
Abstract
Background
While immune checkpoint inhibitors such as anti-PD-L1 antibodies have revolutionized cancer treatment, only subgroups of patients show durable responses. Insight in the relation between clinical response, PD-L1 expression and intratumoral localization of PD-L1 therapeutics could improve patient stratification. Therefore, we present the modular synthesis of multimodal antibody-based imaging tools for multiscale imaging of PD-L1 to study intratumoral distribution of PD-L1 therapeutics.
Results
To introduce imaging modalities, a peptide containing a near-infrared dye (sulfo-Cy5), a chelator (DTPA), an azide, and a sortase-recognition motif was synthesized. This peptide and a non-fluorescent intermediate were used for site-specific functionalization of c-terminally sortaggable mouse IgG1 (mIgG1) and Fab anti-PD-L1. To increase the half-life of the Fab fragment, a 20 kDa PEG chain was attached via strain-promoted azide-alkyne cycloaddition (SPAAC). Biodistribution and imaging studies were performed with 111In-labeled constructs in 4T1 tumor-bearing mice. Comparing our site-specific antibody-conjugates with randomly conjugated antibodies, we found that antibody clone, isotype and method of DTPA conjugation did not change tumor uptake. Furthermore, addition of sulfo-Cy5 did not affect the biodistribution. PEGylated Fab fragment displayed a significantly longer half-life compared to unPEGylated Fab and demonstrated the highest overall tumor uptake of all constructs. PD-L1 in tumors was clearly visualized by SPECT/CT, as well as whole body fluorescence imaging. Immunohistochemistry staining of tumor sections demonstrated that PD-L1 co-localized with the fluorescent and autoradiographic signal. Intratumoral localization of the imaging agent could be determined with cellular resolution using fluorescent microscopy.
Conclusions
A set of molecularly defined multimodal antibody-based PD-L1 imaging agents were synthesized and validated for multiscale monitoring of PD-L1 expression and localization. Our modular approach for site-specific functionalization could easily be adapted to other targets.
Graphical Abstract

Similar content being viewed by others


Background
Over the past decade, immunotherapy, in particular blockade of immune checkpoint molecules such as Programmed Death 1 (PD-1) and its ligand PD-L1 using antibodies, has revolutionized the field of cancer therapeutics due to unparalleled responses in a range of cancers [1,2,3]. Despite these successes, a large number of patients does not benefit from this treatment [4]. Moreover, anti-PD-L1 (aPD-L1) treatment is associated with significant immune-related side effects [5,6,7,8] and comes with a high economic burden on society [9]. PD-L1 immunohistochemistry (IHC) has been shown to be a partial predictor for response [10]. However, not all patients selected using this biomarker actually respond to treatment. Therefore, there is a clear need to better understand the dynamics of response to PD-L1 blockade. Currently, IHC on tumor biopsies is the gold standard for characterization of the tumor microenvironment even though it has significant downsides. First of all, IHC of biopsy material yields limited spatial information as PD-L1 expression can be heterogeneous within and between tumor lesions and does not provide information about the accessibility for PD-L1 therapeutics [11]. In contrast, non-invasive whole-body nuclear imaging of PD-L1 allows for assessment of multiple lesions concurrently and if desired over time also taking into account target accessibility. However, the resulting images have a limited spatial resolution, which does not allow for tissue analysis on a cellular level to differentiate between cell populations that express the targeted molecule [12]. This is relevant because in some tumors therapeutic outcome has been linked to intratumoral PD-L1 expression by tumor-infiltrating immune cells, whereas PD-L1 was not expressed by tumor cells [13, 14]. However, others have reported the reverse, indicating a context dependency for this correlation [15, 16]. Therefore, whole-body imaging would ideally be supplemented with an imaging modality suitable to assess the distribution of PD-L1 therapeutics at a cellular level, such as a fluorescent dye. This allows microscopic imaging at high spatial resolution [17].
Several preclinical [18,19,20,21] and clinical [22, 23] imaging studies have been performed using different PD-L1-targeting agents. In most of these studies, imaging moieties were coupled to the targeting antibody using non-selective protein modification methods, without spatial control. However, such random conjugation can interfere with the antigen-binding region and consequently alter the binding affinity [24] and in vivo pharmacokinetics [25, 26]. In contrast, site-specific labeling does not interfere with the antigen-binding site and yields a more controlled, homogeneous and therefore reproducible product. Recently, we developed an approach to modify antibodies site-specifically by applying the CRISPR/HDR technology to PD-L1 hybridoma cells [27]. We generated Fab fragments and chimeric antibodies with switched mouse Fc isotypes, while at the same time introducing a sortag [28, 29] at the C-terminus of the heavy chain. This tag enables site-specific chemo-enzymatic functionalization of these antibody products. Isotype switching is relevant in the context of imaging, since multiple doses of a foreign antibody can elicit an immune response directed against the Fc region [30,31,32,33], impacting longitudinal monitoring of a target. Moreover, small antibody formats like Fab fragments lack the Fc region entirely and can be of interest due to their relatively short half-life [34,35,36,37,38] and potentially increased ability to penetrate tumor tissue [39,40,41]. Unfortunately, a fast clearance rate can also lead to low tumor retention [35], which can be attenuated by conjugation to a polyethyleneglycol (PEG) chain [42,43,44,45].
Here, we demonstrate that combining our CRISPR/HDR hybridoma engineering technology with chemical synthesis provides a plug-and-play platform to create molecularly defined antibody-based multimodal, multiscale PD-L1 imaging agents. To this end, we functionalized engineered mouse IgG1 (mIgG1) and Fab aPD-L1 site-specifically with a peptide containing both the radionuclide chelator diethylenetriaminepentaacetic acid anhydride (DTPA) and near-infrared (NIR) dye sulfo-Cy5 (Fig. 1). Additionally, an azide was included in the peptide to allow for further functionalization through strain-promoted azide-alkyne cycloaddition (SPAAC). This way, we modified our Fab fragment with a 20 kDa PEG chain to increase the half-life in vivo. We show that our agents allow for whole-body SPECT imaging as well as tissue-level fluorescence microscopy imaging. To the best of our knowledge, this is the first example of multimodal and multiscale imaging to assess the intratumoral distribution of PD-L1 therapeutics.

