
Abstract
Achondroplasia, Duchenne muscular dystrophy, and osteogenesis imperfecta are among the most frequent rare genetic disorders affecting the musculoskeletal system in children. Rare genetic disorders are severely disabling and can have substantial impacts on families, children, and on healthcare systems. This literature review aims to classify, summarize and compare these non-medical impacts of achondroplasia, Duchenne muscular dystrophy and osteogenesis imperfecta.
French abstract
L’achondroplasie, la dystrophie musculaire de Duchenne et l’ostéogenèse imparfaite sont des maladies génétiques rares du système musculo-squelettique pédiatrique. On assiste à un intérêt croissant pour l’étude des maladies génétiques rares dont la plupart sont handicapantes et ont des conséquences néfastes sur familles, les patients et les systèmes de santé. Malgré leurs spécificités, certaines conséquences des maladies génétiques rares sont communes et gagneraient à être étudiées de façon générique. Cependant, plusieurs travaux continuent d’adopter une approche spécifique à une maladie. Par ailleurs, on assiste à un foisonnement de termes qui évaluent les conséquences des maladies génétiques rares (qualité de vie, impact, effet, fardeau etc.…). Si ce pléthore de termes reflète la complexité du sujet, il rend difficile la synthèse et la comparaison entre maladies. Dans cet article, les auteurs classifient puis effectuent une revue narrative et comparative des impacts de trois maladies génétiques musculo-squelettiques pédiatriques: l’achondroplasie, la dystrophie musculaire de Duchenne et l’ostéogenèse imparfaite.
Similar content being viewed by others


Introduction
A rare disease is defined as one that affects less than one in 2000 individuals in Europe and one in 1250 in the United States [1]. Most rare diseases are severely disabling genetic disorders that can have substantial impacts on families and children, and on healthcare systems [2],[3]. Despite their specific biomedical features, many rare genetic diseases (RGDs) share several non-medical characteristics, in particular, their psychosocial consequences [4]. While it is acknowledged that research on these non-medical common features may benefit from a non-categorical approach, a disease-specific approach remains common. In addition, the literature assessing these issues is sometimes difficult to summarize due to the inconsistent use of terminology. Several terms are commonly used in the literature to examine the impacts of RGDs: burden of care, quality of life, impacts, consequences, meaning of leaving, and coping strategies [5]-[7]. This proliferation of concepts, which depends on research interest and disciplinary tradition (e.g., biomedical, psychological, economic, social and nursing) reflects the complexity of the field, but may also lead to a fragmented version of ‘the same reality’. A clear and synthetized conceptualization of the impacts of RGDs with a non-disease-specific approach is warranted. Scoping reviews, an increasingly popular knowledge synthesis approach in health care, can help in such conceptualization [8]. Moreover, scoping reviews can yield a framework for collating and summarizing results that can empower patients and families, raise awareness among health care professionals, identify knowledge gaps and priorities for future research, and advocate for policies to develop support services for families [8],[9].
This paper reports a scoping review that describes the literature on non-medical impacts for patients and families. Due to the exploratory nature of the review and to reflect the research areas of our team, we delineated the focus of this review to three common RGDs in the pediatric orthopedic context: achondroplasia, Duchenne muscular dystrophy (DMD) and osteogenesis imperfecta (OI). These three RGDs are single-gene musculoskeletal diseases characterized by physical disability and little or no impairment of mental ability. Table 1 provides their clinical and genetic characteristics.
The three specific objectives of this review research were: i) to categorize the types of non-medical impacts of achondroplasia, DMD and OI; ii) to summarize these impacts; and iii) to discuss findings on these impacts across the three diseases.
Methods
While systematic reviews synthesize the complete nature of a particular field, outlining what approaches are effective and where further research is required, scoping reviews are exploratory projects that map the literature available on a topic, identifying the key concepts, theories, sources of evidence, and gaps in the research [10]. We therefore opted for a scoping review to allow for a quick mapping of the “key concepts underpinning a research area and the main sources and types of evidence available” [11]. We adopted a broad definition of impact that includes consequences of, but also reactions to, a disease. We followed the five stages suggested for a quality scoping review: i) identification of the research question; ii) identification of relevant studies; iii) selection of studies to include in the review; iv) charting of information and data within the included studies; and v) collating, summarizing and reporting results of the review [11].
