
Abstract
Aβ42 is one of the most extensively studied blood and Cerebrospinal fluid (CSF) biomarkers for the diagnosis of symptomatic and prodromal Alzheimer’s disease (AD). Because of the heterogeneity and transient nature of Aβ42 oligomers (Aβ42Os), the development of technologies for dynamically detecting changes in the blood or CSF levels of Aβ42 monomers (Aβ42Ms) and Aβ42Os is essential for the accurate diagnosis of AD. The currently commonly used Aβ42 ELISA test kits usually mis-detected the elevated Aβ42Os, leading to incomplete analysis and underestimation of soluble Aβ42, resulting in a comprised performance in AD diagnosis. Herein, we developed a dual-target lateral flow immunoassay (dLFI) using anti-Aβ42 monoclonal antibodies 1F12 and 2C6 for the rapid and point-of-care detection of Aβ42Ms and Aβ42Os in blood samples within 30 min for AD diagnosis. By naked eye observation, the visual detection limit of Aβ42Ms or/and Aβ42Os in dLFI was 154 pg/mL. The test results for dLFI were similar to those observed in the enzyme-linked immunosorbent assay (ELISA). Therefore, this paper-based dLFI provides a practical and rapid method for the on-site detection of two biomarkers in blood or CSF samples without the need for additional expertise or equipment.
Graphical Abstract

Similar content being viewed by others
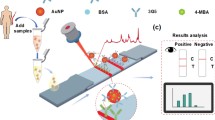

Introduction
Alzheimer's disease (AD) is a chronic neurodegenerative disorder, which may begin to develop 20–30 years before clinical onset, accompanied by progressive neuropathology, brain atrophy, and ultimately lead to cognitive decline [1,2,3]. By 2050, the number of AD worldwide will increase from the estimated 50 million reported in 2018 to 152 million [4]. Current treatment strategies for AD are limited to symptom relief [5, 6]. There is an urgent need to develop practical diagnostic tools and conduct large-scale early prevention and screening of high-risk groups to solve the public health crisis caused by AD. Presently, the diagnosis of AD is still mainly based on clinical cognitive assessment and physical examination. Histopathological examination of brain autopsy specimens is the only way to diagnose definite AD. Although the exact mechanism of AD pathogenesis remains elusive, amyloid β (Aβ) has been proposed to be an essential biomarker and therapeutic target for AD [7,8,9]. To date, magnetic resonance imaging (MRI) for measuring brain volume and neuronal connections, and Aβ- or tau-based positron-emission tomography (PET) for detecting pathological protein deposition in the brain have been used in AD diagnosis [10,11,12,13]. Although MRI and PET imaging tools with good diagnostic performances of brain diseases are approved for clinical use, the economic burden of imaging hinders their wide application in the identification of AD [14]. In addition, limited MRI or PET equipment is difficult to meet the growing number of AD patients. Expect for imaging diagnosis tools, it has also developed the detection of cerebrospinal fluid (CSF) biomarkers, such Aβ1-40 (Aβ40), Aβ1-42 (Aβ42), and the phosphorylation of tau (p-Tau) for AD diagnosis. Among them, the Aβ-based ELISA methods have been used as a reference for clinical diagnosis [15,16,17].
Aβ is a 38–43 amino acid polypeptide derived from amyloid precursor protein that is continuously cleaved by β- and γ-secretase. The predominant Aβ subtypes are Aβ40 and Aβ42, of which Aβ42 is easier to form fibrils and has a stronger tendency to aggregate. Increasing evidence has implicated that soluble Aβ42 oligomers (Aβ42Os) are the most critical toxic species causing AD-related synapse/neuron loss [18, 19] and memory decline [20]. Aβ42 monomers (Aβ42Ms) can modulate the Aβ self-assembly process to maintain intracellular signal transduction to achieve synaptic plasticity and homeostasis [21]. Therefore, both Aβ42Os and Aβ42Ms are related to the pathogenesis of AD. Abnormal Aβ42 levels can be detected in CSF and amyloid-β PET neuroimaging (amyloid-PET) [22,23,24]. However, there are some limitations, such as high testing costs, limited equipment, and invasiveness [25]. Compared with CSF measurements that may cause lumbar puncture and back pain, blood-related diagnosis tools are more acceptable due to their ease of collection, less invasiveness, and low cost. Multiple evidence shows that Aβ appearing in human blood [26,27,28] may be transmitted by peripheral monocytes [29], axonal [30, 31], exosomes [32, 33] from the brain over the blood–brain barrier. Furthermore, changes in the increased β-sheet structure of Aβ in human blood are associated with AD biomarkers in cerebrospinal fluid (CSF) and amyloid-PET [27]. Dynamic monitoring of Aβ42Ms and Aβ42Os levels in the blood helps to accurately diagnose AD before clinical symptoms appear.
Many attempts have been made to explore new diagnostic tools for several core biomarkers, including enzyme-linked immunosorbent assay (ELISA) [34,35,36], nanoparticle-based immunoassays [37], electrochemistry [38], surface-enhanced Raman spectroscopy [39, 40], fluorescence [41], and electrochemical biosensors [42, 43]. However, due to the heterogeneity and transient nature of Aβ oligomers, most of these assays not only cannot distinguish between Aβ42Os and Aβ42Ms but also require equipment or/and expertise. The development of technologies that can dynamically detect changes in the levels of Aβ42Ms and Aβ42Os in the blood is essential for the accurate diagnosis of AD.
Paper-based lateral flow immunoassay (LFI) has been widely used for the rapid diagnosis of blood biomarkers due to its simplicity, portability, cost-effectiveness, and rapid detection of target biomarkers [44,45,46,47]. However, the accuracy and sensitivity of LFIS to a single biomarker still need to be improved, especially in the face of complicated pathological diseases such as AD with multiple biomarkers, including Aβ42Ms, Aβ42Os, p-Tau181, and p-Tau217. Currently, diagnostic tools for AD mainly target one biomarker, such as Aβ, p-Tau, or total tau, with compromised sensitivity and specificity. Previous studies reported that the average specificities of AD diagnostic methods based on Aβ, t-tau, or p-tau are 84.1%, 82.3%, and 83%, while their average sensitivities were 79.4%, 80.5%, and 78.3% [48,49,50]. However, the average specificity and sensitivity of multi-biomarker-based detection methods are over 86% and 83.5%, which are relatively higher than those based on a single biomarker Aβ, p-Tau, or total tau [48,49,50]. Recently, we screened the preferred antibody pairs 1F12/1F12 and 1F12/2C6 to achieve accurate detection of Aβ42Ms and Aβ42Os. The strategy used in our previous study for the detection of oligomeric and monomeric Aβ was to use the antibody 1F12 with a single epitope to capture Aβ42Ms and Aβ42Os and then incubated with the detection antibody 1F12 with the same epitope for detecting only oligomers because oligomers were aggregated by multiple monomers, exposing several identical epitopes that can be recognized by the same detection antibody. While for Aβ42Ms, they were only detected by the antibody 2C6 with different epitopes from the capture antibody 1F12 to form a sandwich structure [51]. In addition, our analysis of the levels of Aβ42Ms and Aβ42Os in brain and blood samples was closely related to the progression of AD, indicating that simultaneous detection of Aβ42Ms and Aβ42Os may improve the specificity and sensitivity of AD detection [51]. Given that, we look forward to developing a test strip that can accurately detect the levels of Aβ42Ms and Aβ42Os in the blood to achieve a rapid and accurate diagnosis of AD. In this study, we developed a dual-target lateral flow immunoassay (dLFI) by adopting Aβ42-specific monoclonal antibodies 1F12 and 2C6 with unique/overlapping epitopes to create steric hindrance between antibody capture and detection. Herein, 1F12 was labeled with gold nanoparticles (1F12-AuNP) as the capture antibody. Then, 1F12 and 2C6 were used as detection antibodies to be immobilized on nitrocellulose (NC) membrane as two test lines, and goat anti-mouse IgG was immobilized on one end of the NC membranes as a control line. The clinical manifestations of dLFI were tested in blood samples of 5xFAD mice and AD patients. We aimed to develop a multi-objective LFI for rapid and high-performance diagnosis of AD.
