
Abstract
Background
Colon adenocarcinoma (COAD) is a gastrointestinal tumor with a high degree of malignancy. Its deterioration process is closely related to the tumor microenvironment, and transcription factors (TF) play a regulatory role in this process. Currently, there is a lack of exploration between the genes related to the COAD tumor microenvironment and the survival prognosis of patients. Models composed of multiple genes usually predict the survival prognosis of patients more accurately than single genes. We can analyze the multigene models that can predict the prognosis of COAD from the current database.
Methods
The limma package of the R programming language is used for gene differential expression analysis. Kaplan-Meier curve is used to analyze the relationship between the patient risk score model and survival data. The hazard model is used to analyze the relationship between the risk score and the clinical data of COAD patients. The information of immune genes and immune cells is obtained from IMMPORT database and TIMER database. Receiver operating characteristic (ROC) curve is used to judge the stability of the model.
Results
We found 7 immune genes, which can built a risk score model to predict the survival prognosis of COAD. According to univariate and multivariate analysis, the risk score can be used as an independent predictor. The content of some immune microenvironment cells will also increase as the risk score increases.
Conclusions
We found 7 immune genes, such as SLC10A2 (solute carrier family 10 member 2), CXCL3 (C-X-C motif chemokine ligand 3), IGHV5-51 (immunoglobulin heavy variable 5-51), INHBA (inhibin subunit beta A), STC1 (stanniocalcin 1), UCN (urocortin), and OXTR (oxytocin receptor), can constitute a model for predicting the prognosis of COAD. They may provide potential therapeutic targets for clinical treatment of COAD.
Similar content being viewed by others

Background
Colon adenocarcinoma (COAD) is a type of malignant tumor of the digestive tract, which can be subdivided into right COAD and left COAD according to location. According to a WHO report in 2018, COAD is the third most common adenocarcinoma worldwide, and 1.8 million COAD cases were diagnosed in 2018 (10% of all tumors). Adenocarcinoma of the colon is relatively common in both men and women. There were 881,000 patients who died of COAD in 2018 [1,2,3,4]. Recently, many studies have shown that the immune microenvironment plays an important role in the process of tumors [5], such as the CCR (cinnamoyl-CoA reductase) family and the CCL (CCR-like protein) famil y[6], and transcription factors can regulate this process [7, 8], but the research on the immune microenvironment in the field of COAD still needs further exploration. Compared with the single genes predicting the prognosis of cancer patients, multigene models can more accurately predict the prognosis of cancer patients, so the building of a multigene model related to the tumor microenvironment has become the focus of this research [9].
With the development of various network databases [10], such as clinical database TCGA (https://portal.gdc.cancer.gov/) [11], immune gene database IMMPORT (https://www.immport.org/shared/home) [12], TIMER (https://cistrome.shinyapps.io/timer/) [13], and transcription factor database Cistrome (http://www.cistrome.org/) [14], we can find the immune genes related to the prognosis of COAD through data analysis methods such as limma package (http://www.bioconductor.org/packages/release/bioc/html/limma.html) [15]. Cox regression analysis is used to build a risk score model of immune genes related to prognosis. Seven immune genes building this model are SLC10A2 (solute carrier family 10 member 2), CXCL3 (C-X-C motif chemokine ligand 3), IGHV5-51 (immunoglobulin heavy variable 5-51), INHBA (inhibin subunit beta A), STC1 (stanniocalcin 1), UCN (urocortin), and OXTR (oxytocin receptor). We define the sample with the highest risk score of 50% as the high-risk group and the sample with the lowest risk score of 50% as the low-risk group. Subsequent studies will combine relevant clinical data to further compare the differences between the two groups.
The result of this study is we built a multigene model related to the immune microenvironment, which can predict the prognosis of COAD patients. These seven immune genes may provide potential therapeutic targets for clinical treatment of COAD.
Methods
Gets the relevant data from the network database
Obtaining COAD gene expression data and clinical data from the TCGA (https://portal.gdc.cancer.gov/) database, immune gene names were obtained from the IMMPORT (https://www.immport.org/shared/home) database, and immune genes were screened from the downloaded data. Transcription factor data from the Cistrome (http://www.cistrome.org/) database and tumor microenvironment-related gene infiltration data were obtained from the TIMER (https://cistrome.shinyapps.io/timer/) database.