Molecular toolbox of site-specific functionalizable anti-PD-L1 antibody formats and imaging peptides. A Application of the CRISPR/HDR strategy to anti-PD-L1 hybridoma MIH5 created a sortaggable Fab fragment and chimeric mouse IgG1 monoclonal antibody against PD-L1. B Molecular structure of imaging peptides IH20 and IH18
Results
The effect of isotype switching and site-specific conjugation on the in vivo behavior of aPD-L1 imaging agents
First, using our molecular toolbox (Fig. 1A and B) we set out to determine the consequence of isotype switching of the PD-L1 targeting antibody (clone MIH5) from rIgG2a to mIgG1 [27], as well as random versus site-specific radiolabeling and additional functionalization with a fluorescent dye, on in vivo biodistribution of these compounds (Fig. 2A). To facilitate the incorporation of imaging moieties in a site-specific way, we synthesized the DTPA equipped nuclear imaging peptide IH20 and the multimodal nuclear and NIR imaging peptide IH18, both containing an N-terminal triglycine motif (Fig. 1B and Additional file 1: Fig. S1). After production of the ‘wild type’ rIgG2a (rIgG2a WT) and the c-terminal sortag-histag carrying mIgG1 chimeric aPD-L1 (aPD-L1 mIgG1-srt-his), we site-specifically conjugated peptides IH20 and IH18 to the latter antibody to obtain aPD-L1 mIgG1-IH20 and mIgG1-IH18 (70% isolated yield, Fig. 2b). For non-site-specific DTPA conjugation we exposed aPD-L1 mIgG1-srt-his and rIgG2a WT to an excess of S-2-(4-Isothiocyanatobenzyl)-DTPA (p-SCN-Bn-DTPA). Having the four conjugates in hand, we labeled them with 111In and determined the biodistribution in 4T1 tumor-bearing mice. At 24 h p.i., tumor uptake was similar for all constructs (± 15% ID/g, Fig. 2C, Additional file 1: Table S1). Radiolabeled isotype switched mIgG1 PD-L1-srt-his antibody showed a twofold increase in hepatic uptake compared with the WT rIgG2a antibody (20.65 ± 3.67% ID/g vs 6.56 ± 0.51% ID/g, p < 0.0001), while blood concentration and kidney uptake were significantly lower (2.91 ± 0.88% ID/g and 10.94 ± 2.86% ID/g for blood; 5.44 ± 0.43% ID/g and 8.19 ± 1.21% ID/g for kidneys, p < 0.0001). Site-specifically conjugated isotype-switched mIgG1 with either DTPA alone (mIgG1-IH20) or with the additional fluorophore (mIgG1-IH18) did not show a significant change in blood clearance or liver uptake, compared to randomly labeled WT rIgG2a antibody. Spleen uptake of mIgG1-IH18 was increased compared to mIgG1-IH20 (16.80 ± 0.68% ID/g vs 12.11 ± 0.43% ID/g, p = 0.0015). Duodenum uptake was slightly decreased for IH18- and IH20-labeled antibodies (7.42 ± 0.64% ID/g, p = 0.0009, 6.53 ± 1.69% ID/g, p < 0.0001) compared to the MIH5 WT antibody (10.94 ± 0.52% ID/g). Taken together, isotype switching and site-specific conjugation of PD-L1 targeting antibody with IH18 and IH20 was feasible without major changes in the tumor targeting.

Biodistribution of anti-PD-L1 monoclonal antibody panel. A WT MIH5 antibody and mouse (mIgG1) anti-PD-L1 antibodies were labeled non-site-specifically with DTPA. mIgG1 was labeled site-specifically with IH20 and IH18, where IH20 contains DTPA alone and IH18 contains an additional sulfo-Cy5. B SDS-PAGE analysis of purified site-specifically labeled antibodies. An analytical fraction of mIgG1-IH18 and mIgG1-IH20 was reacted with 5 kDa mPEG-DBCO to analyze purity and azide functionality. This resulted in the near-quantitative conversion to a product of higher molecular weight. (C) Mice bearing orthotopic 4T1 tumors were injected with one of four different 111In-labeled anti-PD-L1 antibodies. Biodistribution was determined ex vivo 24 h after injection. Values are presented as percentage injected dose per gram (%ID/g) and shown as mean ± SD, n = 5
Site-specific multimodal PD-L1 imaging conjugates with different pharmacokinetics
Having demonstrated the ability to produce site-specifically functionalized aPD-L1 mIgG1 chimera with IH18, we next aimed to use our platform to produce multimodal imaging conjugates with different pharmacokinetic properties. In addition to mIgG1-IH18, we produced c-terminal sortag-histag carrying aPD-L1 Fab fragments [27] to conjugate IH18 in a sortase catalyzed reaction to obtain Fab-IH18 (75% isolated yield, Fig. 3A and Additional file 1: Fig. S4). To prolong the anticipated short half-life, we reacted the azide functionality in Fab-IH18 with 20 kDa mPEG-DBCO to obtain Fab-IH18-PEG (60% isolated yield, Fig. 3A and Additional file 1: Fig S3C).

In vitro characterization of multimodal PD-L1 imaging tools. A SDS-PAGE analysis of the antibody formats before and after sortagging with IH18. After purification using NiNTA beads and SEC, an analytical fraction of mIgG1-IH18 and Fab-IH18 was reacted with 5 kDa mPEG-DBCO to analyze purity and azide functionality. This resulted in the near-quantitative conversion to a fluorescent product of higher molecular weight. Fab-IH18-PEG was produced through large-scale reaction of Fab-IH18 with 20 kDa mPEG-DBCO and subsequently purified using cation exchange chromatography. B Competition assay. Renca cells were incubated with a serial dilution of unlabeled anti-PD-L1 MIH5 (WT) or different antibody-conjugate (mIgG1-IH18, Fab-IH18, Fab-IH18-PEG) concentrations, as well as commercially available anti-PD-L1-PE. Data are presented as mean ± SD, n = 3
In vitro characteristics
To validate preservation of antigen specificity, we carried out a competitive binding assay for mIgG1-IH18, Fab-IH18 and Fab-IH18-PEG. All site-specifically conjugated constructs and the WT rIgG2a competed with a commercially available fluorescently labeled MIH5 aPD-L1 antibody for target binding in a concentration-dependent manner, indicating post-conjugation antigen specificity (Fig. 3B). Moreover, IC50 values were similar for mIgG1 PD-L1-IH18 (180 pM) and the unconjugated WT antibody (88 pM). The IC50 values of the Fab fragments were ± 100-fold higher compared to mIgG1-IH18 (18.3 nM and 27.8 nM for Fab-IH18 and Fab-IH18-PEG, respectively). Direct monitoring of binding to target cells measuring sulfo-Cy5 fluorescence reflected the results of the competitive binding assay (Fig. S5A). Finally, all 111In radiolabeled antibody-conjugates showed target-specific binding and internalization over time (Additional file 1: Fig. S5B-D). These data suggest a relatively high internalization rate for mIgG1-IH18 and Fab-IH18-PEG compared to Fab-IH18.
Biodistribution in tumor-bearing mice
The biodistribution of the radiolabeled multimodal aPD-L1 site-specific conjugates was assessed in 4T1 tumor-bearing mice 4 h, 24 h and 72 h p.i. for mIgG1-IH18 and 1 h, 4 h and 24 h p.i. for Fab-IH18 and Fab-IH18-PEG (Fig. 4 and Additional file 1: Table S2). For mIgG1-IH18, tumor uptake was highest at 24 h p.i. (13.24 ± 2.93%ID/g). However, of the timepoints measured, the optimum tumor/blood ratio (TBR) was achieved 72 h p.i. (20.67 ± 8.60) due to high concentrations in blood at the 24 h time point (6.13 ± 1.81%ID/g). Fab-IH18 was cleared from the circulation more rapidly, demonstrated by a low concentration in the blood 24 h p.i. (0.31 ± 0.08%ID/g). Highest TBR and tumor accumulation was observed at 24 h p.i. (32.68 ± 7.95; 9.67 ± 0.84%ID/g respectively). Fab-IH18 displayed a high kidney accumulation (78.36 ± 10.31%ID/g), due to renal clearance of the low molecular weight conjugate. In contrast, Fab-IH18-PEG showed increased circulation time compared with Fab-IH18 (the concentration 24 h p.i. was 2.03 ± 0.30%ID/g), causing the TBR to remain lower than for the other two constructs (8.22 ± 1.32, p = 0.0005 and p < 0.0001 compared to mIgG1 after 72 h and Fab after 24 h, respectively), while tumor accumulation for Fab-IH18-PEG after 24 h was significantly higher than Fab-IH18 (16.55 ± 2.80%ID/g, p < 0.0001). Taken together the different conjugates demonstrated high tumor uptake with TBR in accordance with clearance kinetics of the antibody formats used.