Materials and methods
Materials
Aβ42 (DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA), Aβ40 (DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV), P-Tau396,404 (RENAKAKTDHGAEIVYK-[Ser(P)]PVVSGDT[Ser(P)]PRHL), Cis-Tau (KVAVVRpT(5,5-dimethyl-l-proline)PKSPS), and P-Tau231 (KVAVVRpTAPKSPS) were custom-synthesized as lyophilized powders by Royo Biotech Co., Ltd (Shanghai, China) with a purity of > 95%. The detailed information of synthesized peptides, including HPLC and mass spectrometry results, are shown in Additional file 1: Fig. S1–3. The Aβ42-specific monoclonal antibodies (mAbs) 1F12 and 2C6 were produced in our laboratory, of which the epitope of 1F12 is Aβ3–9, while the epitopes of 2C6 are Aβ3–9, Aβ13–19, Aβ18–25, Aβ29–36, and Aβ36–42 [51]. Trisodium citrate dehydrates (HAuCl4) and bovine serum albumin (BSA) were purchased from Sigma Aldrich (St. Louis, MO, USA). NaCl, polyvinyl pyrrolidone (PVP), K-40, sucrose, Casein, NaN3, PEG, Tris, MES, and Tween-20 were obtained from Beijing Biotopped Science and Technology Co., Ltd. (Beijing, China). Filter paper and semi-rigid polyvinyl chloride (PVC) sheets were purchased from Jieyi Biological Technology Co., Ltd. (Shanghai, China). Glass fibre membranes and nitrocellulose membranes were obtained from Millipore (Billerica, MA, USA). Goat anti-mouse IgG and protein A resins were purchased from GenScript (Nanjing, China). N-Hydroxysuccinimide (NHS) modified magnetic nanoparticles were obtained from LinkedIn Biotechnology Co., Ltd. (Shanghai, China).
Preparation of gold nanoparticles (AuNP)
AuNP was synthesized following the HAuCl4 reduction scheme with citric acid [52]. Briefly, 99 mL of ultrapure water was added to a purified bottle, and 1 mL of 1% HAuCl4·3H2O was then added with continuous stirring and heating. The solution was heated until the solution started to boil slightly, and 1 mL of filtered 1% trisodium citrate solution was then added. After heating for a few minutes, the solution gradually changed from colorless to grey, black, purple, and red. The heating was then stopped, and the solution was continuously shaken slowly until it cooled.
Preparation of 1F12-conjugated AuNP and magnetic nanoparticles
AuNP were synthesized using the citrate reduction as per the HAuCl4 protocol [52]. First, 1F12 antibody was purified with protein A immunoaffinity column, and conjugated to AuNP according to the following steps: (1) 1 mL of AuNP was added to a 1.5 mL centrifuge tube, followed by the addition of 16 μL of 0.1 M K2CO3 with thorough mixing; (2) 25 μg of 1F12 was added to the mixture for 15 min, keeping it undisturbed for another 15 min at 25°C; (3) 10 μL of 10% BSA was added to the mixture with shaking for 15 min and the mixture was then kept undisturbed for 15 min at 25°C to achieve a sealing effect on the surface of AuNP; (4) the mixture was centrifuged at 13,500 rpm for 10 min, and the precipitate was collected to resuspend in 1 mL of PBS (0.01 M, pH 7.4, including 0.03% Tris, 2% Sucrose, 0.2% Casein, 1% BSA, 0.1% PVP, 1% NaN3, 0.1% PEG, 0.05% Tween-20) and stored at 4°C for further use.
Further, 1F12-conjugated magnetic nanoparticles (1F12-MNPs) were prepared by making the amino group of 1F12 react with the carboxyl group of MNPs, as we previously described [53]. Briefly, 1 mL MNPs (10 mg) was washed twice with 1 mL deionized water followed to wash with 4-Morpholineethanesulfonic acid (MES) buffer (0.02 M MES, pH5.0). Then, the MNPs were resuspended in 200 μL of MES buffer and then 300 μg of 1F12 (200 μL) was added for 30 min reaction at 25°C in shaking. Next, 100 μL of freshly prepared EDC-HCl buffer (52.2 mM, dissolved in 0.02 M MES, pH5.0) was added to react in shaking overnight, followed to be blocked with blocking buffer (1% BSA dissolved in 0.02 M MES, pH5.0). Finally, the prepared 1F12-MNPs were stored in the storage buffer (PBS containing 0.1% Tween-20 and 0.02% NaN3, pH 7.4) at 4°C for further use. The prepared antibody-modified MNPs were evaluated via reduced tris-tricine SDS–polyacrylamide gel electrophoresis (SDS-PAGE), ELISA, and immunoprecipitation followed with Western blotting (IP-Western blotting).
Characterization of AuNP, MNP, Aβ42Ms, and Aβ42Os
The prepared AuNP, magnetic nanoparticle (MNP), Aβ42Ms, and Aβ42Os were confirmed by transmission electron microscopy (TEM) and scanning electron microscopy (SEM), respectively. In short, a small drop of samples (5 μL) was deposited onto a copper grid, and the excess liquid was removed by blotting using a filter paper, thus leaving a thin film of the solution on the grid. Subsequently, Tecnai G20 transmission electron microscope (FEI Ltd., USA) and Phenom Pharos scanning electron microscope (SEM) (Phenom Ltd., NLD) were used to characterize the morphology of the abovementioned samples. The particle size distribution and Zeta-potential of all nanoparticles were measured via dynamic light scattering (photon correlation spectroscopy) on a Zetasizer Nano-ZS90 system (Malvern Instruments, Worcestershire, UK).
Immunoprecipitation and Western blotting
The prepared Aβ42 samples, including Aβ42Ms and Aβ42Os, were incubated with 1F12-MNPs for 30 min at 25°C, and then the enriched Aβ42 was eluted with 0.1 M glycine (pH 3.0) and denatured in loading buffer (Boster Biotech, USA) for 10 min at 95°C, followed to run on a 12% reducing SDS-PAGE. The proteins were transferred onto a polyvinylidene fluoride membrane at 160 mA for 1 h. The membrane was blocked with 5% skimmed milk dissolved in PBS-T buffer, and then incubated with the primary antibody (1F12 or 2C6) and the secondary antibody HRP-conjugated goat anti-mouse IgG (H + L) in an order. The membrane was washed three times with PBS-T buffer (KCl 2.7 mM, KH2PO4 2 mM, NaCl 137 mM, Na2HPO4 10 mM, 0.05% Tween-20, pH 7.4) and the immunological signals were detected using the ECL-substrate (Vazyme, China) on Tanon 5200 Muiti (Shanghai, China).
ELISA
For indirect ELISA, the wells of a 96-well plate (Corning Inc., USA) were coated with 0.5 μg/well of Aβ42 overnight at 4°C. The plate was blocked with 5% skimmed milk, and then incubated with primary antibodies (1F12, AuNP-1F12, or 1F12-MNPs) and the secondary antibody HRP-conjugated goat anti-mouse IgG (H + L) in an order. The 96-well plate was washed four times with PBS-T in each step. The immunoreaction was visualized by TMB substrate solution (Tiangen Biotech, Beijing, China) and detected with an Epoch Microplate Spectrophotometer (Bio Tek, USA) at 450 nm.
