
Abstract
Background
Inflammatory bowel disease (IBD), which includes ulcerative colitis (UC) and Crohn’s disease (CD), is a chronic, idiopathic, repeated inflammatory disease. Colorectal cancer (CRC) that develops in patients with IBD is known as colitis-associated colorectal cancer (CAC), but the underlying carcinogenic mechanism remains unclear. Genomic analysis of sporadic CRC has been well described based on next-generation sequencing (NGS) data. Using NGS, we compared all exons of 415 cancer-associated genes in patients in Japan and the USA who had CRC and found similar genomic alteration patterns among the two populations. However, genomic analysis of CAC has not been thoroughly investigated.
Main body
The molecular pathogenesis of CAC shares many features with sporadic CRC, but there are distinct variations in the time and frequency of some alterations. Gene alterations in CAC are gradually being elucidated using genomic sequencing analyses. Some studies have shown that gene alteration patterns differ between UC and CD. The carcinogenesis of CAC depends on unique environmental, genetic, and immunological factors.
Conclusions
In this review, we have discussed the differences in genomic alterations between sporadic CRC and CAC. NGS in patients with IBD has the potential to detect early CAC and to suggest therapeutic targets.
Similar content being viewed by others

Background
Inflammatory bowel disease (IBD) is a chronic, idiopathic, repeated inflammatory disease that includes ulcerative colitis (UC) and Crohn’s disease (CD) [1]. IBD has distinct pathological and clinical characteristics; however, its etiology has not been completely elucidated [2].
IBD with colonic involvement markedly increases the risk of colorectal cancer (CRC). However, the detection of dysplastic or early malignant lesions on colonoscopy in patients with UC can be difficult, thus making the investigation of colitis-associated cancer (CAC) challenging.
Recently, the genomic analysis of sporadic CRC has been well-demonstrated using whole-exome sequencing or clinical target sequencing by next-generation sequencing (NGS) technology. Although these analyses have not been thoroughly utilized for analyzing CAC, limited studies have demonstrated that the mutations in CAC are similar to those in sporadic CRC. On the contrary, the timing of acquisition of mutations in particular genes may differ between CAC and sporadic CRC. This review focused on the results of genomic analysis of CAC, particularly on the differences in genomic alterations between sporadic CRC and CAC.
This study was approved by the ethics committee of the Niigata University Medical and Dental Hospital (No. 772).
Ulcerative colitis
UC, which is a form of IBD which affects the mucosal layer of the colon, has become increasingly prevalent worldwide [1]. Recently, the prevalence of UC has been increasing worldwide, with the highest rates in Europe (505 per 100,000), Canada (248 per 100,000), and the USA (241 per 100,000) [1]. In Japan, the prevalence rate of UC was 121.9 per 100,000 in 2013 [3]. UC is characterized by relapsing and remitting colonic mucosal inflammation. The therapy for UC aims to resolve clinical symptoms, such as rectal bleeding and diarrhea [4] and to induce and maintain endoscopically confirmed remission, with the long-term goal of preventing disability and CAC [1].
Crohn’s disease
CD is an inflammatory disease that affects the entire digestive tract from the mouth to the anus [5]. Its pathogenesis is not completely understood, but it is thought to develop due to environmental triggers in genetically susceptible patients [6]. The incidence of CD is also increasing worldwide. Kalla et al. reported that the incidence of CD was 29.3 per 100,000 in Australia, 20.2 per 100,000 in Canada, and 10.6 per 100,000 in northern Europe [6]. In Japan, the number of patients with CD has markedly increased. The prevalence rate in Japan was 2.9 per 100,000 in 1986 and increased to 13.5 per 100,000 by 1998 [7]. Patients with active CD often have poor quality of life which is secondary to abdominal pain, bowel obstruction, and fistula. Therapeutic agents for CD aim to treat or prevent these complications. Therefore, it is very important to ensure the best outcomes using a multidisciplinary approach toward treatment [6].
Epidemiology of colitis-associated cancer
CAC was first recognized as a complication of UC by Crohn and Rosenberg in 1925 [8, 9], with malignancy developing in the colon or rectum in areas with active inflammation. It is listed as the cause of death in 10–15% of all patients with IBD [10,11,12,13]. The main risk factors for CAC include onset of IBD in young age, family history of sporadic CRC, longer duration of IBD, greater extent of colitis, existence of primary sclerosing cholangitis, increased severity of colitis, pseudopolyps, and dysplasia [14], whereas protective factors include use of folic acid, ursodeoxycholic acid, and 5-ASA, total proctocolectomy, and compliance with CRC surveillance (Table 1). The potential risk of CAC in sufferers with IBD is 1.5–2 times greater than that in the general population [14,15,16].