Biodistribution results of anti-PD-L1 multimodal imaging tools. Biodistribution is shown at several time points after injection (p.i.) of 111In-labeled A mIgG1-IH18, B Fab-IH18 or C Fab-IH18-PEG. D Tumor uptake values (left graph) and tumor/blood ratios (right graph) of the three constructs are compared directly at each time point. Data are shown as mean ± SD, n = 5
Multiscale multimodal PD-L1 imaging
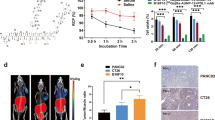
Next, we performed SPECT/CT and IVIS imaging in mice bearing orthotopic 4T1 tumors using our 111In-labeled PD-L1 multimodal imaging conjugates (Fig. 5, Additional file 1: Fig. S6). Based on the previous biodistribution results (Fig. 4), imaging was performed at 24 and 72 h for mIgG1-IH18, and at 4 and 24 h for Fab-IH18. For Fab-IH18-PEG we added a 48 h timepoint based on the slower clearance kinetics. All constructs were detectable in the tumor. For Fab-IH18 and Fab-IH18-PEG, SPECT/CT images showed a clear tumor-background signal. As expected, high accumulation of Fab-IH18 was observed in the kidneys. SPECT/CT images of Fab-IH18-PEG after 48 h were comparable to 24 h. The IVIS data show a clear signal at the tumor site, indicating that IVIS imaging can additionally be used to detect tracer accumulation in the tumor (Fig. 5). After the last scan, tumor and healthy tissues were collected for ex vivo biodistribution studies (Table S3). These results correlate with the findings from SPECT/CT and fluorescence imaging. Finally, we set out to visualize the localization of the multimodal imaging agent mIgG1-IH18 on a tissue and cellular level. We analyzed tumor sections using immunohistochemistry (IHC) and performed autoradiography and fluorescence imaging of adjacent sections. This showed a large degree of co-localization of PD-L1 expression with the radioactive and fluorescent signal (Fig. 6A), with only a small amount of signal in PD-L1 negative regions. Overall, these data suggest in vivo tracer binding is target-specific and that the conjugates remain intact (Fig. 6A). Importantly, NIR fluorescence microscopy allowed us to determine the localization of the multimodal PD-L1 imaging agent and its co-localization with PD-L1 expression, even at cellular resolution (Fig. 6B, Additional file 1: Fig. S7).

SPECT/CT and IVIS imaging of labeled antibody distribution in mice bearing orthotopic 4T1 tumors. Representative examples of SPECT/CT and IVIS scans of mice bearing 4T1 tumors, acquired at different time points after injection of 111In-labeled mIgG1-IH18, Fab-IH18 or Fab-IH18-PEG. Tumors are indicated with white arrows. Fluorescence intensity is indicated as radiant efficiency [(p/s/cm2/sr)/(μW/cm2)]

Analysis of tumor sections shows distribution of mIgG1-IH18 radiographic and fluorescent signal and PD-L1 expression. A Physically adjacent tissue sections of a tumor from a mIgG1-IH18 treated mouse are stained with H&E, PD-L1 immunostaining, or where fluorescent and radiographic signal is acquired. PD-L1, autoradiography and fluorescence patterns are overlapping, demonstrating co-localization of these signals. B Fluorescence microscopy shows localization of mIgG1-IH18 with cellular resolution (DAPI in blue, Cy5 fluorescence in purple and PD-L1 immunofluorescent staining in green in the merge image)
Discussion
While PD-L1 blockade yields durable responses in subgroups of cancer patients, many patients do not benefit from this treatment. As such, there is a clear need to better understand the biomarkers and dynamics of treatment response in order to improve patient stratification. To gain more insight into the fundamentals of anti-PD-L1 treatment, we developed multimodal, multiscale PD-L1 imaging agents to image PD-L1 drug distribution in vivo.
In this work we took advantage of the genetically engineered MIH5 derived hybridoma lines we created via our CRISPR/HDR approach to produce isotype switched mIgG1 chimeric PD-L1 targeting antibodies, as well as Fab fragments equipped with a sortag and a histag at the c-terminus of the heavy chain [27]. We synthesized a sortaggable peptide (IH20) carrying a DTPA metal chelator, as well as a cysteine and azidolysine for further functionalization. We attached a sulfo-Cy5 fluorophore as a proof of concept (IH18), but in principle any fluorescent and/or photodynamic dye could be attached. Sortase mediated chemoenzymatic conjugation enabled us to produce a panel of site-specific conjugates in good yields. The applicability of our CRISPR/HDR platform to any hybridoma, in combination with the modular conjugation strategy, makes our approach adaptable to a wide range of targets, as well as imaging modalities and other cargo molecules to produce molecularly defined conjugates in a reproducible manner.
Our in vivo results showed that isotype switching and site-specific modification of MIH5 aPD-L1 antibodies does not significantly change tumor uptake. However, hepatic accumulation was increased twofold for the mIgG1 PD-L1-srt-his antibody compared to the other antibodies, most likely caused by the presence of the Histag [46, 47]. Although previous studies showed that random DTPA conjugation via lysine side chains can influence affinity [24], in vivo pharmacokinetics [25, 48] and therapeutic index [49], we did not observe differences in blood clearance or tumor uptake for site-specifically versus randomly labeled mIgG1 aPD-L1 with DTPA or IH20. Finally, no statistical differences in biodistribution were observed for IH20- and IH18-conjugated chimeric mIgG1 aPD-L1, except for the spleen, where uptake of mIgG1-IH18 was significantly higher, potentially caused by the apparent higher blood concentration 24 h p.i. Taken together, site-specific conjugation with IH18 and IH20 was feasible without major changes in the tumor targeting.
Side-by-side comparison of the different multimodal imaging formats showed that antigen specificity was retained for mIgG1-IH18, Fab-IH18 and Fab-IH18-PEG. The IC50 value of mIgG1-IH18 was in the same range as that of the WT MIH5 antibody. In accordance with literature, the Fab’s IC50 was 100-fold higher, due to its monovalency [27, 42, 50, 51]. Previously, it has been shown that non-site-specific PEGylation strongly decreases affinity [44], while site-specific PEGylation leads to a (largely) preserved affinity [42, 52,53,54]. The slight decrease in affinity for Fab-IH18-PEG could be caused by transient intramolecular blocking of the binding site by the long PEG chain, resulting in a slower on rate [55]. After labeling with 111In, all three site-specific multimodal imaging agents displayed target-specific binding and internalization (Additional file 1: Fig. S5b-d). In vivo, 111In-labeled conjugates accumulated in the tumor and other PD-L1-expressing tissues such as lymph nodes, brown fat and duodenum. Most notably, the rate of clearance was different for each of these three constructs, with mIgG1-IH18 reaching its optimum TBR at 72 h. In contrast, Fab-IH18 displayed a similar tumor uptake to mIgG1-IH18 but a higher TBR already after 24 h. In translation to the clinic, this would mean faster consecutive imaging, more insight into the pharmacokinetics, as well as a shorter wait period between injection and imaging, making Fab-IH18 superior to mIgG1-IH18. Compared to Fab-IH18, Fab-IH18-PEG showed increased tumor retention, which is likely caused by the increased circulation time: blood concentration is the driving force for initial diffusion into the tumor, where the antibody (fragment) can subsequently be retained by target binding [41].
SPECT/CT and IVIS imaging demonstrated non-invasive multimodal detection of the aPD-L1 imaging agents, correlating with the biodistribution results. Subsequent analysis of tumor sections showed a very strong co-localization between PD-L1 expression and the fluorescent and radioactive signals. However, especially for Fab PD-L1-IH18-PEG some additional tracer accumulation was observed in areas that were not positive for PD-L1 based on IHC. This could indicate a small degree of unspecific tracer uptake caused by the enhanced permeability and retention effect, or could be attributed to comparing adjacent sections. Together, these data indicate that in vivo tracer binding is largely target-specific and that our conjugates remain intact. Small antibody formats such as Fab fragments have been linked to improved tissue penetration in vitro [56] and due to their high diffusion rate [57], they have been hypothesized to have higher mobility in the extracellular matrix (ECM) of tumor cells [41]. Moreover, their small size causes less molecular crowding and they are therefore less likely to display the so-called “binding site barrier effect” [41]. However, we did not see any evidence that this applies to our multimodal imaging conjugates. The overall performance of our multimodal conjugates is comparable to previously published PD-L1 imaging agents [58, 59].