For competitive ELISA, 96-well plates were coated with 0.5 μg Aβ42 in each well and blocked with 5% skimmed milk. Triplicates of biotinylated 1F12 or 2C6 (250 ng/mL) were mixed with serially diluted Aβ preparations of Aβ40, Aβ42Ms, and Aβ42Os diluted in PBS with the final concentration from 25 μM to 5 pM. After 1 h pre-incubation at 4°C in 1.5 mL tubes, the antibody-antigen mixtures were incubated on the Aβ42 antigen-coated plates for 1 h at 25°C. After incubating with streptavidin-coupled poly-HRP, the immunoreaction was visualized and detected as above described.
Immunofluorescence assay
The 15-μm coronal frozen sections of the brain tissue samples were permeabilized with 0.2% Triton X-100 for 20 min at 25°C, then blocked with 3% bovine serum albumin for 2 h, followed to incubate with Cy3-conjugated mAb 1F12, 2C6 and Iba 1 for overnight at 4°C. Besides, the slides were counterstained with thioflavin S and 6-diamino-2-phenylindole (DAPI; Thermo Fisher Scientific). The fluorescence signals were detected by Zeiss LSM710 confocal microscope.
Optimization of key parameters
To keep the dLFI with the optical working condition, we detail to optimize the volume of K2CO3 and the mAb 1F12 concentration of conjugation. For the volume of K2CO3 optimization, the 1 mL of the AuNP solution was adjusted to 4, 8, 12, 16, 20, 24, 28, and 32 μL of 0.1 M K2CO3. While for mAb 1F12 concentration optimization, the mAb 1F12 was added dropwise to 1 mL of AuNP solution at final concentrations of 5, 10, 15, 20, 25, and 30 μg/mL.
Preparation of dLFI
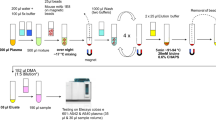
Scheme 1b describes the main components of a one-piece dLFI, which mainly includes sample pads, conjugation pads, signal pads, and absorbent pads assembled on a plastic backplane in sequence. The sample pad was impregnated with a buffer solution (0.01 M PBS, pH 7.4, including 1% BSA, 2% Sucrose, 0.1% PEG, and 0.05% Tween-20) and air-dried overnight before use. The goat anti-mouse IgG antibody, 1F12, and 2C6 were coated on the NC membrane to form a control line and two test lines. The coated NC membrane was dried for 30 min and blocked with 1% (w/v) BSA dissolved in 0.01 M PBS for 2 h at 25°C. After blocking, the membrane was washed three times with PBS containing 0.05% [v/v] Tween-20 (PBS-T) and dried at 25°C. Following this, the dLFI was assembled in the following order: the sample pad was first placed on the PVC plate, overlapping the anti-Aβ42 mAb 1F12 conjugate pad by 2 mm, and the bottom of the antibody-coated NC membrane was then overlapped by 2 mm. Next, the absorbent pad was overlapped on the top of the NC membrane by 2 mm. After assembly, the plate was evenly cut into 3.5 mm wide strips, sealed with a desiccant, and stored at 25°C. The concentration of 1F12 coated on the first test line and 2C6 coated on the second test line was 1 mg/mL. The concentration of goat anti-mouse IgG antibody coated on the control line was 0.5 mg/mL.

The principle and test procedure of dLFI. a The blood samples were enriched by the mAb 1F12-modified MNPs and then eluted for dLFI analysis. Schematic representation of the working principle of the dLFI for the detection of Aβ42Os (b), Aβ42Ms (c), Aβ42Ms and Aβ42Os (d), or without Aβ42Ms or/and Aβ42Os (e)
Detection principle and test procedure of dLFI
Our multicomponent biosensor system developed for rapidly and simultaneously detecting Aβ42Ms and Aβ42Os was based on two groups of paired mAbs. Due to Aβ42Os was aggregated by several Aβ42Ms, we used the group of 1F12/1F12 with the same epitope that ensures only Aβ42Os was detected but not Aβ42Ms, which need a pair of antibodies with different epitopes just liking our described as the group of 1F12/2C6. The following steps were included in dLFI assay process: the antibody-modified MNPs were used for the enrichment of Aβ peptides dissolved in the samples and then eluted with 0.1 M glycine (pH 3.0), followed by immediate neutralization to pH 7.4 using a neutralization buffer (1 M Tris–HCl, pH 8.5). Each collected solution (approximately 50 μL) was dripped onto the sample pad. Aβ42Ms and/or Aβ42Os in samples were first recognized by 1F12–AuNP conjugate mixture on the conjugate pad and migrated along the NC membrane via capillary action. The Aβ42Os–1F12–AuNP complex was first solubilized by 1F12 coated on the NC membrane to form the test line 1, whereas Aβ42Ms–1F12–AuNP complex continued to migrate until it was solubilized by mAb 2C6 coated on the NC membrane to form the test line 2. The remaining 1F12–AuNP were captured by goat anti-mouse IgG coated on the NC membrane to form the control line. The test results were evaluated within 5 min.
Preparations of oligomeric and monomeric Aβ
The detailed steps of oligomeric and monomeric Aβ preparations were described in our previous report [51]. In brief, Aβ42Ms and Aβ40Ms were obtained by dissolving lyophilized Aβ42 or Aβ40 peptides in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) for overnight incubation at 25°C. HFIP was evaporated with nitrogen to form a thin film, and Aβ was re-dissolved in dimethyl sulfoxide. Aβ42 oligomers (Aβ42Os) and Aβ40 oligomers (Aβ40Os) were obtained from 50 μM Aβ42 or Aβ40 monomer solution after 24 h incubation at 37°C in the dark, respectively.
Specificity and sensitivity of the dLFI
Several different compounds such as Aβ40Ms, Aβ40Os, p-Tau396,404, Cis-Tau, p-Tau231, and BSA were detected to evaluate the specificity of the dLFI. For sensitivity evaluation, a series of synthetic Aβ42Ms and Aβ42Os dilutions, ranging from 625 ng/mL to 154 pg/mL, were prepared by diluting with PBS. Each dilution was applied to the dLFI test, and the detection limit was determined. The visual limit of detection is defined as the minimum concentration of Aβ42Ms and Aβ42Os that leads to the complete disappearance of the test line.
Analysis of blood samples using dLFI
All procedures involving animal studies have been reviewed and approved by the Institutional Animal Care and Use Committee of Huazhong University of Science and Technology. Human blood samples were obtained from the patients/participants in Union Hospital of Huazhong University of Science and Technology who have been provided with their written informed consent to participate in this study.
The blood samples of 5xFAD mice (Stock No: 34848-JAX) aged 3 months (n = 4) and 9 months (n = 4) were collected and used to evaluate the analytical performance and applicability of the dLFI. A mixture of Aβ42Ms and Aβ42Os was used as a positive control, while blood samples from C57BL/6 J mice at 3 months (n = 3) and 9 months old (n = 3) were used as negative controls. In addition, blood samples collected from HC (n = 7) and AD patients (n = 8) were analyzed with dLFI. The detailed information of the participant in this manuscript is summarized in Additional file 1: Table S1.
Validation and analysis with sandwich ELISA
The blood samples of 5xFAD or C57BL/6 J mice at 3 months (n = 4) and 9 months of age (n = 4), and HC (n = 7) and AD patients (n = 8) were analyzed via a prepared sandwich ELISA for Aβ42 monomers and Aβ42 oligomers as our previous described [51].
Statistical analyses
The data are presented as means ± SD. Unpaired t-test was used for two-group comparisons. One-way analysis of variance (ANOVA) was used for multigroup comparisons. Statistical significance is represented in the figure by *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and n.s. (indicating no significance). All statistical analyses were performed with GraphPad Prism 8.0 software.