CAC in patients with UC only accounts for 1% of all CRC cases [10]. However, it is a serious sequela of the disease and accounts for one sixth of all deaths in patients with UC [17]. There is a consensus that the risk of UC-associated cancer is highest in patients with extensive disease for a long duration [9]. Teenagers with pancolitis have a lifetime risk of CRC that exceeds 15% [14]. Fumery et al. reported that, in patients with UC with low-grade dysplasia, the pooled annual incidence of CRC was 0.8% and that of advanced neoplasia was 1.8% [18]. The high-risk features associated with dysplasia progressing to CAC in patients with UC include concomitant primary sclerosing cholangitis (odds ratio, 3.4; 95% confidence interval, 1.5–7.8), distal location (2.0; 1.1–3.7), and multifocal dysplasia (3.5; 1.5–8.5) [14]. Advanced-stage CAC in Japanese patients with UC has a worse prognosis than that of sporadic CRC. However, no difference was found in survival when CAC is at an early stage [19]. In Japan, the proportion of patients with UC who underwent surgery for CAC increased from 13.8% in 2008 to 20% in 2013 [20].
The risk of CRC in CD was reported to be 1.7–2.4 times greater than in the general population [6, 14]. Moreover, there exists a high risk of small intestine cancer in patients with CD [6]. In Japan, the incidence of CD-associated cancer has been on the rise, and the commonest site of occurrence is anorectal lesion. Sasaki et al. reported that the 5-year survival rate for CD-associated cancer was only 46.2%, compared with 89% in UC-associated cancer because of delayed diagnosis in patients with CD [21].
Carcinogenesis
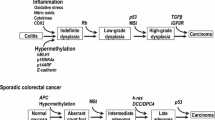
Carcinogenesis of CAC involves transition from low- to high-grade dysplasia and comprises various gene alterations [12]. The molecular pathogenesis of CAC shares many features with sporadic CRC, but there are differences in the time and frequency of some alterations in the dysplasia–carcinoma sequence [14] (Fig. 1). As an illustration, the decline of the adenomatous polyposis coli gene takes place at the beginning with the development of sporadic CRC, although it is generally a delated event within the progression of UC-associated cancer. In addition, p53 mutations appear as important early events in CAC, even prior to the development of dysplasia, yet they occur late in the progression of sporadic CRC [22]. The loss of heterozygosity of p53 is correlated with malignant progression in UC. In a study by Burmer et al., it was detected in 6% of biopsy samples without dysplasia, 33% with low-grade dysplasia, 63% with high-grade dysplasia, and 85% with adenocarcinoma [23]. Furthermore, p53 mutations were found in areas of mucosal inflammation, indicating that chronic inflammation may be mutagenic [24]. On the other hand, the reported rate and timing of microsatellite instability (MSI) are similar in CAC and sporadic CRC [25, 26].

Differences in molecular pathogenesis of sporadic CRC and CAC. The loss of APC occurs early in the development of sporadic CRC, whereas it is usually a late event in CAC. TP53 mutations appear early in CAC, even prior to the development of dysplasia, but late in sporadic CRC. CRC colorectal cancer, CAC colitis-associated cancer, APC adenomatous polyposis coli, MSI microsatellite instability, COX2 cyclooxygenase 2, KRAS Kirsten rat sarcoma viral oncogene homolog, DCC deleted in colorectal carcinoma, SMAD4 SMAD family member 4, CIN chromosomal instability, TGF-β transforming growth factor-β
In IBD, the pathogenesis of CAC is related to chronic inflammatory changes with subsequent genetic alterations via mechanisms, such as the production of mucosal inflammatory mediators, oxidative stress, and changes in immune receptor expression on epithelial cells [12]. Moreover, it has been demonstrated in animal models that inflammatory mediators, such as IL-6, NF-κβ, and Stat3, play important roles in the progression of CAC [27]. Interestingly, a bioactive lipid mediator sphingosine-1-phosphate plays a pivotal role in the progression of CAC in a mouse model [28]. Further investigation is required to examine the roles of these mediators in human patients with CAC.