Identification of relevant publications
From March to June, 2013, we conducted a search of the literature in three electronic databases: Web of Science, CINAHL, and MEDLINE. The following two string combinations of keywords were used: [Impact, burden, quality of life, living with, coping, adjustment, well-being, quality of life, effects, impacts, responses, reactions, psychosocial] AND [Rare genetic disease/disorder, rare childhood disease, osteogenesis imperfecta, brittle bone disease, Duchenne muscular dystrophy, achondroplasia, chronic illness, musculoskeletal system, physical disorders]. We complemented our search for publications with a systematic screening of the table of contents of specific journals dedicated to rare diseases (e.g. Orphanet Journal of Rare Diseases) and a screening of references of relevant publications. Opinions, commentaries, letters, editorials, and publications without an abstract were immediately initially excluded. The initial search yielded the identification of 845 publications. After removing 8 duplicates, two authors independently selected publications addressing the non-medical impacts of Achondroplasia, DMD and OI, based on the titles and the abstracts. Publications that addressed a rare genetic disease other than the three retained by the research group were excluded from this review, as were publications focussed on the identification of genes. In the case of disagreement among authors, the full-text publications were reviewed by a third senior author. A total of 65 full-text publications remained at this stage of the review.
Selection of studies to include in the review
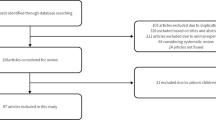
For inclusion in our study, publications had to be i) published between 1980 and 2013; ii) in English or French; iii) about non-medical effects and responses to OI, achondroplasia and DMD; and iv) have a well-defined methodology. Because a scoping review is not intended to assess the quality of studies, emphasis was not placed on methodological rigor of the retained studies. At this second stage, 8 publications were excluded as they examined medical impacts of a treatment or intervention, and 57 publications were retained. Figure 1 displays the study selection flow diagram. We used a data extraction grid to gather the following items: i) general information (e.g., type of study, country, focus); and ii) the relevance of the article to our study and the scientific information such as concepts, theoretical orientation, methodology, and research tradition.

Study selection flow diagram.
Collating, summarizing and reporting results of the review
The 57 retained publications were coded using qualitative structure coding [12] with Nvivo 10 (QSR International) and Endnote X6. From this, we identified main study themes and grouped based on similarity. We iteratively constructed a working framework (Figure 2) to examine separately the extent, range and research activity for OI, achondroplasia and DMD. The adequacy of the framework to review question was tested independently by two authors (MJD and ED) on 10 selected articles. Disagreements were solved by discussion among these authors or by the adjudication of a senior author (FR).

Framework redefining impacts of rare genetic diseases.
To ensure consistent mapping and analysis of data, we developed a glossary of codes used to classify the impacts as follows: i) ‘scope’ refers to whether the article addresses one or several pediatric rare musculoskeletal disorders; ii) ‘direction’ was used to capture whether the article studied the ‘Effect of Disease’ (Direction 1) or ‘Response to the Disease’ (Direction 2); iii) ‘target’ refers to three distinct groups of focus: ‘individuals’ such as patients, adults, carers, or siblings; ‘groups’ such as physicians, caregivers, or school teachers; and ‘societies’ such as communities or society as a whole; iv) ‘timeline’ and ‘trend’ refer to the temporal analysis of impacts; and v) ‘type’ refers to positive or negative consequences; finally vi) ‘domain’ refers to the aspect of QOL being studied (psychological, functional and so on) or the research tradition or academic discipline used to study the impacts. Once the 57 publications were categorized, we performed a narrative review of the findings on the impacts separately for each disease, and then critically compared the findings across the three diseases. An overview of the distribution of the publications using the components of our working framework is presented, as well as a summary of the major findings of the publications achondroplasia, DMD and OI. Finally, we discuss how research compares across the three diseases.
Results
A total of 57 publications on non-medical impacts are included in this review study: 3 about achondroplasia [13]-[15], 39 about DMD [16]-[54] and 15 about OI [55]-[69].
Overview of the publications
The majority of the publications (50) were original research focused on a single disease, while 6 publications concerned two diseases (DMD and other diseases), and 1 publication was a literature review.
The target group assessed was spread between the experiences of the individual with the disease (26), the impact of the disease on families (18), the experience of caregivers only (8), or were focused on the impacts of other groups or tool development and reviews of the literature (5; physicians’ attitudes, the effects on community).
The direction of the impacts was explicitly stated in 44 of the 57: 31 addressed the effects and impacts of living with achondroplasia, DMD or OI, 13 addressed responses to living with the disease on individuals, groups or society (such as resilience, adjustment, coping strategies and attitudes). In these 44 papers, the occurrence of a RGD was conceptualised as a source of stress to which people react. The direction was either a combination of directions, or not explicitly stated, in the remaining 13 papers. Table 2 outlines the studies according to the scope, target, direction, timeline, trend, and type of impacts.
Papers were also spread over several domains (as defined above), with 15 examining the quality of life (QOL) through a normative and validated measure of function, psychosocial well-being, and perception of health (2 achondroplasia, 7 DMD, and 6 OI). Psychosocial impacts on their own were addressed in 18 publications (14 DMD, 4 OI), and functional status on its own was addressed in 10 publications (1 achondroplasia, 6 DMD, 3 OI). The remaining 14 publications assessed the domains of illness experience, utilisation of healthcare services, palliative care, medical staff reactions to the disease, or newborn screening. Tables 3, 4 and 5 report the general information and major findings of the publications for the domain.