Results and discussion
Mechanism of the proposed biosensor
In this study, we combined the biomarker measurement of Aβ42Ms and Aβ42Os to obtain a more accurate diagnosis of AD. To further verify the results, we developed dLFI using two matched antibody pairs 1F12/1F12 and 1F12/2C6 to obtain a rapid on-site response and accurate detection tests and to determine the changes of Aβ42Ms and Aβ42Os in the blood for clinical validation of AD diagnosis. As shown in Scheme 1a, the blood samples were firstly enriched with MNPs modified by mAb 1F12, and then the Aβ42Ms or/and Aβ42Os were eluted for LFI analysis. The eluted Aβ42Ms or Aβ42Os were added to the sample pad and then recognized by the 1F12–AuNP conjugate mixture on the conjugate pad and migrated along the NC membrane via capillary action. The Aβ42Os–1F12–AuNP complex was first fixed by 1F12 coated on the NC membrane to form the test line 1, due to Aβ42Os were aggregated by multiple monomers, exposing several same epitopes that could be recognized by the same detection antibody (Scheme 1b). This strategy is only used for the detection of Aβ42Os. The Aβ42Ms–1F12–AuNP complex continued to migrate and was solubilized by mAb 2C6 with different epitopes toward Aβ42 (specific to amino acids 3–9, 13–19, 18–25, 29–36, and 36–42 of Aβ42 sequence). Compared to 1F12 (specific to amino acids 3–9), Aβ42Ms were only detected by a pair of antibodies with different epitopes (Scheme 1c). If Aβ42Ms and Aβ42Os are present in the sample, the 1F12–AuNP conjugates will be combined with the two test lines and the control line (Scheme 1d). If there is no Aβ42 in the sample solution, the 1F12–AuNP conjugates will not bind to the two test lines but will show the control line, forming a red band (Scheme 1e).
Characterization of mAb 1F12 and 2C6
We first evaluated whether the two antibodies could stain Aβ plaques in the brains of 5xFAD mice. The results of confocal fluorescence images showed that Aβ plaques were stained by Cy3-labeled 1F12 or 2C6 and co-localized with thioflavin S, and its functional characteristic was to bind Aβ plaques (Fig. 1a) [54,55,56]. The binding selectivities of 1F12 and 2C6 for different Aβ species were detected by competitive ELISA. The morphology of the prepared Aβ42Ms and Aβ42Os was first confirmed by TEM. The results showed that the morphology of Aβ42Ms was coil structures (Fig. 1b, left), while Aβ42Os presented a β-sheet and typical fibril structure (Fig. 1b, right). A 12% reduced SDS-PAGE gel was used to confirm the molecular weight and components of the prepared Aβ42Ms and Aβ42Os (Fig. 1c). Western blot results showed that both Aβ42Ms and Aβ42Os were well recognized by 1F12 or 2C6 (Fig. 1d). Competitive ELISA was used to evaluate the binding selectivity of 1F12 and 2C6 to different Aβ species. Figures 1e and f show that 1F12 and 2C6 have high binding selectivity to Aβ42 species rather than Aβ40. The IC50 values of 1F12 for Aβ42Ms and Aβ42Os were 180.8 nM and 18.1 nM, respectively. While for 2C6, the IC50 values for Aβ42Ms and Aβ42Os were 90.3 nM and 7.5 nM, respectively (Fig. 1g). The IC50 values plus the Kd values of 1F12 (Kd = 1.66 ± 0.09 nM for Aβ42Ms and Kd = 0.38 ± 0.04 nM for Aβ42Os) and 2C6 (Kd = 3.59 ± 0.27 nM for Aβ42Ms and Kd = 0.61 ± 0.03 nM for Aβ42Os) that were reported in our previous study indicated [51] that 1F12 and 2C6 have high binding affinity and selectivity for both Aβ42Ms and Aβ42Os.

Characterization of conformation-specific antibodies for Aβ42Ms and Aβ42Os. a Confocal fluorescence images of mouse 5xFAD brain sections using Cy3-labeled anti-Aβ42 monoclonal antibody 1F12 or 2C6 and thioflavin S. (Scale bar: 500 μm). b Parallel analysis of the morphology of Aβ42Ms and Aβ42Os by cryo-transmission electron microscopy. (Scale bar: 500 nm). The SDS-PAGE (c) and Western blotting (d) analysis of the prepared Aβ42Ms and Aβ42Os using 1F12 or 2C6. The binding selectivity of 1F12 (e) and 2C6 (f) to Aβ42 and Aβ40 was determined by competitive ELISA. g The logIC50 value of 1F12 or 2C6 to Aβ42 and Aβ40. The data are presented as means ± SD, n = 3 in e and f. One-way analysis of variance (ANOVA) was used for multigroup comparisons. Statistical significance is indicated in the figures by ****p < 0.0001
Characterization of AuNP and AuNP-1F12 conjugates
The synthesized AuNP had good dispersion and uniformity with wine red, and a characteristic single absorption peak was found at 525 nm (Additional file 1: Fig. S4a). The morphology and size of prepared AuNP were characterized using a Tecnai G20 transmission electron microscope and dynamic light scattering with an average size of 40 nm (FEI Ltd., USA) (Fig. 2a and b). After being conjugated with 1F12, the size distribution and ζ-potential of AuNP–1F12 increased significantly with the size from 40 nm (before conjugation) to approximately 80 nm (after conjugation) (Fig. 2b) and ζ-potential from − 38 (before conjugation) to approximately − 23 (after conjugation) (Additional file 1: Fig. S4b). Besides, the absorption spectrum of 1F12 was measured, and the results showed a sharp decrease in the absorption peak at 280 nm after conjugation with AuNP (Additional file 1: Fig. S4c). Coupled with the results of the 12% reduction SDS-PAGE gel, they jointly confirmed the successful combination of 1F12 to AuNP (Fig. 2c). The ELISA was used to evaluate the bioactivity of AuNP-1F12 conjugate and results showed that both 1F12 and AuNP-1F12 reacted well with Aβ42 (Fig. 2d).

Characterization of AuNP–1F12 conjugates. a TEM images of synthetic AuNP. (Scale bar: 200 nm). b The size distributions of AuNP before and after modification. c A 12% reduced SDS-PAGE gel analysis of AuNP–1F12 and 1F12. d ELISA was used to determine the bioactivity of AuNP–1F12 conjugates. The optimal volume of K2CO3 for the conjugation of 1F12 with AuNP was evaluated by the color (e) and OD525 value (g). The color (f) and OD525 value (h) were used to evaluate the optimal concentration of 1F12 of synthetic AuNP-1F12. For Fig. 3b, d, g, and h, the data are presented as mean ± SD, n = 3. Statistical significance is indicated in the figures by n.s. (indicating no significance)
Optimization of the dLFI
To improve the sensitivity, accuracy, and reproducibility of the dLFI test, the capture antibody 1F12–AuNP was optimized during the preparation process. We first optimized the volume of K2CO3, which is a commonly used buffer to adjust the pH of AuNPs solution to better bind the antibody. Figure 2e and g show the effect of K2CO3 volume on the 1F12–AuNP conjugates. As the volume of K2CO3 increased from 4 to 16 μL, the color, optical density (OD) value, and absorption spectrum at 525 nm of the 1F12–AuNP solution gradually increased, the color of the solution changed from lavender to wine red (Fig. 2e), and the OD525 value increased from 0.23 to 0.71 (Fig. 2g). With a further increase from 20 to 32 μL, the OD525 value (Fig. 2g) and the absorption spectrum (Fig. S4d) decreased slightly. To obtain a higher colorimetric ratio, 16 μL was selected as the optimal volume of K2CO3 of antibody coupling, and the pH value of the AuNPs solution was approximately 8.5. When the pH value is equal to or slightly high than the isoelectric point (pI) of the antibody (pI = 8), the antibody is electrically neutral, resulting in a small electrostatic interaction between the antibody and AuNPs, so that the antibody is more easily to adsorb on the surface of AuNPs [57,58,59]. When the pH value is less than the pI, the antibody is positively charged. Since the AuNP is negatively charged, the antibody is easily adsorbed to form large polymers, leading to the aggregation of AuNPs [57,58,59]. On the contrary, once the pH is higher than the pI, the antibody will be negatively charged and repel the negatively charged AuNPs, causing them to fail to bind to each other [57,58,59].
