA study. Diabetes Care. 2009;32:1904–6.
Morton S, Thangaratinam S. Statins in pregnancy. Curr Opin Obstet Gynecol. 2013;25:433–40.
Jiang M, Strand DW, Franco OE, Clark PE, Hayward SW. PPAR gamma: A molecular link between systemic metabolic disease and benign prostate hyperplasia. Differentiation. 2011;82:220–36.
Smith SR, De Jonge L, Volaufova J, Li Y, Xie H, Bray GA. Effect of pioglitazone on body composition and energy expenditure: a randomized controlled trial. Metabolism. 2005;54:24–32.
Cheng S, Massaro JM, Fox CS, Larson MG, Keyes MJ, McCabe EL, et al. Adiposity, cardiometabolic risk, and vitamin D status: the Framingham Heart Study. Diabetes. 2010;59:242–8.
Asemi Z, Samimi M, Tabassi Z, Shakeri H, Esmaillzadeh A. Vitamin D supplementation affects serum high-sensitivity C-reactive protein, insulin resistance, and biomarkers of oxidative stress in pregnant women. J Nutr. 2013;143:1432–8.
Scott-Pillai R, Spence D, Cardwell CR, Hunter A, Holmes VA. The impact of body mass index on maternal and neonatal outcomes: a retrospective study in a UK obstetric population, 2004–2011. BJOG. 2013;120:932–9.
Garabedian MJ, Williams CM, Pearce CF, Lain KY, Hansen WF. Extreme morbid obesity and labor outcome in nulliparous women at term. Am J Perinatol. 2011;28:729–34.
Jensen H, Agger AO, Rasmussen KL. The influence of prepregnancy body mass index on labor complications. Acta Obstet Gynecol Scand. 1999;78:799–802.
Abenhaim HA, Benjamin A. Higher cesarean section rates in women with high body mass index: are we managing differently? J Obstet Gynaecology Canada. 2011;33:443–8.
Vahratian A, Siega-Riz AM, Savitz DA, Zhang J. Maternal Pre-pregnancy Overweight and Obesity and the Risk of Cesarean Delivery in Nulliparous Women. Ann Epidemiol. 2005;15:467–74.
Vinturache A, Moledina N, McDonald S, Slater D, Tough S. Pre-pregnancy Body Mass Index (BMI) and delivery outcomes in a Canadian population. BMC Pregnancy Childbirth. 2014;14:422.
Green C, Shaker D. Impact of morbid obesity on the mode of delivery and obstetric outcome in nulliparous singleton pregnancy and the implications for rural maternity services. Aust N Z J Obstet Gynaecol. 2011;51:172–4.
Cedergren MI. Non-elective caesarean delivery due to ineffective uterine contractility or due to obstructed labour in relation to maternal body mass index. Eur J Obstet Gynecol Reprod Biol. 2009;145:163–6.
Sukalich S, Mingione MJ, Glantz JC. Obstetric outcomes in overweight and obese adolescents. Am J Obstet Gynecol. 2006;195:851–5.
Wendremaire M, Mourtialon P, Goirand F, Lirussi F, Barrichon M, Hadi T, et al. Effects of leptin on lipopolysaccharide-induced remodeling in an in vitro model of human myometrial inflammation. Biol Reprod. 2013;88:45.
Higgins CA, Martin W, Anderson L, Blanks AM, Norman JE, McConnachie A, et al. Maternal Obesity and its Relationship With Spontaneous and Oxytocin-Induced Contractility of Human Myometrium In Vitro. Reprod Sci. 2009;17:177–85.
Acknowledgements
The authors acknowledge Dr. Nancy Lowe for her mentorship of NC through studies on labor dystocia, and David Carlson, for the original medical illustration. Our appreciation to Drs. Thomas Jansson, Peggy Neville, Andy Bradford, Elizabeth Holt, Emily Su, Gosia Skaznik-Wikiel, and Nancy Lowe for discussions and helpful feedback on the manuscript.
Grant support: NIH National Institute of Nursing Research, Grant # 1F31NR014061-01A1, March of Dimes Graduate Nursing Grant, and The University of Colorado School of Nursing ‘Touched by a Nurse’ Scholarship (NC); R01-DK101659 (TLH); and March of Dimes Basil O’Connor award, #5-FY12-37, NIH/NCATS Colorado CTSA Grant #UL1TR001082, and UCDenver WRHR #2K12HD001271 (KJH).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NC and KJH conceived of and designed the approach to the review and the manuscript summary. NC conducted literature search with TLH (obesity metabolism) and KJH (labor mechanism). NC drafted the manuscript with critical revision by TLH and KJH. All authors read, revised, and approved the submitted version.
Additional file
Additional file 1: Table S1.
Studies Located in Comprehensive Review of Mechanisms of Parturition Dysfunction in Obesity (PDF 956 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Carlson, N.S., Hernandez, T.L. & Hurt, K.J. Parturition dysfunction in obesity: time to target the pathobiology. Reprod Biol Endocrinol 13, 135 (2015). https://doi.org/10.1186/s12958-015-0129-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12958-015-0129-6