Narrative review
The non-medical impacts of achondroplasia
Evidence from the publications on achondroplasia suggests that affected individuals have an impaired QOL when compared to first-degree relatives [14]. The lower QOL could mainly be linked to psychosocial limitations including lower self-esteem and social stigmatization, to the extent that serial lower limb lengthening appeared a good option to patients despite its numerous complications [13],[15]. The functional status of patients with achondroplasia was impaired by back pain and pain in the lower extremity, resulting in some cases in complete cessation of work. However, the functional limitation and psychological distress remained unchanged over time [13].
The non-medical impacts of DMD
The overall QOL of parents and their children who are affected with DMD was reported to be lower than in the general population, particularly due to impairments in physical functioning [19],[42]. Boys with DMD reported significantly lower QOL than healthy peers in physical and psychosocial domains. Physical limitations increased with age as respiratory problems and muscle weaknesses occurred. However, despite the progressive course of the disease, the psychosocial QOL tended to be higher in adolescents with DMD than in their younger counterparts, suggesting the development of effective coping strategies over time [41],[50],[52]. For parents, the psychosocial impacts were reported to be great, and quality of life decreased even more around the period of patient transition to wheelchair [22],[23]. Finally, parental reports of QOL did not consistently match that of their children, with many underestimating their child’s QOL [23].
The 14 studies that solely examined the psychological impacts of DMD reported higher levels of anxiety, depression, and guilt in parents, particularly in mothers [25],[26],[46]. However, early diagnosis and family hardiness (the energy resources of the family such as commitment, challenge, and control) were reported to positively influence parental psychological adjustments, the patients’ level of resilience and the reactions of siblings [20],[24],[26],[29],[34],[43],[48],[49],[51],[53].
In addition to the psychological impacts, parents, particularly mothers, reported caring for their children with DMD to be burdensome (help for bathing, toileting), costly in terms of time, and contributing to increased social isolation [44]. Altogether, participation in daily activities and social life was reduced for patients. Siblings of adolescents with DMD seemed more negatively affected than siblings of healthier counterparts.
The non-medical impacts of OI
There was little difference in the overall QOL of those diagnosed with OI as compared to the general population. Functional limitations were more important in patients with severe forms of OI, and higher life satisfaction, resilience, low depression and higher social achievements were common [55],[64],[68]. Parents reported several accusations of child abuse, lack of information on the disease, disruptions of family activities due to the occurrence of fractures, and social isolation [56]-[58].
Other findings
Besides these psychosocial impacts of achondroplasia, DMD and OI on families, the impacts on groups and the society were broached in few studies. Two publications examined the attitudes of medical staff about a condition or related treatments options [17],[40]. These publications concluded that physicians could promote some experimental treatment options provided that there was a shared-decision making with families. The medical staffs were also found to support newborn and prenatal screening whenever available. Finally, it should be noted that the impacts on the use of health services and economic factors were not assessed in the papers included in this review.
Discussion
The impacts of achondroplasia, DMD and OI on families reported in this review are derived from in-depth qualitative analysis of subjective illness experiences or objective normative measures of QOL. These methodological specificities resulted in variations in the impacts between and within similar categories of publications that may limit the comparison across the three diseases. However, caring for children particularly with DMD and OI was burdensome because of daily practical problems (bathing, toileting), increased time costs and social isolation, as reported in studies on other RGDs [70]-[72]. Consequently, carers reported lower QOL particularly in critical periods of loss of ambulation in boys with DMD and during the occurrence of fractures in OI as recently confirmed [73].
As for the QOL in patients with these three diseases, findings seem contradictory with some publications reporting higher QOL and other reporting lower QOL when compared to healthier unaffected controls [20],[22]. The severity of functional and physical impairments follow the severity of the disease, and could be seen as increasing from small functional impairment, such as back pain in achondroplasia, to restricted ambulation from bone deformities and numerous fractures in OI, to progressively reduced ambulation and confinement to a wheelchair in DMD [22]. On the contrary and strikingly, the psychological aspect of QOL in patients did not parallel the severity of the disease. For example, DMD is life-threatening and its psychosocial impacts are expected to be more severe than for achondroplasia and OI; however, patients with OI did not seem to experience less psychological distress than those with DMD [41],[69]. As for the course of the disease, the stable (unchanged) level of psychological distress in patients with achondroplasia reflects the non-progressive course of the disease [13]. The “up and down” pattern of psychological distress in patients with severe OI also confirms the occurrence of fractures as critical periods over the chronic course of OI [73]. While functional limitations increased, the self-reported psychosocial QOL of boys with DMD did not decrease with age. They may have developed effective coping strategies that ought to have been better documented [52]. This finding was confirmed in previous studies. Indeed the level of impact on family including psychological stress was high for families who had children affected by rare genetic metabolic conditions even when the child’s function was not impaired and the disease was not severe [5].