In UC, the colonic epithelium undergoes recurrent cycles of inflammation and tissue repair, resulting in the accumulation of reactive oxidative species which can cause further epithelial damage and lead to dysplasia [29]. The release of proinflammatory cytokines including NF-κβ is also a well-known mechanism [25]. The histopathogenesis of UC-associated CRC involves a step-wise progression from inflamed and hyperplastic epithelia to flat dysplasia and finally to adenocarcinoma [30].
Carcinogenesis of cancers associated with CD remains unclear compared with that of UC [31]. In CD, dysplasia occurs more often in areas close to other primary tumors [22]. Sigel et al. found that dysplasia was found adjacent to a carcinoma in 87% of cases and distant in 41% of cases [32]. Moreover, NF-κβ is thought to be associated with the pathogenic mechanism of CD [33].
Genomic alterations in sporadic colorectal cancer
CRC is the third most common cancer worldwide [34]. In the USA, it is the second most common cause of cancer-related deaths [35]. Also, in Japan, the incidence of CRC has increased dramatically in the last decade. CRC is the leading cause of death in females, and the third leading cause of death in males among malignant neoplasms [19]. The well-known risk factors for CRC include male sex, older age, personal history of CRC, high body mass index, and lower activity [14]. Sporadic CRC typically develops from a premalignant adenoma through mutations in genes, such as APC, KRAS, DCC, and TP53 (Fig. 1). The Cancer Genome Atlas (TCGA) Network reported the results of whole-genome sequencing for 224 patients with CRC, with 16% found to be hypermutated [36] and 84% to be non-hypermutated. Excluding the hypermutated cancers, 24 genes were significantly mutated, of which the eight most frequently reported were APC, TP53, KRAS, PIK3CA, FBXW7, SMAD4, TCF7L2, and NRAS. Other altered pathways in CRC may include mutations in the PI3K and RAS-MAPK pathways, deregulation of TGF-β signaling, and changes in WNT signaling pathway, which occurred in 93% of all tumors [36]. TCGA data showed that non-hypermutated CRCs cannot be distinguished at the genomic level; on the other hand, right-sided CRCs were more likely to be hypermethylated than those of CRCs at other sites.
Genomic alterations in Japanese patients with colorectal cancer
Although ethnic and geographical differences may exist in the genomic alterations in CRC, most genomic data has been obtained from western countries, with data lacking from Asian countries. Recently, we investigated all exons of 415 cancer-associated genes, including major driver genes, in patients with CRC from Japan and the USA using NGS to compare the western and Asian populations [37]. After data analysis, we correlated the mutation burden with DNA mismatch repair status, obtained clear genomic mutational signatures, and identified genomic alteration patterns in CRC patients in Japan and the USA, which were similar to whole-exome sequencing data by TCGA.
We found hypermutated tumors in 8% of Japanese patients with CRC and 2% of American patients with CRC, both of which are commonly correlated with DNA mismatch repair deficiency (MMR-D) evaluated by immunohistochemical staining of MMR proteins, such as MLH1, MSH2, MSH6, and PMS2 [37]. We also identified overall similarities in the detection of actionable oncogenic driver genes in Japanese and American patients with CRC. For instance, genomic changes in oncogenic pathways such as the cell cycle, RAS/RAF, PI3K, and WNT were identical in Japanese and American patients; however, there are some distinct differences between the two populations. We found significant differences in ERBB2, APC, TP53, CDKN2A, and NRAS mutations between Japanese and American patients, which may show epidemiological distinctions between the two populations.
We further compared our results with the data obtained from TCGA [37] and found a difference in the BRAF mutation patterns between the two studies. In the TCGA-CRC cohorts, BRAF mutations were predominantly seen in the V600E hotspot, which is often restricted to hypermutated tumors. TCGA data showed that in non-hypermutated tumors, the rate of BRAF mutations of right-sided tumors were also significantly higher than that of tumors at sites. In contrast, our results showed that both Japanese and American patients with CRC had many types of non-V600E mutations inside and outside the kinase domain, including D594G, a kinase-dead BRAF that can drive tumor development through interactions with CRAF. Furthermore, we showed that BRAF mutations were recognized in both right- and left-sided tumors [37].