As shown in Fig. 2f and h, as the concentration of 1F12 increases from 5 to 25 μg/mL, the color gradually changes from purple to wine red (Fig. 2f), and the OD525 value (Fig. 2h) increases from 0.41 to 0.64. However, when the concentration of 1F12 was as high as 30 μg/mL, the OD525 value (Fig. 2h) and absorption spectrum (Additional file 1: Fig. S4e) only slightly increased. Therefore, the optimal concentration of 1F12 was 25 μg/mL. The significant change in the color of the AuNP solution during the antibody coupling process could be explained by the fact that the AuNPs cannot be fully labeled when no antibody is added or the amount of added antibody is insufficient. The unlabeled AuNPs were aggregated under the action of 10% NaCl. On the contrary, only when the amount of antibody reaches or slightly exceeds the stable amount, the AuNPs can remain stable [60, 61].
Specificity and sensitivity of the dLFI
The specificity of dLFI was evaluated among six compounds. As shown in Fig. 3a, dLFI can detect a mixture of Aβ42Ms and Aβ42Os, and does not cross-react with the tested peptides, indicating that dLFI based on 1F12/1F12 and 1F12/2C6 antibody pairs could accurately detect both Aβ42Os and Aβ42Ms (Fig. 3a and c). Therefore, dLFI can be used to effectively evaluate multiple analytes. Each test line in the strip has a cut-off value, which is the minimum Aβ42Ms or Aβ42Os concentration required to make the test line visible in samples. For Aβ42Ms or Aβ42Os, the cut-off value of the dLFI was 154 pg/mL (Fig. 3b). Different concentrations of Aβ42Ms and Aβ42Os were used in dLFI to generate two standard curves. The color intensity of the test line is directly proportional to the concentration of the analyte in samples. Figure 3d shows two standard curves of dLFI. The linear relationship of dLFI is y = 1.37972 + 0.32557x, and the LOD of Aβ42Os is 154 pg/mL (Fig. 3d, inserted Figure, red square). The linear relationship of dLFI is y = 0.80853 + 0.4771 × with LOD of 154 pg/mL for Aβ42Ms (Fig. 3d, inserted Figure, blue circle).

The performance of the dLFI. The visual result (a) and gray value (c) on the T line of the dLFI in the specificity assay, n = 3. b The sensitivity of dLFI for the simultaneous detection of Aβ42Ms and Aβ42Os. d The plotted linear curve of different Aβ42 conformations, n = 3. e, f Correlation between results from dLFI (Y-axis) and sandwich ELISA (X-axis) in spiked samples, n = 12. Data are presented as means ± SD. One-way analysis of variance (ANOVA) was used for multigroup comparisons. Statistical significance is represented in the figures by ****p < 0.0001 and n.s. (indicating no significance)
The reliability and practicability of the proposed dLFI
The reliability and practicability of the proposed dLFI sensor in spiked samples were verified and compared by sandwich ELISA. The spiked concentration conformed to the linear range. Therefore, the results (Fig. 3e and f) showed that the two methods were well consistent. The correlation coefficients of Aβ42Ms and Aβ42Os were 0.9969 and 0.9833, respectively. Although the sensitivity of dLFI is relatively low, the dLFI sensor requires less than 30 min of enrichment and immunoreaction time to complete sample analysis, while traditional sandwich ELISA requires about 3 h.
The performance of the dLFI in detecting Aβ42Os and Aβ42Ms in 5xFAD mice
To check the performance of the dLFI in the actual tests, blood samples of 5xFAD (AD model mice) or C57BL/6 J mice (control mice) at 3 and 9 months old were collected for dLFI detection. The mixture of Aβ42Ms and Aβ42Os was used as a positive control. The results showed that in the blood of C57BL/6 J mice at 3 and 9 months old, dLFI could hardly detect Aβ42Ms or Aβ42Os (Fig. 4a, left, black and blue box, and Fig. 4b-c). Interestingly, the dLFI results showed that Aβ42Ms were the main form of Aβ42 in 3-month-old 5xFAD mice (Fig. 4a, right, purple box, and Fig. 4b), but the results were different in 9-month-old 5xFAD mice. In 9-month-old 5xFAD mice, the color of Aβ42Os in the first test line was significantly enhanced, but Aβ42Ms were hardly observed in three of the four 5xFAD mice (Fig. 4a, right, red box, and Fig. 4c). Only one of the four 9-month-old 5xFAD mice showed high levels of Aβ42Ms and Aβ42Os. These results revealed that Aβ42Ms appeared in the peripheral blood of 5xFAD mice at an early stage [e.g., 3-month-old 5xFAD mice had less Aβ plaque load and Iba 1-positive cells staining (Fig. 4f, up)]; as the disease progresses, high levels of Aβ42Os and insoluble Aβ plaques were the main forms in the brain. As Aβ42Ms in the brain gradually aggregated into Aβ42Os, soluble Aβ42 could enter the blood, resulting in a decrease in Aβ42Ms level and an increase in Aβ42Os level [e.g., 9-month-old 5xFAD mice had more Aβ plaque load and Iba 1-positive cells staining (Fig. 4f, down)]. Of note, this elevated Aβ42Os level was closely related to the Aβ plaque area (Fig. 4d, p < 0.0001) and soluble Aβ42 level (Fig. 4e, p < 0.0001) in the brain of 5xFAD mice, indicating that its level may reflect the progress of the disease. The phenomenon observed in 5xFAD mice was that the accumulation of Aβ42Os is directly accompanied by the decrease of Aβ42Ms, consistent with the typical clinical symptoms of AD patients [62,63,64,65,66,67]. Altogether, our results showed that Aβ42Os or Aβ42Ms that are mis-detected by commonly used ELISA are valuable biomarkers for AD diagnosis when they are accurately distinguished and detected.

Simultaneous detection of Aβ42Ms and Aβ42Os in the blood of 3- and 9-month-old 5xFAD and C57BL/6 J mice. The visual results (a) and grey value (b, c) on the T-line of the dLFI in blood samples from 5xFAD (n = 8) and C57BL/6 J (n = 6) mice at 3 and 9 months old. d, e Correlation between blood Aβ42Os level and Aβ plaque area (d, p < 0.0001) or soluble Aβ42 level (e, p < 0.0001) in the brain of 5xFAD mice, a total of n = 25, of which n = 5 in each age group, including 1, 3, 6, 9, and 12 months old. f Confocal fluorescence images of Aβ plaques and Iba 1-positive cell staining in 3 or 9-month-old 5xFAD mice. (Scale bar: 100 μm). Data are presented as means ± SD. Unpaired t-test was used for two-group comparisons. Statistical significance is indicated in the figures by **p < 0.01, ***p < 0.001 and n.s. (indicating no significance)
The performance of the dLFI in detecting Aβ42Os and Aβ42Ms in AD patients
To further improve the sensitivity and accuracy of the dLFI in the analysis of human blood samples, immunocapture magnetic beads were used to enrich the Aβ42Os and Aβ42Ms in samples (Fig. 5a). The unmodified MNPs were characterized and found to be approximately 190 nm (Additional file 1: Fig. S5a). After conjugation with 1F12, compared with bared MNPs, the size and Zeta potential of MNPs showed significant changes (Additional file 1: Fig. S5b–d). Furthermore, a 12% reduced SDS-PAGE gel showed two typical bonds, including the light and heavy chains observed in the lanes of 1F12 and 1F12-MNPs, confirming the successful conjugation of 1F12 with MNPs (Fig. 5b). The biological activity of 1F12-MNPs was assessed by ELISA and IP-Western blotting. The results of ELISA (Fig. 5c) and IP-Western blotting (Fig. 5d) confirmed that 1F12-MNPs inherited the binding affinity of 1F12 for Aβ42Ms and Aβ42Os. After using dLFI to detect blood samples of HC (n = 7) and AD patients (n = 8), the results showed that the Aβ42Ms level of HC was significantly higher than that of AD patients (Fig. 6a, left, black box, and Fig. 6b, p = 0.034). In contrast, Aβ42Os were observed in AD patients, but not in HC (Fig. 6a, right, red box, and Fig. 6b, p = 0.039). In addition, a sandwich ELISA test was performed using the enriched blood samples to evaluate the levels of Aβ42Ms and Aβ42Os. As shown in Fig. 6c, compared with blood samples from HC, samples from AD patients showed a significant decrease in Aβ42Ms (p = 0.0265) and an increase in Aβ42Os (p = 0.086). This phenomenon is consistent with the clinical symptoms reported by multiple studies [28, 66, 68, 69].