Acquisition of differentially expressed genes of interest
Using the R script (Gene.diff.R) to obtain differentially expressed genes of interest, then, the differential expression analysis of immune genes uses R script (immuneGene.immuneDiff.R), and the screening condition is LogFC (log fold change) > 2 and FDR (false discover rate) < 0.05. Then, differentially expressed transcription factors were analyzed using R script (immuneGene.TFdiff.R), and the screening conditions were LogFC > 1 and P < 0.05.
Obtain immune genes related to the survival prognosis of COAD patients
Univariate Cox regression analysis was performed on the differentially expressed immune genes obtained by the above method and the clinical data of COAD patients, and then, the immune genes related to the prognosis of COAD patients were obtained. The screening conditions are P < 0.01 and hazard ratio ≠ 1(immuneGene.uniCox.R).
Calculation of risk score and independent prognostic analysis
First, through univariate Cox regression analysis, the immune genes related to the prognosis of COAD are obtained and then through multivariate Cox regression to find the immune genes that can build the risk score model (immuneGene.multiCox.R). Risk score = ExpmRNA1 × coefmRNA1 + ExprmRNA2 × coefmRNA2 +···+ ExpmRNAn × coefmRNA. “Exp” indicates the expression level of the gene; “coef” indicates the correlation coefficient of the gene. Finally, we combined clinical data and used immuneGene.uniIndep.R and immuneGene.multiIndep.R for univariate independent prognostic analysis and multivariate independent prognostic analysis.
Immune genes will draw the interaction network of transcription factors
Mapping the interaction network of immune genes and transcription factors using R script (immuneGene.TFcor.R) and Cytoscape [16], the screening conditions are Cor = 0.4 and P = 0.01.
Analysis of clinical correlation of 7 genes (building of risk scoring model)
We correlate the risk score with clinical data and analyze and make the corresponding models. Building of Kaplan-Meier curve (K-M) uses R script (immuneGene.survial.R); the building of receiver operating characteristic (ROC) model uses R script (immuneGene.ROC.R); the building of risk curve uses R script (immuneGene.riskPlot.R); clinical correlation was built using R script (immuneGene.clincialCor.R), and the immune cell correlation graph was built using R script (immuneGene.immuneCor.R). The K-M curve is used to express the relationship between risk scores and patient survival data. P < 0.05 is considered statistically significant. The ROC curve is used to indicate the sensitivity of the model. 0.5–0.7 means the sensitivity is acceptable, 0.7–0.9 means the sensitivity is good, and > 0.9 means the sensitivity is excellent.
Correlation analysis between samples and tumor microenvironment
Twenty samples from the top 10 and the bottom 10 of the risk score were selected to analyze the cells composed of the tumor microenvironment. The TIMER database was used for this analysis and draw (Fig. 9). Figure 8 is drawn using R script (immuneGene.immuneCor.R).
Result
Acquisition of immune DEGs and differentially expressed transcription factors
The research team downloaded clinical data and gene expression data of 385 COAD patients from the TCGA (https://portal.gdc.cancer.gov/) database and obtained differentially expressed genes (DEGs) through screening (screening conditions: LogFC > 2 and FDR < 0.05) (Figs. 1 and 2a, b). The IMMPORT (https://www.immport.org/shared/home) database contains the names of a large number of immune genes. We obtain the differentially expressed immune genes through the intersection of the immune gene names and DEGs (Fig. 2c, d). The transcription factor names were obtained from the Cistrome (http://www.cistrome.org/) database and then screened for eligible transcription factors from DEGs. The screening conditions for transcription factors are LogFC > 1 and P < 0.05. The volcano and heat maps were drawn (Fig. 2e, f).