Contradicting correlations have been found between clinical whole-body PD-L1 PET imaging and treatment response [22, 23, 60, 61]. Our multimodal platform enabled us to image aPD-L1 tracer localization with cellular resolution with NIR fluorescence microscopy. This could provide valuable insight, because the cell type expressing PD-L1 has been linked to therapeutic outcome. For example, PD-L1 expressed by immune cells correlated with response for aPD-L1 therapy in some tumor types, whereas PD-L1 expressed by tumor cells did not [13, 14], while others have reported the reverse [15, 16]. Therefore, direct visualization of the cellular localization of the PD-L1 fraction which can be targeted with multimodal theranostic antibody-based imaging agents and hence antibody therapeutics could provide valuable insight into the correlation of PD-L1 expression with response to aPD-L1 therapy.
Conclusion
We demonstrate that our modular platform allows for site-specific functionalization of aPD-L1 formats with peptides containing sulfo-Cy5 as a fluorophore and DTPA for radiolabeling, without significantly impacting the pharmacokinetic properties. We showed that fluorescent and radiolabeled mIgG1-IH18, Fab-IH18 and Fab-IH18-PEG aPD-L1 display different pharmacokinetic properties, especially in terms of clearance. Our multimodal imaging agents allowed for whole-body SPECT/CT and IVIS imaging, as well as microscopy imaging, and we show that tracer accumulation is mainly target specific. Together, these data demonstrate that site-specific chemo-enzymatic conjugation of multiple imaging moieties to aPD-L1 antibodies is feasible and gives rise to molecularly defined multimodal PD-L1 imaging tools. Furthermore, the methodology described in our paper could be easily adapted for other targets. These agents can be employed to investigate immunotherapy in a preclinical setting, using whole-body imaging as well as analysis on tissue level and could potentially be translated for clinical applications.
Materials and methods
General methods and materials
Chemicals were used without further purification. Amino acids were purchased from Bachem (Bubendorf, Switzerland) or Novabiochem (EMD Chemicals, Gibbstown, USA), except for Fmoc-NH-PEG3-COOH (Iris Biotech, Marktredwitz, Germany). Solvents were obtained from ThermoFisher Scientific, Merck Millipore and Biosolve. LC–MS data were recorded on a Thermo Finnigan LCQ Fleet system, which consists of a Shimadzu LC-20A Prominence system (Shimadzu, ‘s-Hertogenbosch, The Netherlands) with a Gemini NX-C18 column, 150 × 2.1 mm, particle size 3 μm (Phenomenex, Utrecht, The Netherlands) with PDA detector coupled to a Thermo LCQ Fleet mass spectrometer. For LC–MS analysis, an acetonitrile/water gradient was used of 5–100% in 60 min, flow rate 0.2 mL/min. For peptide purification, preparative HPLC was performed on a Shimadzu LC-20A Prominence system (Shimadzu, ‘s-Hertogenbosch, The Netherlands) equipped with a Gemini NX-C18 column, 150 × 21.20 mm, particle size 10 μm (Phenomenex, Utrecht, The Netherlands) or on a Waters HPLC system equipped with an XBridge Prep-C18 column, particle size 5 µm, 150 × 30 mm and an Equity QDa Mass Detector. On the Shimadzu, the gradient used was acetonitrile/water 5–40% in 35 min, flow rate 10 mL/min. On the Waters HPLC system, the acetonitrile/water gradient was 5–25% in 10 min, flow rate 40 mL/min. Analytical HPLC was performed on a Shimadzu LC-20A Prominence system (Shimadzu, ‘s-Hertogenbosch, The Netherlands) equipped with a Gemini NX-C18 column, 150 × 3 mm, particle size 3 µm (Phenomenex, Utrecht, The Netherlands). For both the analytical and preparative HPLC, peptides were monitored at 254 and 215 nm.
Fluorescence analysis was carried out using the Typhoon Trio Variable Mode Imager System from GE Healthcare (Eindhoven, The Netherlands). SDS-PAGE coomassie analysis was performed using a Bio-rad ChemiDoc XRS + system. For size-exclusion chromatography (SEC) and cation-exchange chromatography (CEX), a Bio-rad NGC Quest Plus System with 4-wave UV–Vis detector and a 10 mL/min pump (Bio-rad, Veenendaal, The Netherlands). For SEC, The NGC system was equipped with a Superdex 75 Increase 10/300 GL Size-exclusion column (GE Healthcare, Eindhoven, The Netherlands). For CEX, a 1 mL HiTrap SP FF (GE Lifesciences, 17505401) cation exchange column was used on the same NGC Quest Plus Bio-Rad system, operating at 0.5 mL/min. Antibody concentration was determined using the NanoDrop 2000c (Thermo Fisher Scientific). FACS measurements were performed on FACSVerse (BD Biosciences), and analysis was performed using FlowJo V10 software.
Cell culture
Previously, MIH5 hybridoma cells were engineered to express mIgG1, mIgG2asilent or Fab fragments against PD-L1 with a Sortag and a Histag at the C-terminus of the heavy chain(s) (plasmids available at www.addgene.org/, ID: 124802, 124807 and 124810) [27]. Other cell lines that were used throughout this study were 4T1 (ATCC CRL-2539) and Renca (ATCC CRL-2947). All cells were cultured in RPMI-1640 (Gibco, 11875-093, Thermo Fisher Scientific) supplemented with 10% heat-inactivated fetal calf serum (FCS), 2 mM UltraGlutamine (BE16-605E/U1, Lonza) and 1 × antibiotic–antimycotic (15240-062, Thermo Fisher Scientific). In addition, Hybridoma medium contained 50 μM Gibco 2-mercaptoethanol (2-ME) (21985-023, Thermo Fisher Scientific). Renca cell medium additionally contained 0.1 mM non-essential amino acids (NEAA) (11140-035, Thermo Fisher Scientific) and 1 mM Sodium Pyruvate (Gibco, 11360-070), Thermo Fisher Scientific). (Semi-) adherent cells were washed with phosphate-buffered saline (PBS) (Fresenius Kabi) and detached by incubation with 0.025% trypsin and 0.01% ethylenediaminetetraacetic acid (EDTA) in PBS (TE) (Thermo Fisher, R001100) for 10 min. at 37 °C, or by using a cell scraper.
Production of antibody conjugates
Production and isolation of sortaggable antibodies
After expansion, Hybridoma cells were seeded in a CELLine Disposable Bioreactor (Corning, 353137). The medium compartment contained 900 mL Hybridoma medium with 1% FBS, the cell compartment contained 15 mL Hybridoma medium with 10% FBS. Medium was harvested from cell compartment every 7 days and separated from the cells by centrifugation (5 min, 1500 rpm), filtered through a 20 µm filter (Whatman) and stored at − 20 °C until the moment of antibody purification. Pellet was resuspended in 30 mL medium and live cells were separated from dead cells by using Ficoll density centrifugation (Lymphoprep; Axis-Shield PoC AS, Oslo, Norway). Subsequently, 30 million live cells were reseeded into the CELLine bioreactor. For each flask, multiple seeding—harvesting cycles were performed. For antibody purification, the supernatant was thawed and incubated for 20 min at room temperature (RT) with 1–2 mL Ni–NTA beads (Qiagen, 30210). Subsequently, the suspension was transferred to an Econo-Pac Chromatography Column (Bio-Rad, 7321010). The column was washed with 10 column volumes (CV) of wash buffer (50 mM NaH2PO4.H2O, 300 mM NaCl, 20 mM imidazole, 0.05% Tween 20, pH 8.0), 100 CV of 0.1% Triton X-114 (Merck, 9036-19-5) in sterile PBS at 4 °C, and 20 CV sterile PBS. These three steps were repeated twice. Finally, the antibody was eluted with elution buffer (50 mM NaH2PO4.H2O, 300 mM NaCl, 250 mM imidazole, 0.05% Tween 20, pH 8.0). The resulting elution fractions were concentrated with Amicon Ultra-15 Centrifugal 10 kDa MWCO filter units (Merck, Z717185) for Fab fragments and 50 kDa MWCO filter units for monoclonal antibodies. Buffer exchange was performed using sortase buffer (50 mM Tris, 150 mM NaCl, pH 7.5). Antibody concentration was determined using the NanoDrop 2000c (Thermo Fisher Scientific) using the protein program and dividing by 1.4 for monoclonal antibodies and by 1.35 for a Fab fragment. Protein purity was assessed under reducing conditions using SDS-PAGE gel electrophoresis (12% acrylamide) and Sypro Ruby Protein Gel stain (S12000, Thermo Fisher Scientific). Typical yields per week were around 2–2.5 mg for Fab and 1.5–2 mg for mIgG1.