Genomic alterations in colitis-associated carcinoma
Several previous reports have described the histopathological and genetic features as well as the roles of immune response and cytokines in CAC compared with sporadic CRC [12]. Kinugasa demonstrated that patients with high-grade dysplasia and those with CAC have increased β-catenin transcriptional activity that may contribute to increased claudin CL1 expression [2]. An association with MSI has been demonstrated in patients with UC, and the higher rate of MSI in long-standing UC is likely concerned with the genomic instability generated by chronic inflammatory stimulation [12]. Robles et al. reported whole-exome sequencing data, using human CAC samples [38]. Components of the Rho pathway that are responsible for cell motility and cytoskeleton remodeling include genes such as RAC1, DOCK2, DOCK3, PREX2, and RADIL, which were found to be frequently mutated in CAC compared with sporadic CRC. EP300 and TRRAP, which are epigenetic regulators and chromatin modifiers, were found to be more frequently mutated in CAC than in sporadic CRC. ERBB2, 3, and 4 pathways were found to be upregulated/amplified more often in CAC, but the c-MYC pathway was equally expressed in CAC and sporadic CRC [39]. Robles et al. also noted that, despite CAC and sporadic CRC having similar missense mutations within the DNA-binding domain of p53, the identity and molecular distribution of single substitution mutations were different [38]. Specifically, in patients with CAC, there were no mutations at the R273 hotspot, and just one mutation was discovered at each of the R248, G245, and R175 hotspots, which are more commonly mutated in patients with sporadic CRC. Conversely, patients with CAC had more mutations in the R282, R158, H179, and R342 hotspots, which are rarely identified in patients with sporadic CRC. They showed that the Rho and Rac pathways were affected in 10 patients with UC-associated cancer but in only 3 with CD-associated cancer (p = 0.025), indicating that these pathways may be preferentially activated in UC. WNT pathway genes were altered in almost all sporadic CRCs, most commonly APC [38]. Conversely, a lower rate of APC inactivation and higher rate of SOX9 transcription factor inactivating mutation were seen in the CAC cohort [38]. This indicates that CAC has a unique molecular profile which differs inherently from sporadic CRC, potentially providing clues to the etiology of CAC [38].
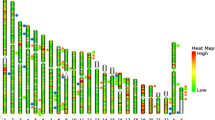
Yaeger analyzed genomic alterations in over 300 cancer-related genes in 47 CACs: 29 UC-associated and 18 CD-associated [40]. Using NGS analysis, they found 6.2 genomic alterations per tumor. Also, they showed that genomic alterations in TP53, IDH1, and MYC were significantly more frequent and mutations in APC were less frequent than those reported in sporadic CRCs in TCGA database. The frequency of gene alterations in CAC were TP53 (89%), APC (21%), KRAS (40%), SMAD4 (17%), MYC (26%), GNAS (13%), and IDH (11%) (Fig. 2). The activation of RTK/RAS signaling was common in CAC (UC, 57% and CD, 72%). WNT pathway components such as TGF-β and MYC were altered in approximately half of patients with CAC (UC, 41% and CD, 56%). IDH mutations in CAC were more common than in sporadic CRC. APC and IDH alterations were significantly more common in CD than in UC (Fig. 3). The specificity of IDH mutations in CD suggests its potential as a therapeutic target. The unique genetic mutations in CAC may also enable new diagnostic and screening tools to detect early CAC in patients with IBD [39].

Comparison of the frequency of genetic alterations in CAC and sporadic CRC. TP53 mutations are the most commonly occurring mutations in CAC. However, the mutations in APC are less frequent in CAC than in sporadic CRC. Genomic alteration patterns in Japanese and American patients are similar. CAC colitis-associated cancer, TCGA The Cancer Genome Atlas, FM Foundation Medicine, APC adenomatous polyposis coli, KRAS Kirsten rat sarcoma viral oncogene homolog, SMAD4 SMAD family member 4, IDH1 isocitrate dehydrogenase

Comparison of the frequency of genetic alterations in UC and CD. The frequency of genetic alterations shares many features between UC and CD, but there are also some differences between the two. TP53 mutations are the most commonly occurring mutations in UC and CD. APC and IDH alterations are significantly more frequent in CD than in UC. UC ulcerative colitis, CD Crohn’s disease, APC adenomatous polyposis coli, KRAS Kirsten rat sarcoma viral oncogene homolog, SMAD4 SMAD family member 4
Conclusions
IBD is associated with an increased risk of CRC in which the carcinogenesis of CAC depends on unique environmental, genetic, and immunologic factors compared with sporadic CRC [41]. Although genomic sequencing for patients with IBD using NGS technology has not been thoroughly investigated, recent studies regarding CAC have suggested that there are several mutational differences between patients with sporadic CRC and those with CAC. Genomic sequencing for patients with IBD has the potential to identify specific genomic alterations in CAC which may lead to early detection and may identify potential molecular targets for the treatment of CAC.