Characterization of antibody-modified NHS-magnetic nanoparticles (MNPs). a Schematic illustration of the working steps for the use of antibody-modified MNPs for the enrichment of Aβ42Os and/or Aβ42Ms. b A 12% reduced SDS-PAGE gel analysis of 1F12-MNPs. 1F12 and NMNs were used as controls. ELISA (c) and IP-Western blotting (d) analysis for the bioactivity of 1F12-MNPs. Data are presented as means ± SD, n = 3 in c

Detection of Aβ42Ms and/or Aβ42Os in human blood samples by dLFI. The visual results (a) and grey value (b) of the Aβ42Ms and/or Aβ42Os level in the blood of AD patients (n = 8) or healthy controls (n = 7) were analyzed by dLFI. c The Aβ42Ms and/or Aβ42Os levels in the blood of AD patients (n = 8) or healthy controls (n = 7) were detected by sandwich ELISA. d Total Aβ42 levels in human blood samples were detected by sandwich ELISA (n = 8 for AD patients and n = 7 for healthy controls). Data are presented as means ± SD. Unpaired t-test was used for two-group comparisons. Statistical significance is indicated in the figures by *p < 0.05, **p < 0.01 and n.s. (indicating no significance)
To illustrate the ability of the dLFI in Aβ detection, a horizontal comparison of various available technologies in Table 1, such as ELISA [36, 70], nanoparticles-based immunoassays [71], surface-enhanced Raman spectroscopy [39], fluorescence [41], electrochemical biosensors [37], etc. ELISA is mainly used as a stopgap measure with the highest sensitivity in Aβ detection, but it cannot effectively distinguish monomers and oligomers. As shown in Fig. 6d, total Aβ42 levels detected by sandwich ELISA using a pair of antibodies recognizing different epitopes of Aβ42 showed no significant difference between AD and HC groups (p = 0.4837). Considering that the level of soluble Aβ42Os in AD patients is elevated, it is easy to be mis-detected in the measurement of Aβ42, resulting in the underestimation of Aβ42Ms level and poor performance in assessing the progression of AD [36, 72].
For nanoparticle-based immunoassays, including surface-enhanced Raman spectroscopy, fluorescence, and electrochemical biosensors, except for lacking the ability to distinguish monomers and oligomers, some special equipment and professional skills are required. On the contrary, dLFI can effectively detect monomers and oligomers by the naked eye within 30 min, which is not only high sensitivity but also simple, user-friendly, and instant detection without special equipment and professional skills. Altogether, our results indicate that dLFI can simultaneously detect Aβ42Ms and Aβ42Os in patient blood samples, with high sensitivity and specificity (Table 2).
Conclusion
This study provided a new method for the simultaneous detection of Aβ42Ms and Aβ42Os in AD blood using effective and rapid multiple techniques. The dLFI could detect the levels of Aβ42Ms and Aβ42Os in the blood quickly (within 30 min, including a 25-min enrichment step and a 5-min dLFI test step) and semi-quantitatively by naked eyes. In this system, the pre-incubation step can ensure high sensitivity and stability of the strip sensor. In conclusion, the dLFI allows high-throughput testing of small samples and has the potential to become a powerful tool for the rapid and accurate diagnosis of AD.
References
McDade E, Bateman RJ. Stop Alzheimer’s before it starts. Nature. 2017;547:153–5.
Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol. 2018;25:59–70.
Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403.
Ittner LM, Gotz J. Amyloid-beta and tau—a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci. 2011;12:65–72.
Citron M. Strategies for disease modification in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:677–85.
Brunden KR, Trojanowski JQ, Lee VM. Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat Rev Drug Discov. 2009;8:783–93.
Lee D, Kim SM, Kim HY, Kim Y. Fluorescence chemicals to detect insoluble and soluble amyloid-β aggregates. ACS Chem Neurosci. 2019;10:2647–57.
Duan Y, Chen J, Jin Y, Tu Q, Wang S, Xiang J. Antibody-free determinations of low-mass, soluble oligomers of Aβ(42) and Aβ(40) by planar bilayer lipid membrane-based electrochemical biosensor. Anal Chem. 2021;93:3611–7.
Jia Y, Yang L, Feng R, Ma H, Fan D, Yan T, Feng R, Du B, Wei Q. MnCO(3) as a new electrochemiluminescence emitter for ultrasensitive bioanalysis of β-amyloid(1–42) oligomers based on site-directed immobilization of antibody. ACS Appl Mater Interfaces. 2019;11:7157–63.
Jack CR Jr. Alzheimer disease: new concepts on its neurobiology and the clinical role imaging will play. Radiology. 2012;263:344–61.
Jack CR Jr, Wiste HJ, Schwarz CG, Lowe VJ, Senjem ML, Vemuri P, Weigand SD, Therneau TM, Knopman DS, Gunter JL, et al. Longitudinal tau PET in ageing and Alzheimer’s disease. Brain. 2018;141:1517–28.
Mueller A, Bullich S, Barret O, Madonia J, Berndt M, Papin C, Perrotin A, Koglin N, Kroth H, Pfeifer A, et al. Tau PET imaging with (18)F-PI-2620 in patients with Alzheimer disease and healthy controls: a first-in-humans study. J Nucl Med. 2020;61:911–9.
Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, Fripp J, Tochon-Danguy H, Morandeau L, O’Keefe G, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31:1275–83.
Lee JC, Kim SJ, Hong S, Kim Y. Diagnosis of Alzheimer’s disease utilizing amyloid and tau as fluid biomarkers. Exp Mol Med. 2019;51:1–10.
Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, Chai X, Proctor NK, Eichenlaub U, Zetterberg H, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med. 2020;26:379–86.
Thijssen EH, La Joie R, Strom A, Fonseca C, Iaccarino L, Wolf A, Spina S, Allen IE, Cobigo Y, Heuer H, et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer’s disease and frontotemporal lobar degeneration: a retrospective diagnostic performance study. Lancet Neurol. 2021;20:739–52.
Kang JH, Korecka M, Toledo JB, Trojanowski JQ, Shaw LM. Clinical utility and analytical challenges in measurement of cerebrospinal fluid amyloid-β(1–42) and τ proteins as Alzheimer disease biomarkers. Clin Chem. 2013;59:903–16.
Zhong Y, Shobo A, Hancock MA, Multhaup G. Label-free distribution of anti-amyloid D-AIP in Drosophila melanogaster: prevention of Abeta42-induced toxicity without side effects in transgenic flies. J Neurochem. 2019;150:74–87.
Hughes C, Choi ML, Yi JH, Kim SC, Drews A, George-Hyslop PS, Bryant C, Gandhi S, Cho K, Klenerman D. Beta amyloid aggregates induce sensitised TLR4 signalling causing long-term potentiation deficit and rat neuronal cell death. Commun Biol. 2020;3:79.
He N, Jin WL, Lok KH, Wang Y, Yin M, Wang ZJ. Amyloid-beta(1–42) oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis. 2013;4: e924.
Hampel H, Vassar R, De Strooper B, Hardy J, Willem M, Singh N, Zhou J, Yan R, Vanmechelen E, De Vos A, et al. The beta-secretase BACE1 in Alzheimer’s disease. Biol Psychiatry. 2021;89:745–56.