Peptide synthesis
The backbone of multimodal imaging peptides IH20 and IH18 (Fig. 1b) was synthesized on Rink resin (500 mg, 0.29 mmol) using standard Fmoc-based Solid-Phase Peptide Synthesis (SPPS). Coupling and deprotection steps were followed to completion using a Kaiser test. For each coupling step, 3 eq. of amino acid (AA) was used. For most coupling steps, 3.3 eq. of 1 M DIPCDI and 3.6 eq. of 1 M HOBt in DMF were used. Coupling steps were incubated for 45 min to overnight. For coupling of H2N-PEG3-COOH, 1.5 eq. HATU and 3 eq. DIPEA were added and this mixture was agitated overnight. Upon completion of each coupling step, as indicated by the Kaiser test, the resin was washed three times with DMF and the remaining free amines were capped using a 3:2 mixture of Acetic anhydride and pyridine in DMF. After capping, the resin was washed three times again with DMF. Subsequently, the Fmoc group on the last AA in the chain was removed by incubating the resin with 20% piperidine for 20 min. After each deprotection, the next AA was coupled as described. For the N-terminal glycine, Boc-protected glycine was used instead of Fmoc-protected to allow for easy removal of all protective groups when cleaving the peptide from the resin. Upon completion of the backbone, the resin was washed subsequently with DMF (3×), DCM (3×) and diethyl ether (3×). Peptide on resin was then dried and stored at − 20 °C.
To allow the chelation of radionuclides, the chelator DTPA was attached to the lysine side chain of the peptide. The resin (300 mg, 120 µmol) was swelled in DCM, before multiple cycles of treatment with 1.2% TFA in DCM for 2 min (optimal deprotection after 15 cycles), After which the resin was washed extensively with DCM and subsequently with DMSO. Then, S-2-(4-Isothiocyanatobenzyl)-diethylenetriamine pentaacetic acid (p-SCN-Bn-DTPA, Macrocyclics, B-305) (236 mg, 360 µmol, 3 eq.) was dissolved in DMSO and DIPEA (0.73 mL, 4.20 mmol, 35 eq.) was added. This mixture was agitated with the resin overnight at RT. Finally, the peptide was fully deprotected and cleaved off the resin by incubating with a 92.5/2.5/2.5/2.5 mixture of TFA/H2O/TIS/Thioanisole for 2–3 h. The peptide was precipitated in ice cold diethyl ether and air-dried. After drying, the resulting off-white solid was dissolved in 80/20 H2O/ACN and lyophilized.
The resulting IH20 peptide was purified using RP-HPLC (10–40% ACN/H2O in 35 min), and analyzed using LC–MS. HPLC rt: 20.67. LC–MS(ESI +): m/z calcd for C52H84N16O20S22+ ([M + 2H]2+) 659.28, found 659.30. C52H84N16O20S23+ ([M + 3H]3+): 439.85, found 439.94. After purification and lyophilization, IH20 was obtained as a white powder (yield 43%).
To yield IH18 by modification of IH20 with sulfo-Cy5, IH20 (10.8 mg, 7.54 µmol) was dissolved in DMF (60 mg/mL) and mixed with 1.1 eq. (6.7 mg, 8.30 µg) sulfo-cyanine-5-maleimide (Lumiprobe, 13380) in PBS (pH 6.9, 9.5 mg/mL). This mixture was agitated overnight at RT. The resulting IH18 peptide was purified using RP-HPLC (5–40% ACN/H2O in 35 min), and analyzed using LC–MS. HPLC rt: 27.27. LC–MS (ESI +) m/z calcd for C90H128N20O29S42+ ([M + 2H]2+) 1041.40, found 1041.80. C90H128N20O29S43+ ([M + 3H]3+): 694.60 found: 694.84. After lyophilization, IH18 was obtained as a blue powder (10.6 mg, 4.83 µmol, yield 64%).
Site-specific enzymatic conjugation
After optimization, batch sortagging was carried out using 0.5–1.0 mg of antibody (fragment) and 25 eq of IH18 or IH20 for mIgG1 PD-L1 and 50 eq of IH18 or IH20 for Fab PD-L1. After termination of the reaction with EDTA (final concentration of 10 mM), the reaction mixture was incubated with 200 µl Ni–NTA beads for 20 min at RT, in order to remove unreacted antibody and Sortase. Beads were separated from the reaction mixture using empty spin columns (Jena Bioscience, AC-552-25), and washed twice with wash buffer (50 mM NaH2PO4.H2O, 300 mM NaCl, 20 mM imidazole, 0.05% Tween 20, pH 8.0) and twice with PBS. Reaction mixture and wash fractions were combined and purified using size-exclusion chromatography (SEC), with 1 mM EDTA in PBS as buffer (10 mL/min). Fractions containing the product were combined and concentrated using Amicon Ultra-15 Centrifugal Filter Units, MWCO 10 kDa (Merck, Z717185). Buffer exchange was performed using sterile PBS. Antibody concentration was determined on NanoDrop, using the UV–vis program and measuring at 280 nm for IH20 and at 280 and 646 nm for IH18. Protein purity was assessed under reducing conditions using SDS-PAGE gel electrophoresis (12% acrylamide), comparing starting material, product and product clicked with 5 kDa mPEG-DBCO to verify functionalization of all heavy chains. Per construct, several batches were produced and these batches were combined before performing in vitro and in vivo experiments to guarantee batch uniformity. Quantification of protein purity was achieved using densitometry. Densitometry was performed in ImageJ, and defined as (product bands − background)/(non-product bands − background) × 100%. Purity mIgG1 PD-L1-IH18 = 85%, Fab PD-L1-IH18 = 96%.
PEGylation and purification of FabPD-L1-IH18
Fab PD-L1-IH18 was conjugated to 20 kDa PEG by adding 10 Eq. (94.0 nmol) of mPEG-DBCO (Click Chemistry Agents, A120) in PBS (3.33 mM) directly to Fab PD-L1-IH18 in PBS (470 µg, 9.40 nmol, 3.0 mg/mL). This mixture was incubated for 90 min at 37 °C, buffer exchange was performed with 0.02 M acetate buffer (pH 4.5, buffer A) using Amicon Ultra-4 Centrifugal Filter Units, MWCO 10 kDa (Merck, UFC801024) and subsequently purified using a 1 mL HiTrap SP FF (GE Lifesciences, 17505401) cation exchange column on an NGC Quest Plus Bio-Rad system operating at 0.5 mL/min. After equilibrating with buffer A for 8 column volumes, a linear gradient of 0.5 M NaCl in buffer A was applied (0–0.5 M NaCl in 30 column volumes). PEGylated Fab PD-L1-IH18 were typically eluted after about 8 column volumes (0.15 M NaCl). Fractions containing the purified product were concentrated using Amicon Ultra-4 Centrifugal 10 kDa MWCO filter units (Merck, UFC 801024) and sterile PBS as a buffer, and analyzed by 12% SDS-PAGE. The concentration was determined using Nanodrop. This method was verified as reliable by use of a BCA assay (Pierce BCA Protein Assay Kit, ThermoFisher Scientific). Quantification of protein purity was achieved by performing densitometry, as described above. Purity Fab PD-L1-IH18-PEG20kDa = 89%.