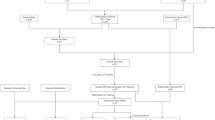
Flow chart of this study

a Heat map of genes expression. b Volcano map of genes expression. c Heat map of immune genes expression. d Volcano map of immune genes expression. e Heat map of transcription factors expression. f Volcano map of transcription factors expression
Building of the prognostic-related immune gene model and the interaction network of prognostic-related immune genes and transcription factors
Univariate Cox regression analysis was used to study the differentially expressed immune genes related to survival prognosis. The results showed that there are 12 immune genes that are closely related to the prognosis of COAD (Fig. 3a). Cor = 0.4 and P = 0.01 screening criteria were used to establish the interaction between immune genes and transcription factors, and network diagrams were made (Fig. 3b). Details of the regulatory relationship between transcription factors and the immune genes associated with COAD prognosis are shown in Table 1. The results showed that 8 immune genes are closely related to the regulation of transcription factors and belong to positive regulation.

a Forest plot of 12 immune genes associated with survival prognosis in patients with COAD. b Regulatory network of immune genes and transcription factors related to the prognosis of COAD. The circles represent immune genes (green represents a downward adjustment, and red represents an upward adjustment), and the triangles represent transcription factors
Calculation of immune gene risk score and building of survival prognosis model
The 12 immune genes (Fig. 3a) related to the survival prognosis of COAD patients obtained by univariate Cox analysis were included. Then, using multivariate Cox regression analysis, the screening conditions were P < 0.05 and hazard ratio (HR) ≠ 1, and 7 immune genes were eligible and included for further calculation of risk score. We define the sample with the top 50% of the risk score as the high-risk group and the sample with the bottom 50% of the risk score as the low-risk group. Subsequent studies will further compare the differences between the two groups. Among the immune genes related to survival prognosis, 7 immune genes are closely related to the composition of risk score, which are SLC10A2, CXCL3, IGHV5-51, INHBA, STC1, UCN, and OXTR (Table 2); this is also the key immune gene that we will study later. According to the median risk score, the risk score is divided into two groups. Survival and surmiser packages in R were used to correlate risk score with survival prognosis and draw the Kaplan-Meier survival curves. The results showed that P = 8.876e−04. The survival prognosis of the high-risk group was significantly worse than that of the low-expression group (Fig. 4a). The survivalROC package of R language is used to draw the ROC curve. The results show that the AUC of the ROC curve = 0.749 (Fig. 4b). Detailed data on the survival rates of high- and low-risk patients are shown in Tables 3 and 4.

a Kaplan-Meier survival curve of high-risk group and low-risk group. b ROC curve of survival prognosis model (0.5–0.7 means the sensitivity is acceptable, 0.7-0.9 means the sensitivity is good, and > 0.9 means the sensitivity is excellent)
Immune gene risk curve mapping and independent prognostic analysis
Using R language related codes to draw related pictures of the risk curve, the results showed that with the gradual increase of the immune gene risk value, the survival time of patients gradually decreased (Fig. 5a, b). Heat map of related immune gene expression is shown in Fig. 5c. Univariate independent prognostic analysis showed that the hazard ratio of risk score was 1.033 (1.018–1.049) and P < 0.001. Multivariate independent prognostic analysis showed that hazard ratio of risk score was 1.026 (1.011–1.042) and P < 0.001 (Fig. 6a, b). Risk scores are clinically and statistically significant.

a Risk curve of risk score growth trend. b Diagram of the relationship between risk score and patient survival time. c Building of a heat map of immune genes in the risk score model

a Forest plot of univariate Cox regression analysis between immune genes and clinical data constituting the risk score model. b Forest plot of multivariate Cox regression analysis between immune genes and clinical data that constitute the risk score model
Correlation analysis of immune genes and clinical data
We analyzed the correlation between the 7 immune genes that make up the immune score and clinical data, using the R language beeswarm package. The results showed that there were statistically significant correlations between seven immune genes and clinical data, namely, CXCL3, OXTR, and STC1 (Fig. 7). Among them, the expressions of CXCL3 were statistically significant in correlation with stage, while the expressions of OXTR and STC1 were statistically significant in correlation with T. CXCL3 also has significant difference in N and M. In T1–2, the tumor invades the submucosa, or the tumor invades the muscularis intestinal wall; in T3–T4, the tumor infiltrates the muscularis laminae and reaches the subserosa, or the tumor has penetrated the peritoneum; in N0, no regional lymph node metastasis; in N1–3, there is metastasis in regional lymph nodes; in M0, the tumor has no distant metastasis; in M1, the tumor has distant metastasis. Stages I, II, and III are early colon cancer, and stage IV is advanced colon cancer.