Abbreviations
- CAC:
-
Colorectal cancer
- CD:
-
Crohn’s disease
- CRC:
-
Colorectal cancer
- IBD:
-
Inflammatory bowel disease
- MMR-D:
-
Mismatch repair deficiency
- MSI:
-
Microsatellite instability
- NGS:
-
Next-generation sequencing
- TCGA:
-
The Cancer Genome Atlas
- UC:
-
Ulcerative colitis
References
Ungaro R, Meandrous S, Allen PB, Peyrin-Biroulet L, Colombel JF. Ulcerative colitis. Lancet. 2017;389:1756–70.
Kinugasa T, Akagi Y. Status of colitis-associated cancer in ulcerative colitis. World J Gastrointest Oncol. 2016;8:351–7.
Ng WK, Wong SH, Ng SC. Changing epidemiological trends of inflammatory bowel disease in Asia. Intest Res. 2016;14:111–9.
Peyrin-Biroulet L, Sandborn W, Sands BE, Reinisch W, Bemelman W, Bryant RV, et al. Selecting therapeutic targets in inflammatory bowel disease (STRIDE): determining therapeutic goals for treat-to-target. Am J Gastroenterol. 2015;110:1324–38.
Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380:1590–605.
Kalla R, Ventham NT, Satsangi J, Arnott ID. Crohn’s disease. BMJ. 2014;349:g6670.
Yao T, Matsui T, Hiwatashi N. Crohn’s disease in Japan: diagnostic criteria and epidemiology. Dis Colon Rectum. 2000;43:S85–93.
Crohn B, Rosenberg H. The sigmoidoscopic picture of chronic ulcerative colitis (non-specific). Am J Med Sci. 1925;170:220–8.
Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–35.
Choi PM, Zelig MP. Similarity of colorectal cancer in Crohn’s disease and ulcerative colitis: implications for carcinogenesis and prevention. Gut. 1994;35:950–4.
Mattar MC, Lough D, Pishvaian MJ, Charabaty A. Current management of inflammatory bowel disease and colorectal cancer. Gastrointest Cancer Res. 2011;4:53–61.
Kusunoki M. Colitis-associated cancer. 1st ed. Tokyo, Heidelberg, New York, Dordrecht, London: Springer; 2015. p. 1–150.
Herszényi L, Barabás L, Miheller P, Tulassay Z. Colorectal cancer in patients with inflammatory bowel disease: the true impact of the risk. Dig Dis. 2015;33:52–7.
Beaugerie L, Itzkowitz SH. Cancers complicating inflammatory bowel disease. N Engl J Med. 2015;373:195.
Beaugerie L, Svrcek M, Seksik P, Bouvier AM, Simon T, Allez M, et al. Risk of colorectal high-grade dysplasia and cancer in a prospective observational cohort of patients with inflammatory bowel disease. Gastroenterology. 2013;145:166–75.
Herrinton LJ, Liu L, Levin TR, Allison JE, Lewis JD, Velayos F. Incidence and mortality of colorectal adenocarcinoma in persons with inflammatory bowel disease from 1998 to 2010. Gastroenterology. 2012;143:382–9.
Gyde S, Prior P, Dew MJ, Saunders V, Waterhouse JA, Allan RN. Mortality in ulcerative colitis. Gastroenterology. 1982;83:36–43.
Fumery M, Dulai PS, Gupta S, Prokop LJ, Ramamoorthy S, Sandborn WJ, et al. Incidence, risk factors, and outcomes of colorectal cancer in patients with ulcerative colitis with low-grade dysplasia: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2017;15:665–74.
Watanabe T, Konishi T, Kishimoto J, Kotake K, Muto T, Sugihara K, et al. Ulcerative colitis-associated colorectal cancer shows a poorer survival than sporadic colorectal cancer: a nationwide Japanese study. Inflamm Bowel Dis. 2011;17:802–8.