Random conjugation of DTPA to PD-L1 antibodies
To prevent contaminants from disturbing radiolabeling, 1 mg of mIgG1 PD-L1-srt-his and rIgG2a WT antibodies were dialyzed against 5 L sterile PBS (metal-free) using Slide-a-Lyzer Cassettes (ThermoFisher Scientific). Subsequently, a 15 eq. of S-2-(4-Isothiocyanatobenzyl)-diethylenetriamine pentaacetic acid (p-SCN-Bn-DTPA, Macrocyclics, B-305) and 1/10 reaction volume 1 M NaHCO3 in PBS, pH 5.5 were added to each antibody and incubated for 1 h at RT. Non-conjugated p-SCN-DTPA was removed from the reaction mixture by dialysis against 5 L 0.25 M NH4Ac (metal-free, pH 5.5) using Slide-a-Lyzer Cassettes (ThermoFisher Scientific). After dialysis, concentration in all samples was determined via spectrophotometer.
Radiolabeling
Antibody-IH18 conjugates were incubated with 111In (Mallinckrodt BV) in 0.5 M MES buffer (pH 5.4) for 20 min at room temperature under metal-free conditions as described previously [62]. Non-chelated 111In was complexed by adding 50 mM EDTA to a final concentration of 5 mM. Labeling efficiency was determined using thin-layer chromatography on silica gel chromatography strips (Agilent Technologies), using 0.1 M citrate buffer (Sigma-Aldrich, pH 6.0) as mobile phase. Samples with a labeling efficiency below 95% were purified using a PD-10 column (GE Healthcare, 17-0851-01) eluted with PBS. Radiochemical purity of 111In-labeled antibody-IH18 conjugates exceeded 95% in all experiments.
In vitro characterization
IC50 of multimodal PD-L1 imaging conjugates
30,000 Renca cells were seeded per well in a 96-well V-bottom plate, and stained with 30 μl of mIgG1 PD-L1-IH18, Fab PD-L1-IH18, Fab PD-L1-IH18-PEG20kDa or a mIgG1 isotype control (BioLegend, 400102). Concentrations of these constructs ranged from 0.1 pM to 1000 nM for antibodies and 0.4 pM to 6000 nM for Fabs. After 20 min of incubation at 4 °C, 30 μL of commercially available MIH5 PD-L1-PE (ThermoFisher Scientific, 12-5982-82) was added (final concentration 6.7 nM). After another 30 min of incubation at 4 °C, cells were washed and resuspended in PBA and measured on FACS. The IC50 was defined as the antibody-conjugate concentration that was required to inhibit binding of the commercially available fluorescently-labeled antibody by 50%.
Internalization kinetics
Renca cells were cultured to confluency in 6 wells plates and incubated for 1, 3 or 24 h with 105 pM radiolabeled mIgG1, 323 pM Fab or 86 pM PEGylated Fab in RPMI-1640 containing 1% BSA at 37 °C in a humidified atmosphere with 5% CO2. Non-specific binding and uptake were determined by co-incubation with 16 nM unlabeled WT PD-L1 (rIgG2a). After incubation, cells were washed and incubated with acid wash buffer (0.1 M AcOH, 0.15 M NaCl, pH 2.8) for 10 min at 37 °C to remove the membrane-bound fraction of the antibody conjugates. Cells were washed with PBS, and the acid wash and PBS were combined into one fraction containing the membrane-bound activity. Finally, cells containing the internalized activity were lysed with 0.1 M NaOH and harvested. Samples were measured in a gamma counter (Wizard2, Perkin-Elmer, Boston MA) to determine the internalized and membrane-bound activity.
Animal studies
All experiments were performed in accordance with the revised Dutch Act on Animal Experimentation (2014) approved by the central authority for scientific procedures on animals and animal welfare body of the Radboud University, Nijmegen. Mice were housed in individually ventilated cages with a filter top (Blue line IVC, Tecniplast, West Chester, USA) under pathogen-free conditions with cage enrichment present (abundant bedding material and transparent dome), and were fed and watered ad libitum. Biotechnicians were blinded to all groups and compounds administered. Studies were performed with 6–8 weeks old BALB/c mice (Janvier, le Genest-Saint-Isle, France). In all experiments mice received 1 × 106 4T1 cells (ATCC) by injection into the mammary fat pad. Mice were block-randomized across the experimental groups based on tumor size. Experiments started when average tumor size reached 0.3 cm3.
Biodistribution study
For each construct, 3 groups of 5 mice received an intravenous injection of 0.4 pmol 111In-labelled tracer (0.4 MBq, 200 µL). At 1, 2 and 24 h after injection with Fab PD-L1-IH18 or Fab PD-L1-IH18-PEG20kDa, or 4, 24 and 72 h for mIgG1, mice were sacrificed using CO2/O2-asphyxiation. To determine the biodistribution of each construct, tumor and normal tissues (blood, lymph node, muscle, lung, spleen, thymus, kidney, liver, duodenum, colon, brown fat, bone marrow and bone) were harvested, weighed and measured in a gamma counter, as well as 1% standards of the injected dose. Values are reported as percentage injected dose per gram (%ID/g).
SPECT/CT imaging
For each construct, mice (n = 5/group) received 0.4 pmol 111In-labelled tracer (8 MBq, 200 µL). For Fab, scans were acquired at 4 h and 24 h post-injection (p.i.), for Fab-PEG at 4 h, 24 h and 48 h, and the full length antibodies were imaged at 24 and 72 h p.i.. Images were acquired for 45 min under general anesthesia (isoflurane in 100% oxygen, 5% for induction, 2% maintenance) with the U-SPECT-II/CT (MILabs, Utrecht, The Netherlands) using a 1.0 mm diameter pinhole mouse high sensitivity collimator, followed by CT scan (615 µA, 65 kV) for anatomical reference. Scans were reconstructed with MILabs reconstruction software using a 16-subset expectation maximization algorithm, with a isotropic voxel size of 0.2 mm and 1 iteration. SPECT/CT scans were analyzed and maximum intensity projections (MIP) were created using Inveon Research Workplace software (Siemens).
Autoradiography & fluorescence imaging of tissue sections
Snap frozen, unstained tumor section (10 µm) were scanned on a fluorescence flat-bed scanner at pixel resolution of 4 µm and with an excitation/filter setting of 690/700 nm, quality setting ‘high’ (Odyssey CLx, LICOR) and subsequent sections were exposed to a Fujifilm BAS cassette 2025 (Fuji Photo Film) for 3 days. Phosphor luminescent plates were scanned using a phosphor imager (Typhoon FLA 7000; GE) at a pixel size of 25 × 25 µm. Images were acquired with Aida Image software.
Immunohistochemistry
PD-L1 expression in tumors was determined by chromogenic detection using fresh frozen tissue sections fixed in PBS 45% acetone 10% formalin. In short, sections were pre-incubated with 10% normal rabbit serum, endogenous biotin and avidin were blocked, and endogenous peroxidase activity was blocked with 3% H2O2. Subsequently, anti–mPD-L1 (0.4 μg/mL, AF1019, R&D systems) was applied and tissues were incubated overnight at 4 °C. Next, sections were incubated with biotinylated rabbit anti-goat IgG (3.75 μg/mL E0466, DAKO) followed by incubation with avidin–biotin-enzyme complexes (dilution 1:50, Vector Laboratories, Burlingame, CA). Finally, 3’,3’-diaminobenzidine (DAB) was used to develop the tumor sections prior to mounting slides.