Correlation analysis of genes (building a risk score model) and clinical data. a Correlation analysis between CXCL3 and M. b Correlation analysis between CXCL3 and N. c Correlation analysis between CXCL3 and COAD stage. d Correlation analysis between OXTR and T. e Correlation analysis between STC1 and T. T1–2, the tumor invades the submucosa, or the tumor invades the muscularis intestinal wall; T3–T4, the tumor infiltrates the muscularis laminae and reaches the subserosa, or the tumor has penetrated the peritoneum; N0, no regional lymph node metastasis; N1–3, there is metastasis in regional lymph nodes; M0, the tumor has no distant metastasis; M1, the tumor has distant metastasis; stage, the stage of colon cancer. Stages I, II, and III are early colon cancer, and IV is advanced colon cancer
Correlation analysis of risk score and tumor microenvironment cells
Correlation analysis was performed between the risk score assessed by our research and immune microenvironment genes, and the results showed that in CD4, CD8, dendritic, macrophage, and neutrophil cells, as the risk score increased, the expression levels of these genes became upward. And it has statistical significance P < 0.05. Correlation analysis of our risk value model with some widely recognized genes that constitute the immune microenvironment showed that CD4, CD8, dendritic, macrophage, and neutrophil cells were positively correlated with the risk score model. As the risk score increases, so does the expression of these genes (Fig. 8).

Correlation analysis between the expression of immune microenvironment cells and risk score. a Correlation analysis between B cells and risk score. b Correlation analysis between CD4 cells and risk score. c Correlation analysis between CD8 cells and risk score. d Correlation analysis between dendritic cells and risk score. e Correlation analysis between macrophage cells correlation and risk score. f Correlation analysis between neutrophil cells and risk score
Correlation between risk score model and tumor microenvironment
To evaluate the difference in immune cell content between samples with high risk score and samples with low risk score, we selected 20 sets of samples, which were selected from the 10 samples with the highest risk score and the 10 samples with the lowest risk score, through the EPIC database. Calculate the difference in the amount of their direct immune cells, the results showed that in 10 samples with high risk score, the content of cancer-associated fibroblasts (CAFs) cells was significantly higher than that with 10 samples with low risk score. From this, we can know that CAFs cells play an important role in risk score (Fig. 9).

a Comparison of immune cell differences between 10 groups of high risk score samples and 10 groups of low risk score samples, each color represents a different cell type. b The expression of immune cells differs between 10 groups of high risk score samples and 10 groups of low risk score samples. c Immune cell correlation matrix, the positive correlation is red, and the negative correlation is blue
Discussion
The flow of this study is shown in Fig. 1. We searched the TCGA (https://portal.gdc.cancer.gov/) database for 385 cases of COAD and downloaded them. The clinical data and gene expression data in the download data were integrated, and the limma package of R language was used to extract differentially expressed genes (DEGs). Using the immune gene names provided by IMMPORT (https://www.immport.org/shared/home), we can easily screen out the differential gene immune genes from DEGs. In the same way, we download the names of transcription factors from the Cistrome (http://www.cistrome.org/) database and screen out the differentially expressed transcription factors from DEGs (screening conditions: LogFC > 2 and FDR < 0.05). We then conduct further analysis of differentially expressed immune genes (screening conditions: LogFC > 2 and FDR < 0.05) and differentially expressed transcription factors (screening conditions: LogFC > 1 and FDR < 0.05). The differentially expressed immune genes were analyzed by univariate Cox regression using the survival package of R language and clinical data to obtain immune genes related to prognosis. Prognostic-related immune genes are as follows: SLC10A2, CXCL3, NOX4, CCL19, IGHG1, IGHV5-51, IGKV1-33, INHBA, STC1, UCN, VIP, and OXTR (Fig. 3a). We performed an interaction network analysis of prognostic-associated immune genes and differentially expressed transcription factors, and the results are shown in Fig. 3b. Analysis results show that the regulatory network functions are mainly concentrated in optic vesicle morphogenesis and regulation of leukocyte adhesion to arterial endothelial cells, but more specific mechanisms need further study [17].