Some practical implications emerged from this review that can benefit families and caregivers. The correlation between improved parental coping strategies and early diagnosis, better emotional functioning of the child and siblings suggests that there have been efforts to obtaining an early diagnosis and promoting family hardiness for people living with these diseases [74],[75]. To that end, interprofessional teams of geneticists, psychologists and social workers in reference centers for these rare diseases could be effective by offering newborn screening, family-centered and life-span psychosocial support whenever possible [76]. Besides, informal support from family and friends but also institutional health and social support such as respite care can contribute to the relief of the burden of care on families.
From the limited number of publications on the impacts of DMD, achondroplasia and OI, we have learned that research about non-medical impacts of achondroplasia, DMD and OI on healthcare systems and society seems very limited and could be further addressed. However, the selection of the keywords used in the searching strategy in this review might have resulted in the exclusion of some publications. For example, challenges in providing adequate updating training to clinicians and staff on these rare diseases as acknowledged in other RGDs could be examined. In addition, research on healthcare-related impacts might compare the use of healthcare resources according to the severity of the disease. Efficient care delivery models organised around primary care whereby family physicians can help to achieve a smooth transition to adult care institutions must also be investigated [56],[77],[78]. In addition more theoretical understanding is needed about the complex and non-linear interrelationships between two aspects of QOL: the available quality of care and the family functioning and economic status [79]-[81]. Such understanding will help to acknowledge the relative impacts of these determinants on overall QOL and will help to prioritize adequate interventions.
We acknowledge that methodological limitations could have compromised the inclusion of publications reviewed here. Thus, further longitudinal studies combining normative approaches with qualitative in-depth investigations and comparing several diseases could enhance the understanding of patients’ effective coping strategies. Simultaneous use of parent-report and patient-report questionnaires could provide increased insights [22]. Furthermore, our in-house working framework that guided our organization and analysis of the scoping review could aid others in mapping and organizing the literature during a scoping review; however, to extend its use beyond this would require further validation.
Conclusion
In general, DMD and OI negatively impacted carers. Some events seem particularly critical such as occurrence of fractures or loss of ambulation in patients. As for patients, functional limitations seemed to follow the severity of the disease, but psychological distress did not, which calls for a better understanding of effective coping strategies. This conclusion is supported by the reported higher life satisfaction in very severely affected patients at different stages of the progressing course of DMD, and the reported resilience characteristic of adolescents with OI. In face of the great difficulties presented to families, life span and family-centered psychological support must be developed in a timely fashion to aid patients, carers, and siblings.
Abbreviations
- DMD:
-
Duchenne muscular dystrophy
- OI:
-
Osteogenesis imperfecta
- QOL:
-
Quality of life
- RGD(s):
-
Rare genetic disease(s)
References
Schieppati A, Henter JI, Daina E, Aperia A: Why rare diseases are an important medical and social issue. Lancet 2008, 371: 2039–2041. 10.1016/S0140-6736(08)60872-7
Ayme S, Kole A, Groft S: Empowerment of patients: lessons from the rare diseases community. Lancet 2008, 371: 2048–2051. 10.1016/S0140-6736(08)60875-2
Kole A, Faurisson F: The Voice of 12,000 Patients: Experiences and expectations of rare disease patients on diagnosis and care in Europe. Eurodis edition. 2009.
Wallander JL, Varni JW: Effects of pediatric chronic physical disorders on child and family adjustment. J Child Psychol Psychiatry 1998, 39: 29–46. 10.1017/S0021963097001741
Anderson M, Elliott EJ, Zurynski YA: Australian families living with rare disease: experiences of diagnosis, health services use and needs for psychosocial support. Orphanet J Rare Dis 2013, 8: 22. 10.1186/1750-1172-8-22
Shahbazi S, Moghaddam-Banaem L, Ekhtesari F, Ala FA: Impact of inherited bleeding disorders on pregnancy and postpartum hemorrhage. Blood Coagul Fibrinolysis 2012, 23: 603–607. 10.1097/MBC.0b013e3283566af9
Boekaerts M, Roder I: Stress, coping, and adjustment in children with a chronic disease: a review of the literature. Disabil Rehabil 1999, 21: 311–337. 10.1080/096382899297576
Levac D, Colquhoun H, O'Brien KK: Scoping studies: advancing the methodology. Implement Sci 2010, 5: 69. 10.1186/1748-5908-5-69
Daudt HM, van Mossel C, Scott SJ: Enhancing the scoping study methodology: a large, inter-professional team's experience with Arksey and O'Malley's framework. BMC Med Res Methodol 2013, 13: 48. 10.1186/1471-2288-13-48
Brien SE, Lorenzetti DL, Lewis S, Kennedy J, Ghali WA: Overview of a formal scoping review on health system report cards. Implementation Science 2010, 5: 2. 10.1186/1748-5908-5-2
Arksey H, O'Malley L: Scoping studies: towards a methodological framework. Int J Soc Res Methodol 2005, 8: 19–32. 10.1080/1364557032000119616
Saldaña J: The Coding Manula for Qualitative Researchers. Sage Publications, Los Angeles; 2013.