Furthermore, fluorescence detection was performed to co-localize mIgG1 PD-L1-IH18 with PD-L1 expression. Fresh frozen tissue sections were fixed using buffered formalin-acetone fixative and pre-incubated with 10% normal donkey serum or 10% normal goat serum. Subsequently, slides were incubated overnight at 4 °C with anti-mPD-L1 (10 µg/mL, AF1019 R&D biosystems) or isotype control goat IgG (10 µg/mL, 005–000-003, Jackson Immunoresearch). Next, donkey-anti-goat-Alexa488 (2.5 µg/mL in glycerol, A11055, Thermo Fisher) was applied for 30 min and DAPI (0.5 µg/mL, D1306, Invitrogen) for 2 min at RT. Sections were mounted with fluoromount (F4680, DAKO) and fluorescence imaging was performed with a Leica DMI6000 epi-fluorescence microscope fitted with a 63 × 1.4 NA oil immersion objective.
Statistical analyses
Statistical analyses were performed using GraphPad Prism version 5.03 (San Diego, CA) for Windows. Data are presented as mean ± standard deviation. Differences in uptake of the radiolabeled tracers were tested for significance using a one-way ANOVA with a Bonferroni test. A p-value below 0.05 was considered significant.
Availability of data and materials
The dataset used and/or analysed during the current study are available from the corresponding author on reasonable request.
Change history
14 May 2022
A Correction to this paper has been published: https://doi.org/10.1186/s12951-022-01306-y
References
Reiss KA, Forde PM, Brahmer JR. Harnessing the power of the immune system via blockade of PD-1 and PD-L1: a promising new anticancer strategy. Immunotherapy. 2014;6:459–75.
Rolfo C, Caglevic C, Santarpia M, Araujo A, Giovannetti E, Diaz Gallardo C, et al. Immunotherapy in NSCLC: a promising and revolutionary weapon. Immunother Adv Exp Med Biol. 2017;995.
Alexander W. The checkpoint immunotherapy revolution: What started as a trickle has become a flood, despite some daunting adverse effects, new drugs, indications, and combinations continue to emerge. P T. 2016;41:185–91.
Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:1–15.
Brahmer JR, Tykodi SS, Chow LQ, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65.
Cho DC, Sosman JA, Sznol M, Gordon MS, Hollebecque A, Hamid O, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with metastatic renal cell carcinoma (mRCC). J Clin Oncol. 2013;31.
Hamid O, Sosman JA, Lawrence DP, Sullivan RJ, Ibrahim N, Kluger HM, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma (mM). J Clin Oncol. 2013;31.
Herbst R, Gordon MS, Fine GD, Sosman JA, Soria J-C, Hamid O, et al. A study of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic tumors. J Clin Oncol. 2013;31.
Nesline MK, Knight T, Colman S, Patel K. Economic burden of checkpoint inhibitor immunotherapy for the treatment of nonesmall cell lung cancer in US clinical practice. Clin Ther. 2020;42:1682-98.e7.
Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2021;18(6):345–62.
McLaughlin J, Han G, Schalper KA, Carvajal-Hausdorf D, Pelekanou V, Rehman J, et al. Quantitative assessment of the heterogeneity of PD-L1 expression in non-small-cell lung cancer. JAMA Oncol. 2016;2:46–54.
Lu F-M, Yuan Z. PET/SPECT molecular imaging in clinical neuroscience: recent advances in the investigation of CNS diseases. Quant Imaging Med Surg. 2015;5:433–47.
Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563.
Powles T, Paul Eder J, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. 2014.
Kowanetz M, Zou W, Gettinger SN, Koeppen H, Kockx M, Schmid P, et al. Differential regulation of PD-L1 expression by immune and tumor cells in NSCLC and the response to treatment with atezolizumab (anti-PD-L1). Proc Natl Acad Sci USA. 2018;115:E10119–26.
Davis AA, Patel VG. The role of PD-L1 expression as a predictive biomarker: an analysis of all US food and drug administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer. 2019;7:278.
Zhao J, Chen J, Ma S, Liu Q, Huang L, Chen X, et al. Recent developments in multimodality fluorescence imaging probes. Acta Pharmaceutica Sinica B. 2018;8:320–38.
Heskamp S, Hobo W, Molkenboer-Kuenen JDM, Olive D, Oyen WJG, Dolstra H, et al. Noninvasive imaging of tumor PD-L1 expression using radiolabeled Anti-PD-L1 antibodies. Can Res. 2015;75:2928–36.
Hettich M, Braun F, Niedermann G. Positron emission tomography imaging of the PD-1/PD-L1 checkpoint in Naïve and irradiated tumor-bearing mice. Int J Radiat Oncol Biol Phys. 2016;96:S128–9.
Josefsson A, Nedrow JR, Park S, Banerjee SR, Rittenbach A, Jammes F, et al. Imaging, biodistribution, and dosimetry of radionuclide-labeled PD-L1 antibody in an immunocompetent mouse model of breast cancer. Cancer Res. 2016;76:472–9.
Truillet C, Ling H, Oh J, Siok Yeo P, Lee C-Y, et al. Imaging PD-L1 expression with ImmunoPET. Bioconjugate Chem. 2018;29:96–103.
Bensch F, van der Veen EL, Lub-de Hooge MN, Jorritsma-Smit A, Boellaard R, Kok IC, et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat Med. 2018;24:1952–2858.
Niemeijer AN, Leung D, Huisman MC, Bahce I, Hoekstra OS, van Dongen GAMS, et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat Commun. 2018;9:1–5.
Sadiki A, Kercher EM, Lu H, Lang RT, Spring BQ, Zhou ZS. Site-specific bioconjugation and convergent click chemistry enhances antibody-chromophore conjugate binding efficiency. Photochem Photobiol. 2020. https://doi.org/10.1111/php.13231.
Strop P, Liu SH, Dorywalska M, Delaria K, Dushin RG, Tran TT, et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol. 2013;20:161–7.
Adumeau P, Sharma SK, Brent C, Zeglis BM. Site-specifically labeled immunoconjugates for molecular imaging-part 1: cysteine residues and glycans. Mol Imaging Biol. 2016;18:1–17.
van der Schoot JMS, Fennemann FL, Valente M, Dolen Y, Hagemans IM, Becker AMD, et al. Functional diversification of hybridoma-produced antibodies by CRISPR/HDR genomic engineering. Sci Adv. 2019;5:eaaw1822.
Ton-That H, Mazmanian SK, Faull KF, Schneewind O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus. Sortase catalyzed in vitro transpeptidation reaction using LPXTG peptide and NH2-GLY3 substrates. J Biol Chem. 2000;275:9876–81.
Popp MW, Antos JM, Grotenbreg GM, Spooner E, Ploegh HL. Sortagging: a versatile method for protein labeling. Nat Chem Biol. 2007;3:707–8.
Shawler DL, Bartholomew RM, Smith LM, Dillman RO. Human immune response to multiple injections of murine monoclonal IgG. J Immunol. 1985;135.
LoBuglio AF, Wheeler RH, Trang J, Haynes A, Rogers K, Harvey EB, et al. Mouse/human chimeric monoclonal antibody in man: Kinetics and immune response. Proc Natl Acad Sci USA. 1989;86:4220–4.
Klee GG, editor State of the art in clinical and anatomic pathology human anti-mouse antibodies. Arch Pathol Lab Med; 2000.
Khazaeli MB, Conry RM, LoBuglio AF. Human immune respone to monoclonal antibodies. J Immunother. 1994;15:42–52.