We excluded samples with a survival time of less than 90 days from the downloaded clinical data and assessed the survival prognosis by the level of risk score. The results showed that patients with high risk score had significantly worse survival prognosis than patients with low risk score, P = 8.876e−04 (Fig. 4a). The ROC curve showed that AUC = 0.749, and the risk score and prognosis model were more reliable (Fig. 4b). This allows us to group patients according to the risk scores in clinical work to predict their prognosis. According to Fig. 5 a and b, we can find that as the risk score increases, the survival time of the patient decreases. The heat map of Fig. 5c also shows that the genes that build the risk score have higher expression levels in the high risk score array. We included clinical data on COAD and the risk score evaluated in this study into the Cox regression analysis. The results showed that stage, T, M, N, and risk score were statistically significant and clinically significant in the survival prognosis of the patients in the univariate Cox regression analysis. However, in the results of multivariate Cox regression analysis, age, stage, T, and risk score have statistical significance and clinical significance. Based on the analysis of the seven genes and clinical data used to build the risk score model, the results show that the expression of CXCL3 gene in M, N, and stage is higher than that in late stage. This is likely to be related to the mechanism of the immune microenvironment [18]. Studies on the immune microenvironment have shown that some genes that build the immune microenvironment can promote tumor progression (Fig. 7). Some cells that make up the tumor microenvironment, such as B, CD4, CD8, dendritic, macrophage, and neutrophil cells, have been shown in research to be correlated with the survival prognosis of many types of tumor patients [9]. We downloaded the data of these cells through the TIMER (https://cistrome.shinyapps.io/timer/) database and performed correlation analysis with the risk score model we built. The results showed that the expression of CD4, CD8, dendritic, macrophage, and neutrophil cells increased with the increase of the risk score. This also confirms on the side that the risk score model we built has a certain predictive ability for the clinical prognosis of patients.
The formation of the tumor microenvironment is closely related to the occurrence and development of tumors [9]. By studying the cells that constitute the tumor microenvironment, we can effectively find many cells or genes that are closely related to the clinical prognosis of patients. To evaluate the difference in immune cell content between samples with high risk score and samples with low risk score, we further evaluated them in the EPIC database. We selected 20 sets of samples, which were selected from the 10 samples with the highest risk score and the 10 samples with the lowest risk score, which passed the EPIC database [19]. Calculate the difference in the amount of their direct immune cells, the results showed that in 10 samples with high risk score, the content of CAFs cells was significantly higher than that with 10 samples with low risk score. From this, we can know that CAFs cells play an important role in risk score. The related literature reports that cancer-associated fibroblasts (CAFs) are the main cell types in the tumor stromal. CAFs usually promote tumor progression by inducing cell proliferation, inflammation, blood vessel growth, and metastasis. We judged that the content of CAFs is also an important indicator to increase the risk score [20].
In this study, we built an immune gene risk score model for 385 COAD patients through correlation analysis. Through a series of analyses of the disease, it was found that the risk score is closely related to the survival prognosis of patients. In future clinical treatments, we can use the risk score model to effectively predict the survival prognosis of patients with COAD, and we can do targeted immunotherapy for 7 immune genes (SLC10A2, CXCL3, IGHV5-51, INHBA, STC1, UCN, and OXTR) that constitute the risk score to improve the prognosis of patients and improve the treatment effect.
Bile acids, especially secondary bile acids, can promote the development of colorectal cancer, and SLC10A2 can promote this process [21, 22]. CXCL3 is related to the occurrence and development of prostate cancer, colon cancer, and breast cancer. There are also reports in the literature that the effect of suppressing the development of colon cancer can be achieved by immunosuppression of CXCL3 [23,24,25,26,27]. INHBA has a significant relationship with the occurrence and development of gastric, esophageal, and ovarian cancers, and studies have reported that the immunosuppressive treatment of INHBA can reduce the rate of deterioration of gastric and ovarian cancers [28,29,30]. STC1 can promote the metastasis of colon cancer [31, 32].