Ain MC, Abdullah MA, Ting BL, Skolasky RL, Carlisle ES, Schkrohowsky JG, Rigamonti D: Progression of low back and lower extremity pain in a cohort of patients with achondroplasia. J Neurosurg Spine 2010, 13: 335–340. 10.3171/2010.3.SPINE09629
Gollust SE, Thompson RE, Gooding HC, Biesecker BB: Living with achondroplasia: attitudes toward population screening and correlation with quality of life. Prenat Diagn 2003, 23: 1003–1008. 10.1002/pd.743
Kim S-J, Balce GC, Agashe MV, Song S-H, Song H-R: Is bilateral lower limb lengthening appropriate for Achondroplasia? Midterm analysis of the complications and quality of life. Clin Orthop Relat Res 2012, 470: 616–621. 10.1007/s11999-011-1983-y
Abi Daoud MS, Dooley JM, Gordon KE: Depression in parents of children with Duchenne muscular dystrophy. Pediatr Neurol 2004, 31: 16–19. 10.1016/j.pediatrneurol.2004.01.011
Acharya K, Ackerman PD, Ross LF: Pediatricians' attitudes toward expanding newborn screening. Pediatrics 2005, 116: E476-E482. 10.1542/peds.2005-0453
Arias R, Andrews J, Pandya S, Pettit K, Trout C, Apkon S, Karwoski J, Cunniff C, Matthews D, Miller T, Davis MF, Meaney FJ: Palliative care services in families of males with Duchenne muscular dystrophy. Muscle Nerve 2011, 44: 93–101. 10.1002/mus.22005
Baiardini I, Minetti C, Bonifacino S, Porcu A, Klersy C, Petralia P, Balestracci S, Tarchino F, Parodi S, Canonica GW, Braido F: Quality of life in Duchenne muscular dystrophy: the subjective impact on children and parents. J Child Neurol 2011, 26: 707–713. 10.1177/0883073810389043
Bendixen RM, Senesac C, Lott DJ, Vandenborne K: Participation and quality of life in children with Duchenne muscular dystrophy using the International Classification of Functioning, Disability, and Health. Health Qual Life Outcomes 2012, 10: 43. 10.1186/1477-7525-10-43
Beresford BA, Sloper P: Chronically ill adolescents' experiences of communicating with doctors: a qualitative study. J Adolesc Health 2003, 33: 172–179. 10.1016/S1054-139X(03)00047-8
Bray P, Bundy AC, Ryan MM, North KN, Burns J: Health status of boys with Duchenne muscular dystrophy: a parent’s perspective. J Paediatr Child Health 2011, 47: 557–562. 10.1111/j.1440-1754.2011.02022.x
Bray P, Bundy AC, Ryan MM, North KN, Everett A: Health-related quality of life in boys with Duchenne muscular dystrophy: agreement between parents and their sons. J Child Neurol 2010, 25: 1188–1194. 10.1177/0883073809357624
Chen JY: Mediators affecting family function in families of children with Duchenne muscular dystrophy. Gaoxiong Yi Xue Ke Xue Za Zhi 2008, 24: 514–522.