Bullich S, Seibyl J, Catafau AM, Jovalekic A, Koglin N, Barthel H, Sabri O, De Santi S. Optimized classification of (18)F-Florbetaben PET scans as positive and negative using an SUVR quantitative approach and comparison to visual assessment. Neuroimage Clin. 2017;15:325–32.
Morris E, Chalkidou A, Hammers A, Peacock J, Summers J, Keevil S. Diagnostic accuracy of 18F amyloid PET tracers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Eur J Nucl Med Mol Imaging. 2016;43:374–85.
Madhavan A, Whitwell JL, Weigand SD, Duffy JR, Strand EA, Machulda MM, Tosakulwong N, Senjem ML, Gunter JL, Lowe VJ, et al. FDG PET and MRI in logopenic primary progressive aphasia versus dementia of the Alzheimer’s type. PLoS ONE. 2013;8: e62471.
Le Ngoc HT, Park J, Chinnadayyala SR, Cho S. Sensitive electrochemical detection of amyloid beta peptide in human serum using an interdigitated chain-shaped electrode. Biosens Bioelectron. 2019;144: 111694.
Le HTN, Kim D, Phan LMT, Cho S. Ultrasensitive capacitance sensor to detect amyloid-beta 1–40 in human serum using supramolecular recognition of β-CD/RGO/ITO micro-disk electrode. Talanta. 2021;237: 122907.
Liu L, Kwak H, Lawton TL, Jin SX, Meunier AL, Dang Y, Ostaszewski B, Pietras AC, Stern AM, Selkoe DJ. An ultra-sensitive immunoassay detects and quantifies soluble Aβ oligomers in human plasma. Alzheimers Dement. 2021. https://doi.org/10.1002/alz.12457.
Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V, Fowler C, Li QX, Martins R, Rowe C, et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature. 2018;554:249–54.
Cintron AF, Dalal NV, Dooyema J, Betarbet R, Walker LC. Transport of cargo from periphery to brain by circulating monocytes. Brain Res. 2015;1622:328–38.
Boluda S, Iba M, Zhang B, Raible KM, Lee VM, Trojanowski JQ. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 2015;129:221–37.
Ye L, Hamaguchi T, Fritschi SK, Eisele YS, Obermüller U, Jucker M, Walker LC. Progression of seed-induced Aβ deposition within the limbic connectome. Brain Pathol. 2015;25:743–52.
Lakshmi S, Essa MM, Hartman RE, Guillemin GJ, Sivan S, Elumalai P. Exosomes in Alzheimer’s disease: potential role as pathological mediators biomarkers and therapeutic targets. Neurochem Res. 2020;45:2553–9.
Jia L, Qiu Q, Zhang H, Chu L, Du Y, Zhang J, Zhou C, Liang F, Shi S, Wang S, et al. Concordance between the assessment of Aβ42, T-tau, and P-T181-tau in peripheral blood neuronal-derived exosomes and cerebrospinal fluid. Alzheimers Dement. 2019;15:1071–80.
Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, Grodstein F, Wright CI, Blacker D, Rosas HD, et al. The cortical signature of Alzheimer’s disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex. 2009;19:497–510.
Stenh C, Englund H, Lord A, Johansson AS, Almeida CG, Gellerfors P, Greengard P, Gouras GK, Lannfelt L, Nilsson LN. Amyloid-beta oligomers are inefficiently measured by enzyme-linked immunosorbent assay. Ann Neurol. 2005;58:147–50.
Yang T, Hong S, O’Malley T, Sperling RA, Walsh DM, Selkoe DJ. New ELISAs with high specificity for soluble oligomers of amyloid β-protein detect natural Aβ oligomers in human brain but not CSF. Alzheimers Dement. 2013;9:99–112.
Li H, Xie H, Cao Y, Ding X, Yin Y, Li G. A general way to assay protein by coupling peptide with signal reporter via supermolecule formation. Anal Chem. 2013;85:1047–52.
Liu L, Xia N, Jiang M, Huang N, Guo S, Li S, Zhang S. Electrochemical detection of amyloid-β oligomer with the signal amplification of alkaline phosphatase plus electrochemical–chemical–chemical redox cycling. J Electroanal Chem. 2015;754:40–5.
Guerrini L, Arenal R, Mannini B, Chiti F, Pini R, Matteini P, Alvarez-Puebla RA. SERS detection of amyloid oligomers on metallorganic-decorated plasmonic beads. ACS Appl Mater Interfaces. 2015;7:9420–8.
Zhu W, Wang Y, Xie D, Cheng L, Wang P, Zeng Q, Li M, Zhao Y. In situ monitoring the aggregation dynamics of amyloid-β protein Aβ42 in physiological media via a Raman-based frequency shift method. ACS Appl Bio Mater. 2018;1:814–24.
Zhu L, Zhang J, Wang F, Wang Y, Lu L, Feng C, Xu Z, Zhang W. Selective amyloid β oligomer assay based on abasic site-containing molecular beacon and enzyme-free amplification. Biosens Bioelectron. 2016;78:206–12.
Rushworth JV, Ahmed A, Griffiths HH, Pollock NM, Hooper NM, Millner PA. A label-free electrical impedimetric biosensor for the specific detection of Alzheimer’s amyloid-beta oligomers. Biosens Bioelectron. 2014;56:83–90.
Veloso AJ, Chow AM, Ganesh HV, Li N, Dhar D, Wu DC, Mikhaylichenko S, Brown IR, Kerman K. Electrochemical immunosensors for effective evaluation of amyloid-beta modulators on oligomeric and fibrillar aggregation processes. Anal Chem. 2014;86:4901–9.
Wang Y, Deng R, Zhang G, Li Q, Yang J, Sun Y, Li Z, Hu X. Rapid and sensitive detection of the food allergen glycinin in powdered milk using a lateral flow colloidal gold immunoassay strip test. J Agric Food Chem. 2015;63:2172–8.
Hong L, Wang K, Yan W, Xu H, Chen Q, Zhang Y, Cui D, Jin Q, He J. High performance immunochromatographic assay for simultaneous quantitative detection of multiplex cardiac markers based on magnetic nanobeads. Theranostics. 2018;8:6121–31.
Gao F, Lei C, Liu Y, Song H, Kong Y, Wan J, Yu C. Rational design of dendritic mesoporous silica nanoparticles’ surface chemistry for quantum dot enrichment and an ultrasensitive lateral flow immunoassay. ACS Appl Mater Interfaces. 2021;13:21507–15.
Goryacheva OA, Guhrenz C, Schneider K, Beloglazova NV, Goryacheva IY, De Saeger S, Gaponik N. Silanized luminescent quantum dots for the simultaneous multicolor lateral flow immunoassay of two mycotoxins. ACS Appl Mater Interfaces. 2020;12:24575–84.
Zhang LLX, Zhang Z, Luo H. Cerebrospinal fluid and blood biomarkers in the diagnostic assays of Alzheimer’s disease. J Innov Opt Health Sci. 2021. https://doi.org/10.1142/S1793545822300014.
Counts SE, Ikonomovic MD, Mercado N, Vega IE, Mufson EJ. Biomarkers for the early detection and progression of Alzheimer’s disease. Neurotherapeutics. 2017;14:35–53.
Blennow K, Hampel H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol. 2003;2:605–13.
Zhang L, Yang C, Li Y, Niu S, Liang X, Zhang Z, Luo Q, Luo H. Dynamic changes in the levels of amyloid-β42 species in the brain and periphery of APP/PS1 mice and their significance for Alzheimer’s disease. Front Mol Neurosci. 2021;14: 723317.
Verma HN, Singh P, Chavan R. Gold nanoparticle: synthesis and characterization. Vet World. 2014;7:72.