Conclusion
We download data for COAD, immune genes, and transcription factors through a series of bioinformatics databases. A risk score model of COAD immune genes was built. Through a series of clinical correlation analysis, it was found that 7 immune genes (SLC10A2, CXCL3, IGHV5-51, INHBA, STC1, UCN, and OXTR) were correlated with clinical prognosis and risk score of patients with COAD. These seven immune genes may provide potential therapeutic targets for clinical treatment of COAD.
Availability of data and materials
The data used to support the findings of this study are included within the article.
Abbreviations
- COAD:
-
Colon adenocarcinoma
- HR:
-
Hazard ratio
- TF:
-
Transcription factor
- Coef:
-
Correlation coefficient of the gene
- Std.err:
-
Std. error difference
- ID:
-
Identity
- cor:
-
Correlation
- LogFC:
-
Log fold change
- FDR:
-
False discover rate
- P :
-
P value
- Exp:
-
Expression level of the gene
- ROC:
-
Receiver operating characteristic
- K-M:
-
Kaplan-Meier curve
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Chen W, Sun K, Zheng R, Zeng H, Zhang S, Xia C, Yang Z, Li H, Zou X, He J. Cancer incidence and mortality in China, 2014. Chin J Cancer Res. 2018;30(1):1–12.
Feng RM, Zong YN, Cao SM, Xu RH. Current cancer situation in China: good or bad news from the 2018 Global Cancer Statistics? Cancer Commun (Lond). 2019;39(1):22.
Fedeli P, Casella C, Buccelli C, Cannovo N, Vincenzo G. Genetic research: the role of citizens, public health and international stakeholders. Open Public Health J. 2019;12:106–13.
Koi M, Carethers JM. The colorectal cancer immune microenvironment and approach to immunotherapies. Future Oncol. 2017;13(18):1633–47.
Wunderlich CM, Ackermann PJ, Ostermann AL, Adams-Quack P, Vogt MC, Tran ML, Nikolajev A, Waisman A, Garbers C, Theurich S, et al. Obesity exacerbates colitis-associated cancer via IL-6-regulated macrophage polarisation and CCL-20/CCR-6-mediated lymphocyte recruitment. Nat Commun. 2018;9(1):1646.
Taylor CT, Colgan SP. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol. 2017;17(12):774–85.
Marinho HS, Real C, Cyrne L, Soares H, Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014;2:535–62.
Frankel T, Lanfranca MP, Zou W. The role of tumor microenvironment in cancer immunotherapy. Adv Exp Med Biol. 2017;1036:51–64.
Chen C, Huang H, Wu CH. Protein bioinformatics databases and resources. Methods Mol Biol. 2017;1558:3–39.
Blum A, Wang P, Zenklusen JC. SnapShot: TCGA-analyzed tumors. Cell. 2018;173(2):530.
Bhattacharya S, Dunn P, Thomas CG, Smith B, Schaefer H, Chen J, Hu Z, Zalocusky KA, Shankar RD, Shen-Orr SS, et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci Data. 2018;5:180015.
Li B, Severson E, Pignon J-C, Zhao H, Li T, Novak J, Jiang P, Shen H, Aster JC, Rodig S, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17(1):174.
Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, Chen C-H, Brown M, Zhang X, Meyer CA, et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019;47(D1):D729–35.
Law CW, Alhamdoosh M, Su S, Dong X, Tian L, Smyth GK, Ritchie ME. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Res. 2016;5.
Otasek D, Morris JH, Bouças J, Pico AR, Demchak B. Cytoscape automation: empowering workflow-based network analysis. Genome Biol. 2019;20(1):185.
Huang da W, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009, 4(1):44-57.
Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–8.
Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife. 2017;6.
Xing F, Saidou J, Watabe K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci (Landmark Ed). 2010;15:166–79.