Chen JY, Chen SS, Jong YJ, Yang YH, Chang YY: A comparison of the stress and coping strategies between the parents of children with Duchenne muscular dystrophy and children with a fever. J Pediatr Nurs 2002, 17: 369–379. 10.1053/jpdn.2002.123525
Chen JY, Clark MJ: Family function in families of children with Duchenne muscular dystrophy. Fam Community Health 2007, 30: 296–304. 10.1097/01.FCH.0000290542.10458.f8
Chen J-Y, Clark M-J: Family resources and parental health in families of children with Duchenne muscular dystrophy. J Nurs Res 2010, 18: 239–248. 10.1097/JNR.0b013e3181fbe37b
Cyrulnik SE, Fee RJ, Batchelder A, Kiefel J, Goldstein E, Hinton VJ: Cognitive and adaptive deficits in young children with Duchenne muscular dystrophy (DMD). J Int Neuropsychol Soc 2008, 14: 853–861. 10.1017/S135561770808106X
Fee RJ, Hinton VJ: Resilience in children diagnosed with a chronic neuromuscular disorder. J Dev Behav Pediatr 2011, 32: 644–650. 10.1097/DBP.0b013e318235d614
Firth M, Gardnermedwin D, Hosking G, Wilkinson E: Interviews with parents of boys suffering from Duchenne muscular-dystrophy. Dev Med Child Neurol 1983, 25: 466–471. 10.1111/j.1469-8749.1983.tb13791.x
Garralda ME, McConachie H, Le Couteur A, Sriranjan S, Chakrabarti I, Cirak S, Guglieri M, Bushby K, Muntoni F: Emotional impact of genetic trials in progressive paediatric disorders: a dose-ranging exon-skipping trial in Duchenne muscular dystrophy. Child Care Health Dev 2013, 39: 449–455. 10.1111/j.1365-2214.2012.01387.x
Garralda ME, Muntoni F, Cunniff A, Caneja AD: Knee-ankle-foot orthosis in children with duchenne muscular dystrophy: user views and adjustment. Eur J Paediatr Neurol 2006, 10: 186–191. 10.1016/j.ejpn.2006.07.002
Hanayama K, Liu M, Higuchi Y, Fujiwara T, Tsuji T, Hase K, Ishihara T: Dysphagia in patients with Duchenne muscular dystrophy evaluated with a questionnaire and videofluorography. Disabil Rehabil 2008, 30: 517–522. 10.1080/09638280701355595
Hendriksen JG, Poysky JT, Schrans DG, Schouten EG, Aldenkamp AP, Vles JS: Psychosocial adjustment in males with Duchenne muscular dystrophy: psychometric properties and clinical utility of a parent-report questionnaire. J Pediatr Psychol 2009, 34: 69–78. 10.1093/jpepsy/jsn067
James CA, Hadley DW, Holtzman NA, Winkelstein JA: How does the mode of inheritance of a genetic condition influence families? A study of guilt, blame, stigma, and understanding of inheritance and reproductive risks in families with X-linked and autosomal recessive diseases. Genet Med 2006, 8: 234–242. 10.1097/01.gim.0000215177.28010.6e
Jarvinen O, Lehesjoki AE, Lindlof M, Uutela A, Kaariainen H: Carrier testing of children for two X-linked diseases: a retrospective study of comprehension of the test results and social and psychological significance of the testing. Pediatrics 2000, 106: 1460–1465. 10.1542/peds.106.6.1460
Jutai J, Rigby P, Ryan S, Stickel S: Psychosocial impact of electronic aids to daily living. Assist Technol 2000, 12: 123–131. 10.1080/10400435.2000.10132018
Kemper AR, Wake MA: Duchenne muscular dystrophy: issues in expanding newborn screening. Curr Opin Pediatr 2007, 19: 700–704. 10.1097/MOP.0b013e3282f19f65
Kenneson A, Bobo JK: The effect of caregiving on women in families with Duchenne/Becker muscular dystrophy. Health Soc Care Community 2010, 18: 520–528. 10.1111/j.1365-2524.2010.00930.x
Kinali M, Manzur AY, Mercuri E, Gibson BE, Hartley L, Simonds AK, Muntoni F: UK physicians' attitudes and practices in long-term non-invasive ventilation of Duchenne Muscular Dystrophy. Pediatr Rehabil 2006, 9: 351–364.
Kohler M, Clarenbach CF, Boeni L, Brack T, Russi EW, Bloch KE: Quality of life, physical disability, and respiratory impairment in Duchenne muscular dystrophy. Am J Respir Crit Care Med 2005, 172: 1032–1036. 10.1164/rccm.200503-322OC
Manzur AY, Kinali M, Muntoni F: Update on the management of Duchenne muscular dystrophy. Arch Dis Child 2008, 93: 986–990. 10.1136/adc.2007.118141
Marini A, Lorusso ML, D'Angelo MG, Civati F, Turconi AC, Fabbro F, Bresolin N: Evaluation of narrative abilities in patients suffering from Duchenne Muscular Dystrophy. Brain Lang 2007, 102: 1–12. 10.1016/j.bandl.2007.02.003