Zhang L, Shi Y, Chen C, Han Q, Chen Q, Xia X, Song Y, Zhang J. Rapid, visual detection of klebsiella pneumoniae using magnetic nanoparticles and an horseradish peroxidase-probe based immunosensor. J Biomed Nanotechnol. 2019;15:1061–71.
Mena R, Edwards P, Pérez-Olvera O, Wischik CM. Monitoring pathological assembly of tau and beta-amyloid proteins in Alzheimer’s disease. Acta Neuropathol. 1995;89:50–6.
Sun A, Nguyen XV, Bing G. Comparative analysis of an improved thioflavin-s stain, Gallyas silver stain, and immunohistochemistry for neurofibrillary tangle demonstration on the same sections. J Histochem Cytochem. 2002;50:463–72.
Luna-Muñoz J, Peralta-Ramirez J, Chávez-Macías L, Harrington CR, Wischik CM, Mena R. Thiazin red as a neuropathological tool for the rapid diagnosis of Alzheimer’s disease in tissue imprints. Acta Neuropathol. 2008;116:507–15.
Bagchi P, Birnbaum SM. Effect of pH on the adsorption of immunoglobulin G on anionic poly(vinyltoluene) model latex particles. J Colloid Interface Sci. 1981;83:460–78.
Demanèche S, Chapel JP, Monrozier LJ, Quiquampoix H. Dissimilar pH-dependent adsorption features of bovine serum albumin and alpha-chymotrypsin on mica probed by AFM. Colloids Surf B Biointerfaces. 2009;70:226–31.
Peng ZG, Hidajat K, Uddin MS. Adsorption of bovine serum albumin on nanosized magnetic particles. J Colloid Interface Sci. 2004;271:277–83.
Ruppert C, Kaiser L, Jacob LJ, Laufer S, Kohl M, Deigner HP. Duplex Shiny app quantification of the sepsis biomarkers C-reactive protein and interleukin-6 in a fast quantum dot labeled lateral flow assay. J Nanobiotechnol. 2020;18:130.
Kim HM, Kim J, An J, Bock S, Pham XH, Huynh KH, Choi Y, Hahm E, Song H, Kim JW, et al. Au–Ag assembled on silica nanoprobes for visual semiquantitative detection of prostate-specific antigen. J Nanobiotechnol. 2021;19:73.
Motter R, Vigo-Pelfrey C, Kholodenko D, Barbour R, Johnson-Wood K, Galasko D, Chang L, Miller B, Clark C, Green R, et al. Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann Neurol. 1995;38:643–8.
Englund H, Sehlin D, Johansson AS, Nilsson LN, Gellerfors P, Paulie S, Lannfelt L, Pettersson FE. Sensitive ELISA detection of amyloid-beta protofibrils in biological samples. J Neurochem. 2007;103:334–45.
Wang MJ, Yi S, Han JY, Park SY, Jang JW, Chun IK, Kim SE, Lee BS, Kim GJ, Yu JS, et al. Oligomeric forms of amyloid-β protein in plasma as a potential blood-based biomarker for Alzheimer’s disease. Alzheimers Res Ther. 2017;9:98.
Babapour Mofrad R, Scheltens P, Kim S, Kang S, Youn YC, An SSA, Tomassen J, van Berckel BNM, Visser PJ, van der Flier WM, Teunissen CE. Plasma amyloid-β oligomerization assay as a pre-screening test for amyloid status. Alzheimers Res Ther. 2021;13:133.
Nabers A, Ollesch J, Schartner J, Kötting C, Genius J, Hafermann H, Klafki H, Gerwert K, Wiltfang J. Amyloid-β-secondary structure distribution in cerebrospinal fluid and blood measured by an immuno-infrared-sensor: a biomarker candidate for Alzheimer’s disease. Anal Chem. 2016;88:2755–62.
Kim Y, Yoo YK, Kim HY, Roh JH, Kim J, Baek S, Lee JC, Kim HJ, Chae MS, Jeong D, et al. Comparative analyses of plasma amyloid-β levels in heterogeneous and monomerized states by interdigitated microelectrode sensor system. Sci Adv. 2019;5: eaav1388.
Zhang J, Peng M, Jia J. Plasma amyloid-β oligomers and soluble tumor necrosis factor receptors as potential biomarkers of AD. Curr Alzheimer Res. 2014;11:325–31.
Verberk IMW, Slot RE, Verfaillie SCJ, Heijst H, Prins ND, van Berckel BNM. Plasma amyloid as pre-screener for the earliest Alzheimer’s pathological changes. Ann Neurol. 2018;138:1328–36.
Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–13.
Jiang LF, Chen BC, Chen B, Li XJ, Liao HL, Huang HM, Guo ZJ, Zhang WY, Wu L. Detection of Aβ oligomers based on magnetic-field-assisted separation of aptamer-functionalized Fe(3)O(4) magnetic nanoparticles and BaYF(5):Yb, Er nanoparticles as upconversion fluorescence labels. Talanta. 2017;170:350–7.
Liu L, Chang Y, Yu J, Jiang M, Xia N. Two-in-one polydopamine nanospheres for fluorescent determination of beta-amyloid oligomers and inhibition of beta-amyloid aggregation. Sens Actuators B. 2017;251:359–65.
Acknowledgements
We thank the associate professor Jinyang Zhang, Kunming University of Science and Technology, for his technical support for the screening of monoclonal antibodies. We also thank the Engineering Research Center of Nano-Geomaterials of the Ministry of Education and the Center for Nanoscale Characterization & Devices (CNCD) of WNLO-HUST for support with data acquisition and the Analytical and Testing Center of HUST for performing spectral measurements.
Funding
This work was supported by the Major Research Plan of the National Natural Science Foundation of China (Grant No. 91749209), the National Natural Science Foundation of China (Grant No. 81971025), the Fundamental Research Funds for the Central Universities in HUST (2019kfyXKJC076), and Hubei Provincial Natural Science Foundation (2020CFB811).
Author information
Authors and Affiliations
Contributions
LH ordinated the writing of the paper and provided writing guidance and manuscript revision. ZL and DX completed all experiments and contributed to writing the first draft. SY provided AD patient blood samples and helped analyze the test results, as well as clinical diagnosis of AD patients and helped analyze the test results, as well as clinical diagnosis of AD patients. LY and NS participated in most experiments. LX was involved in 5xFAD mice breeding and schematic figure preparations. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures involving experimental animals were carried out under guidelines approved by the Institutional Animal Care and Use Committee of Huazhong University of Science and Technology.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest, financial or otherwise.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Information of the participants in this manuscript. Figure S1. The HPLC and mass spectrometry results of synthesized Aβ42 (a, b) and Aβ40 (c, d) peptides. Figure S2. The HPLC and mass spectrometry results of synthesized p-Tau396,404 (a, b) and p-Tau231 (c, d) peptides. Figure S3. The HPLC (a) and mass spectrometry (b) results of synthesized Cis-Tau peptides. Figure S4. Characterization of AuNP–1F12 conjugates. (a) The color and UV − vis absorption spectra of synthetic AuNP. (b) The Zeta-potentials of AuNP before and after mAb 1F12 modification. (c) UV − vis absorption spectra of mAb 1F12 before and after conjugation. The absorption spectrum of different volumes of K2CO3 (d) and concentrations of 1F12 (e) for the conjugation of 1F12 with AuNP. Data are presented as means ± SD. Figure S5. Characterization of 1F12-modified MNPs. (a) The representative SEM image of bare magnetic nanoparticles (MNPs). (Scale bar: 200 nm). (b) The principle of synthetic antibody-modified MNPs. The sizes (c) and Zeta-potentials (d) of MNPs before and after antibody modification. Data are presented as means ± SD.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, L., Du, X., Su, Y. et al. Quantitative assessment of AD markers using naked eyes: point-of-care testing with paper-based lateral flow immunoassay. J Nanobiotechnol 19, 366 (2021). https://doi.org/10.1186/s12951-021-01111-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12951-021-01111-z