Tang Y, Wang J, Scollard DA, Mondal H, Holloway C, Kahn HJ, et al. Imaging of HER2/neu-positive BT-474 human breast cancer xenografts in athymic mice using 111In-trastuzumab (Herceptin) fab fragments. Nucl Med Biol. 2005;32:51–8.
Heskamp S, van Laarhoven HWM, Molkenboer-Kuenen JDM, Bouwman WH, van der Graaf WTA, Oyen WJG, et al. Optimization of IGF-1R SPECT/CT imaging using 111 In-labeled F(ab′) 2 and fab fragments of the monoclonal antibody R1507. Mol Pharm. 2012;9:120713080738005.
Chakravarty R, Goel S, Valdovinos HF, Hernandez R, Hong H, Nickles RJ, et al. Matching the decay half-life with the biological half-life: ImmunoPET imaging with44Sc-labeled Cetuximab Fab fragment. Bioconjug Chem. 2014;25:2197–204.
Shi S, Orbay H, Yang Y, Graves SA, Nayak TR, Hong H, et al. PET imaging of abdominal aortic aneurysm with 64Cu-labeled anti-CD105 antibody fab fragment. J Nucl Med. 2015;56:927–32.
Obonai T, Fuchigami H, Furuya F, Kozuka N, Yasunaga M, Matsumura Y. Tumour imaging by the detection of fibrin clots in tumour stroma using an anti-fibrin Fab fragment. Sci Rep. 2016;6:1–10.
Covell DG, Barbet J, Holton OD, Black CD, Parker RJ, Weinstein JN. Pharmacokinetics of monoclonal immunoglobulin G1, F(ab’)2, and Fab’ in mice. Can Res. 1986;46:3969–78.
Schmidt MM, Wittrup KD. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol Cancer Ther. 2009;8:2861–71.
Xenaki KT, Oliveira S, van Bergen en Henegouwen PMP. Antibody or antibody fragments: Implications for molecular imaging and targeted therapy of solid tumors. Front Immunol. 2017;8.
Selis F, Focà G, Sandomenico A, Marra C, Mauro CD, Jotti GS, et al. Pegylated trastuzumab fragments acquire an increased in vivo stability but show a largely reduced affinity for the target antigen. Int J Mol Sci.
Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev. 2002;54:459–76.
Chapman AP. PEGylated antibodies and antibody fragments for improved therapy: a review. Adv Drug Deliv Rev. 2002;54:531–45.
Gaberc-Porekar V, Zore I, Podobnik B, Menart V. Obstacles and pitfalls in the PEGylation of therapeutic proteins. Curr Opin Drug Discov Devel. 2008;11:242–50.
Hofström C, Orlova A, Altai M, Wangsell F, Gräslund T, Tolmachev V. Use of a HEHEHE purification tag instead of a hexahistidine tag improves biodistribution of affibody molecules site-specifically labeled with 99mTc, 111In, and 125I. J Med Chem. 2011;54:3817–26.
Liver uptake of radiolabeled targeting proteins and peptides: Considerations for targeting peptide conjugate design, (2012).
Adumeau P, Sharma SK, Brent C, Zeglis BM. Site-specifically labeled immunoconjugates for molecular imaging—part 1: cysteine residues and glycans. Mol Imag Biol. 2016;18:1–17.
Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–32.
Leelawattanachai J, Kwon K-W, Michael P, Ting R, Kim J-Y, Jin MM. Side-by-Side comparison of commonly used biomolecules that differ in size and affinity on tumor uptake and internalization. 2015.
Zhou Y, Goenaga A-L, Harms BD, Zou H, Lou J, Conrad F, et al. Impact of intrinsic affinity on functional binding and biological activity of EGFR antibodies. Mol Cancer Ther. 2012;11:1467–76.
Humphreys DP, Heywood SP, Henry A, Ait-Lhadj L, Antoniw P, Palframan R, et al. Alternative antibody Fab’ fragment PEGylation strategies: combination of strong reducing agents, disruption of the interchain disulphide bond and disulphide engineering. Protein Eng Des Sel. 2007;20:227–34.
Balan S, Choi JW, Godwin A, Teo I, Laborde CM, Heidelberger S, et al. Site-specific PEGylation of protein bisulfide bonds using a three-carbon bridge. Bioconjug Chem. 2007;18:61–76.
Pan H, Liu J, Deng W, Xing J, Li Q, Wang Z. Site-specific PEGylation of an anti-CEA/CD3 bispecific antibody improves its antitumor efficacy. Int J Nanomed. 2018;13:3189–201.
Kubetzko S, Sarkar CA, Plückthun A. Protein PEGylation decreases observed target association rates via a dual blocking mechanisms. Mol Pharmacol. 2005;68:1439–54.
Yokota T, Milenic DE, Whitlow M, Schlom J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 1992;52.
Nugent LJ, Jain RK. Extravascular diffusion in normal and neoplastic tissues. Cancer Res. 1984;44.
Wierstra P, Sandker G, Aarntzen E, Gotthardt M, Adema G, Bussink J, et al. Tracers for non-invasive radionuclide imaging of immune checkpoint expression in cancer. EJNMMI Radiopharm Chem. 2019;4.
Broos K, Lecocq Q, Raes G, Devoogdt N, Keyaerts M, Breckpot K. Noninvasive imaging of the PD-1:PD-L1 immune checkpoint: embracing nuclear medicine for the benefit of personalized immunotherapy. Theranostics. 2018;8(13):3559–70.
Smit J, Borm FJ, Niemeijer A-LN, Huisman MC, Hoekstra OS, Boellaard R, et al. PD-L1 PET/CT imaging with radiolabeled durvalumab in patients with advanced stage non-small cell lung cancer. J Nucl Med. 2021. https://doi.org/10.2967/jnumed.121.262473.
Niemeijer A-LN, Oprea Lager DE, Huisman MC, Hoekstra OS, Boellaard R, van de Veen B, et al. First-in-human study of 89 Zr-pembrolizumab PET/CT in patients with advanced stage non-small-cell lung cancer. J Nucl Med. 2021. https://doi.org/10.2967/jnumed.121.261926.
Brom M, Joosten L, Oyen WJG, Gotthardt M, Boerman OC. Improved labelling of DTPA- and DOTAconjugated peptides and antibodies with 111In in HEPES and MES buffer. EJNMMI Res. 2012;2:1–11.
Funding
C.F. is recipient of the NWO Spinoza award and ERC Adv. Grants ARTimmune (834618) and Pathfinder (269019). M.V. is recipient of ERC Starting Grant CHEMCHECK (679921) and a Gravity Program Institute for Chemical Immunology tenure track Grant by NWO. S.H. is recipient of a NWO Veni award (91617039) and Dutch Cancer Society Grant (10099).
Author information
Authors and Affiliations
Contributions
IMH, PJW, SH and MV conceived the study. IH, KS and MV designed and developed the imaging peptides. FS, MV, JMSS designed and developed the antibody formats. IH, PJW, KS, JDMM-K, DD, MB, JS, OI designed and performed the methods for synthesis and analysis. IH and PJW prepared the first draft of manuscript. SH and MV contributed to the writing of the manuscript. All authors reviewed and agreed on the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal experiments were performed in accordance with the revised Dutch Act on Animal Experimentation (2014) approved by the central authority for scientific procedures on animals and animal welfare body of the Radboud University, Nijmegen.
Consent for publication
Not applicable.
Competing interests
The authors have declared that no competing interest exists.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: “the figures are added”
Supplementary Information
Additional file 1.
Supplementary figures and tables.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hagemans, I.M., Wierstra, P.J., Steuten, K. et al. Multiscale imaging of therapeutic anti-PD-L1 antibody localization using molecularly defined imaging agents. J Nanobiotechnol 20, 64 (2022). https://doi.org/10.1186/s12951-022-01272-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12951-022-01272-5