Pangalila RF, van den Bos GA, Stam HJ, van Exel NJ, Brouwer WB, Roebroeck ME: Subjective caregiver burden of parents of adults with Duchenne muscular dystrophy. Disabil Rehabil 2012, 34: 988–996. 10.3109/09638288.2011.628738
Parker AE, Robb SA, Chambers J, Davidson AC, Evans K, O'Dowd J, Williams AJ, Howard RS: Analysis of an adult Duchenne muscular dystrophy population. QJM 2005, 98: 729–736. 10.1093/qjmed/hci113
Parsons EP, Clarke AJ, Hood K, Lycett E, Bradley DM: Newborn screening for Duchenne muscular dystrophy: a psychosocial study. Arch Dis Child Fetal Neonatal Ed 2002, 86: F91-F95. 10.1136/fn.86.2.F91
Pehler SR, Craft-Rosenberg M: Longing: the lived experience of spirituality in adolescents with Duchenne muscular dystrophy. J Pediatr Nurs 2009, 24: 481–494. 10.1016/j.pedn.2008.06.008
Read J, Kinali M, Muntoni F, Garralda ME: Psychosocial adjustment in siblings of young people with Duchenne muscular dystrophy. Eur J Paediatr Neurol 2010, 14: 340–348. 10.1016/j.ejpn.2009.09.011
Read J, Kinali M, Muntoni F, Weaver T, Garralda ME: Siblings of young people with Duchenne muscular dystrophy - A qualitative study of impact and coping. Eur J Paediatr Neurol 2011, 15: 21–28. 10.1016/j.ejpn.2010.07.006
Simon VA, Resende MB, Simon MA, Zanoteli E, Reed UC: Duchenne muscular dystrophy: quality of life among 95 patients evaluated using the Life Satisfaction Index for Adolescents. Arq Neuropsiquiatr 2011, 69: 19–22. 10.1590/S0004-282X2011000100005
Steele M, Taylor E, Young C, McGrath P, Lyttle BD, Davidson B: Mental health of children and adolescents with Duchenne muscular dystrophy. Dev Med Child Neurol 2008, 50: 638–639. 10.1111/j.1469-8749.2008.03024.x
Uzark K, King E, Cripe L, Spicer R, Sage J, Kinnett K, Wong B, Pratt J, Varni JW: Health-related quality of life in children and adolescents with Duchenne muscular dystrophy. Pediatrics 2012, 130: E1559-E1566. 10.1542/peds.2012-0858
van Wijk E, Messelink BJ, Heijnen L, de Groot IJ: Prevalence and psychosocial impact of lower urinary tract symptoms in patients with Duchenne muscular dystrophy. Neuromuscul Disord 2009, 19: 754–758. 10.1016/j.nmd.2009.07.009
Vandervelde L, Van den Bergh PY, Goemans N, Thonnard JL: Activity limitations in patients with neuromuscular disorders: a responsiveness study of the ACTIVLIM questionnaire. Neuromuscul Disord 2009, 19: 99–103. 10.1016/j.nmd.2008.11.004
Balkefors V, Mattsson E, Pernow Y, Saaf M: Functioning and quality of life in adults with mild-to-moderate osteogenesis imperfecta. Physiother Res Int 2012, ᅟ: ᅟ.
Bernehall Claesson I, Brodin J: What families with children with brittle bones want to tell. Child Care Health Dev 2002, 28: 309–315. 10.1046/j.1365-2214.2002.00282.x
Brodin J: Children and adolescents with brittle bones - psychosocial-aspects. Child Care Health Dev 1993, 19: 341–347. 10.1111/j.1365-2214.1993.tb00738.x
Cole DE: Psychosocial aspects of osteogenesis imperfecta: an update. Am J Med Genet 1993, 45: 207–211. 10.1002/ajmg.1320450211
Daly K, Wisbeach A, Sanpera I Jr, Fixsen JA: The prognosis for walking in osteogenesis imperfecta. J Bone Joint Surg Br 1996, 78: 477–480.
Engelbert RH, Uiterwaal CS, Gulmans VA, Pruijs H, Helders PJ: Osteogenesis imperfecta in childhood: prognosis for walking. J Pediatr 2000, 137: 397–402. 10.1067/mpd.2000.107892
Kok DJ, Uiterwaal C, van Dongen AJ, Kramer PPG, Pruijs HEH, Engelbert RHH, Verbout AJ, Schweitzer DH, Sakkers RJB: The interaction between Sillence type and BMD in osteogenesis imperfecta. Calcif Tissue Int 2003, 73: 441–445. 10.1007/s00223-002-2101-7
Montpetit K, Dahan-Oliel N, Ruck-Gibis J, Fassier F, Rauch F, Glorieux F: Activities and participation in young adults with osteogenesis imperfecta. J Pediatr Rehabil Med 2011, 4: 13–22.
Seikaly MG, Kopanati S, Salhab N, Waber P, Patterson D, Browne R, Herring JA: Impact of alendronate on quality of life in children with osteogenesis imperfecta. J Pediatr Orthop 2005, 25: 786–791. 10.1097/01.bpo.0000176162.78980.ed
Suskauer SJ, Cintas HL, Marini JC, Gerber LH: Temperament and physical performance in children with Osteogenesis Imperfecta. Pediatrics 2003, 111: e153-e161. 10.1542/peds.111.2.e153
Szczepaniak-Kubat A, Kurnatowska O, Jakubowska-Pietkiewicz E, Chlebna-Sokol D: Assessment of quality of life of parents of children with Osteogenesis Imperfecta. Adv Clin Exp Med 2012, 21: 99–104.