Ai X, Mao F, Shen S, Shentu Y, Wang J, Lu S. Bexarotene inhibits the viability of non-small cell lung cancer cells via slc10a2/PPARγ/PTEN/mTOR signaling pathway. BMC Cancer. 2018;18(1):407.
Wang W, Xue S, Ingles SA, Chen Q, Diep AT, Frankl HD, Stolz A, Haile RW. An association between genetic polymorphisms in the ileal sodium-dependent bile acid transporter gene and the risk of colorectal adenomas. Cancer Epidemiol Biomark Prev. 2001;10(9):931–6.
Gui SL, Teng LC, Wang SQ, Liu S, Lin YL, Zhao XL, Liu L, Sui HY, Yang Y, Liang LC, et al. Overexpression of CXCL3 can enhance the oncogenic potential of prostate cancer. Int Urol Nephrol. 2016;48(5):701–9.
Hänggi K, Ruffell B. Oncogenic KRAS drives immune suppression in colorectal cancer. Cancer Cell. 2019;35(4):535–7.
Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, Li J, Wang G, Lan Z, Li J, et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell. 2019;35(4):559-572.e557.
See AL, Chong PK, Lu SY, Lim YP. CXCL3 is a potential target for breast cancer metastasis. Curr Cancer Drug Targets. 2014;14(3):294–309.
Xin H, Cao Y, Shao ML, Zhang W, Zhang CB, Wang JT, Liang LC, Shao WW, Qi YL, Li Y, et al. Chemokine CXCL3 mediates prostate cancer cells proliferation, migration and gene expression changes in an autocrine/paracrine fashion. Int Urol Nephrol. 2018;50(5):861–8.
Katayama Y, Oshima T, Sakamaki K, Aoyama T, Sato T, Masudo K, Shiozawa M, Yoshikawa T, Rino Y, Imada T, et al. Clinical significance of INHBA gene expression in patients with gastric cancer who receive curative resection followed by adjuvant S-1 chemotherapy. In Vivo. 2017;31(4):565–71.
Li X, Yang Z, Xu S, Wang Z, Jin P, Yang X, Zhang Z, Wang Y, Wei X, Fang T, et al. Targeting INHBA in ovarian cancer cells suppresses cancer xenograft growth by attenuating stromal fibroblast activation. Dis Markers. 2019;2019:7275289.
Lyu S, Jiang C, Xu R, Huang Y, Yan S. INHBA upregulation correlates with poorer prognosis in patients with esophageal squamous cell carcinoma. Cancer Manag Res. 2018;10:1585–96.
Peña C, Céspedes MV, Lindh MB, Kiflemariam S, Mezheyeuski A, Edqvist PH, Hägglöf C, Birgisson H, Bojmar L, Jirström K, et al. STC1 expression by cancer-associated fibroblasts drives metastasis of colorectal cancer. Cancer Res. 2013;73(4):1287–97.
Rezapour S, Bahrami T, Hashemzadeh S, Estiar MA, Nemati M, Ravanbakhsh R, Feizi MA, Kafil HS, Pouladi N, Ghojazadeh M, et al. STC1 and NF-κB p65 (Rel A) is constitutively activated in colorectal cancer. Clin Lab. 2016;62(3):463–9.
Acknowledgements
Thanks to the Fund (The Key Research and Development Plan Projects of Anhui Province) for supporting this research.
Funding
The Key Research and Development Plan Projects of Anhui Province (No.201904a07020045).
Author information
Authors and Affiliations
Contributions
Sihan Chen is responsible for writing and submitting the papers; Cao GD, Wu wei, and LU Yida are responsible for data analysis and collation; He Xiaobo, Yang Lei, and Chen Ke are responsible for the production of pictures; Bo Chen and MM Xiong are responsible for the manuscript fees and ideas guidance. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
There were no cell, tissue, or animal studies. No ethical requirements are involved.
Consent for publication
All authors agree to publish the paper.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, S., Cao, G.D., Wei, W. et al. Prediction and identification of immune genes related to the prognosis of patients with colon adenocarcinoma and its mechanisms. World J Surg Onc 18, 146 (2020). https://doi.org/10.1186/s12957-020-01921-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12957-020-01921-9