Kimura H, Takahashi K, Futami K, Ikeuchi H, Tatsumi K, Watanabe K, et al. Has widespread use of biologic and immunosuppressant therapy for ulcerative colitis affected surgical trends? Results of a questionnaire survey of surgical institutions in Japan. Surg Today. 2016;46:930–8.
Sasaki H, Ikeuchi H, Bando T, Hirose K, Hirata A, Chohno T, et al. Clinicopathological characteristics of cancer associated with Crohn’s disease. Surg Today. 2017;47:35–41.
Bressenot A, Cahn V, Danese S, Peyrin-Biroulet L. Microscopic features of colorectal neoplasia in inflammatory bowel diseases. World J Gastroenterol. 2014;20:3164–72.
Burmer GC, Rabinovitch PS, Haggitt RC, Crispin DA, Brentnall TA, Kolli VR, et al. Neoplastic progression in ulcerative colitis: histology, DNA content, and loss of a p53 allele. Gastroenterology. 1992;103:1602–10.
Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807–16.
Fleisher AS, Esteller M, Harpaz N, Leytin A, Rashid A, Xu Y, et al. Microsatellite instability in inflammatory bowel disease-associated neoplastic lesions is associated with hypermethylation and diminished expression of the DNA mismatch repair gene, hMLH1. Cancer Res. 2000;60:4864–8.
Suzuki H, Harpaz N, Tarmin L, Yin J, Jiang HY, Bell JD, et al. Microsatellite instability in ulcerative colitis-associated colorectal dysplasias and cancers. Cancer Res. 1994;54:4841–4.
Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13.
Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23:107–20.
Chen R, Lai LA, Brentnall TA, Pan S. Biomarkers for colitis-associated colorectal cancer. World J Gastroenterol. 2016;22:7882–91.
Nishikawa M, Oshitani N, Matsumoto T, Nishigami T, Arakawa T, Inoue M. Accumulation of mitochondrial DNA mutation with colorectal carcinogenesis in ulcerative colitis. Br J Cancer. 2005;93:331–7.
Yashiro M. Molecular alterations of colorectal cancer with inflammatory bowel disease. Dig Dis Sci. 2015;60:2251–63.
Sigel JE, Petras RE, Lashner BA, Fazio VW, Goldblum JR. Intestinal adenocarcinoma in Crohn’s disease: a report of 30 cases with a focus on coexisting dysplasia. Am J Surg Pathol. 1999;23:651–5.
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–6.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86.
Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67:177–93.
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7.
Nagahashi M, Wakai T, Shimada Y, Ichikawa H, Kameyama H, Kobayashi T, et al. Genomic landscape of colorectal cancer in Japan: clinical implications of comprehensive genomic sequencing for precision medicine. Genome Med. 2016;8:136.
Robles AI, Traverso G, Zhang M, Roberts NJ, Khan MA, Joseph C, et al. Whole-exome sequencing analyses of inflammatory bowel disease-associated colorectal cancers. Gastroenterology. 2016;150:931–43.
Grivennikov SI, Cominelli F. Colitis-associated and sporadic colon cancers: different diseases, different mutations? Gastroenterology. 2016;150:808–10.
Yaeger R, Shah MA, Miller VA, Kelsen JR, Wang K, Heins ZJ, et al. Genomic alterations observed in colitis-associated cancers are distinct from those found in sporadic colorectal cancers and vary by type of inflammatory bowel disease. Gastroenterology. 2016;151:278–87.
Wang ZH, Fang JY. Colorectal cancer in inflammatory bowel disease: epidemiology, pathogenesis and surveillance. Gastrointest Tumors. 2014;1:146–54.
Funding
This work was supported by JSPS KAKENHI Grant Number 17K10624. Denka Co., Ltd., Tokyo, Japan, financially supported this study.
Author information
Authors and Affiliations
Contributions
HK and MN designed the research. YS, YT, HI, MN, JS, and TK analyzed the data. SN, KT, and TW wrote the revised paper. All the authors listed participated in the manuscript and have read and approved the final submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kameyama, H., Nagahashi, M., Shimada, Y. et al. Genomic characterization of colitis-associated colorectal cancer. World J Surg Onc 16, 121 (2018). https://doi.org/10.1186/s12957-018-1428-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12957-018-1428-0