Tolboom N, Cats EA, Helders PJ, Pruijs JE, Engelbert RH: Osteogenesis imperfecta in childhood: effects of spondylodesis on functional ability, ambulation and perceived competence. Eur Spine J 2004, 13: 108–113. 10.1007/s00586-003-0574-3
Van Brussel M, Takken T, Uiterwaal C, Pruijs HJ, Van der Net J, Helders PJM, Engelbert RHH: Physical training in children with osteogenesis imperfecta. J Pediatr 2008, 152: 111–116. 10.1016/j.jpeds.2007.06.029
Widmann R, Laplaza J, Bitan F, Brooks C, Root L: Quality of life in osteogenesis imperfecta. Int Orthop 2002, 26: 3–6. 10.1007/s002640100292
Widmann RF, Bitan FD, Laplaza J, Burke SW, DiMaio MF, Schneider R: Spinal deformity, pulmonary compromise, and quality of life in osteogenesis imperfecta. Spine 1999, 24: 1673–1678. 10.1097/00007632-199908150-00008
Curran AL, Sharples PM, White C, Knapp M: Time costs of caring for children with severe disabilities compared with caring for children without disabilities. Dev Med Child Neurol 2001, 43: 529–533. 10.1017/S0012162201000962
van den Tweel XW, Hatzmann J, Ensink E, van der Lee JH, Peters M, Fijnvandraat K, Grootenhuis M: Quality of life of female caregivers of children with sickle cell disease: a survey. Haematologica 2008, 93: 588–593. 10.3324/haematol.11610
Zurynski Y, Frith K, Leonard H, Elliott E: Rare childhood diseases: how should we respond? Arch Dis Child 2008, 93: 1071–1074. 10.1136/adc.2007.134940
Dogba MJ, Bedos C, Durigova M, Montpetit K, Wong T, Glorieux F, Rauch F: The Impact of Severe Osteogenesis Imperfecta on the Lives of Young Patients and Their Parents – a Qualitative Analysis. BMC Pediatr 2013, 13: 153. 10.1186/1471-2431-13-153
Ratliffe CE, Harrigan RC, Haley J, Tse A, Olson T: Stress in families with medically fragile children. Issues Compr Pediatr Nurs 2002, 25: 167–188. 10.1080/01460860290042558
Kazak AE, Reber M, Snitzer L: Childhood chronic disease and family functionning- a study of phenylketonuria. Pediatrics 1988, 81: 224–230.
Battista RN, Blancquaert I, Laberge AM, van Schendel N, Leduc N: Genetics in health care: an overview of current and emerging models. Public Health Genomics 2012, 15: 34–45. 10.1159/000328846
Zurynski Y, Peadon E, Bower C, Elliott EJ: Impacts of national surveillance for uncommon conditions in childhood. J Paediatr Child Health 2007, 43: 724. 10.1111/j.1440-1754.2007.01216.x
Burton H, Sanderson S, Shortland G, Lee P: Needs assessment and review of services for people with inherited metabolic disease in the United Kingdom. J Inherit Metab Dis 2006, 29: 667–676. 10.1007/s10545-006-0374-0
Eiser C, Morse R: A review of measures of quality of life for children with chronic illness. Arch Dis Child 2001, 84: 205–211. 10.1136/adc.84.3.205
Rajmil L, Perestelo-Perez L, Herdman M: Quality of life and rare diseases. Adv Exp Med Biol 2010, 686: 251–272. 10.1007/978-90-481-9485-8_15
Paterson CR, Rosalind MH: Life expectancy in osteogenesis imperfecta. Br Med J 1996, 312: 351. 10.1136/bmj.312.7027.351
Acknowledgements
This study was supported by the Shriners of North America; the Network for Oral and Bone Health Research (RSBO), which receives funding from the Fonds de la Recherche du Québec-Santé (FRQ-S); and the MENTOR program, which receives funding from the Canadian Institutes of Health Research. FR received support from the Chercheur-Boursier Clinicien program of the Fonds de recherche Québec-Santé. These sponsors had no involvement in determining the content of this paper. We also thank Susan Lemprière and Judith Kashul for editorial revision.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Authors declare no competing interests.
Authors’ contributions
MJD conceived the study, performed the literature review, drafted the initial manuscript and approved this version as submitted. FR conceived the study, critically revised the manuscript and approved this version as submitted. ED performed the literature review and approved this version as submitted. CB critically revised the manuscript and approved this version as submitted.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
Cite this article
Dogba, M.J., Rauch, F., Douglas, E. et al. Impact of three genetic musculoskeletal diseases: a comparative synthesis of achondroplasia, Duchenne muscular dystrophy and osteogenesis imperfecta. Health Qual Life Outcomes 12, 151 (2014). https://doi.org/10.1186/s12955-014-0151-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12955-014-0151-y