
Abstract
Background
To investigate the roles of the transcription factors twist family bHLH transcription factor 1 (TWIST1), twist family bHLH transcription factor 2 (TWIST2), and peroxisome proliferator activated receptor gamma (PPARγ) in the progression of nonalcoholic steatohepatitis.
Methods
The protein levels of TWIST1, TWIST2 and PPARγ were determined in the serum of nonalcoholic fatty liver disease (NAFLD) patients and healthy controls by enzyme-linked immunosorbent assay (ELISA). An in vivo model for fatty liver was established by feeding C57BL/6 J mice a high-fat diet (HFD). An in vitro model of steatosis was established by treating LO-2 cells with oleic acid (OA). RNA sequencing was performed on untreated and OA-treated LO-2 cells followed by TWIST1, TWIST2 and PPARγ gene mRNA levels analysis, Gene Ontology (GO) enrichment and pathway analysis.
Results
The TWIST2 serum protein levels decreased significantly in all fatty liver groups (P < 0.05), while TWIST1 varied. TWIST2 tended to be lower in mice fed an HFD and was significantly lower at 3 months. Similarly, in the in vitro model, the TWIST2 protein level was downregulated significantly at 48 and 72 h after OA treatment. RNA sequencing of LO-2 cells showed an approximately 2.3-fold decrease in TWIST2, with no obvious change in TWIST1 and PPARγ. The PPAR signaling pathway was enriched, with 4 genes upregulated in OA-treated cells (P = 0.0018). The interleukin (IL)-17 and tumor necrosis factor (TNF) signaling pathways were enriched in OA-treated cells.
Conclusions
The results provide evidence that the TWIST2 and PPAR signaling pathways are important in NAFLD and shed light on a potential mechanism of steatosis.
Similar content being viewed by others

Introduction
Nonalcoholic fatty liver disease (NAFLD) is a common chronic liver disorder characterized by lipid accumulation in hepatocytes. Currently, NAFLD affects approximately 20–30% of the Western population and 5–18% of the Asian population and is continuing to increase [1]. Moreover, being a metabolic disease, NAFLD can aggravate other metabolic syndromes, including obesity, type 2 diabetes mellitus, dyslipidemia, hypertension and insulin resistance (IR) [2, 3]. However, the pathogenesis and molecular mechanism of NAFLD are still unclear, and further work is undoubtedly necessary.
NAFLD includes simple steatosis as well as nonalcoholic steatohepatitis (NASH), a more serious condition with associated inflammation. It is well known that NASH can progress to more serious conditions, such as fibrosis, cirrhosis and hepatocellular carcinoma [4]. The widely accepted pathogenesis of NAFLD is the “multiple hit theory”, which suggests that in the development of NAFLD, many hits might work together in parallel [5,6,7]. Obesity and IR are reported to play a fundamental role in the progression of NAFLD. High insulin levels promote lipid synthesis by increasing glycolysis in the liver [8]. In addition, high insulin also reduces the expression of ApoB-100 and microsomal triglyceride transfer protein (MTTP), leading to a reduction in very low density lipoprotein (VLDL) output in the liver, eventually leading to lipid deposition in the liver tissue [9]. IR can also cause hepatic steatosis, and it has been proposed that hepatic steatosis further aggravates hepatic insulin resistance, creating a feedback loop between the two conditions [10]. Kim et al. suggested that hepatic steatosis could lead to IR, but some scholars believe that hepatic steatosis is not enough to cause liver IR [11, 12]. Recently, much more is said about molecular mechanisms because the mechanisms that lead to the release of many mediators are known, which then lead to the formation of proliferation and scar tissue after a long inflammatory process, induction of apoptosis and/or necrosis [13]. The relationship between hepatic steatosis and IR is still controversial and needs further study.
The authors’ research team has previously reported that two transcription factors, TWIST1 and PPARγ, have a positive regulatory role in the insulin sensitivity of 3 T3-L1 adipocytes [14]. In IR models of 3 T3-L1 adipocytes and C57/BL6J mice, silencing TWIST1 expression can relieve IR to a certain degree, indicating the potential clinical value of TWIST1 and PPARγ in steatosis-related disease [15]. As hepatocytes are target cells of insulin, the role of TWIST1 and PPARγ in hepatocytes is undoubtedly worth further exploration. At the same time, TWIST2, the other member of the TWIST family, attracted attention for its role in metabolism and metabolic diseases. Overexpression of TWIST2 was reported to ameliorate hepatocellular steatosis and inhibit inflammation [16].
The present study will continue previous work to explore the role and possible mechanism of TWIST1, TWIST2, and PPARγ in the development of hepatocyte steatosis. This work will mainly be based on clinical samples and in vivo and in vitro models of hepatocyte steatosis. The results will provide evidence for the role of TWIST and PPARγ in NAFLD, clarify their function in the adipogenesis process and provide new insights into steatosis-related diseases.
Materials and methods
Patient inclusion and human serum sample collection
A total of 406 individuals who had a health check-up at Shandong Provincial Qianfoshan Hospital from December 2017 to December 2018 were recruited into the present study. Following the inclusion criteria, these NAFLD patients were generally 18–60 years old, had normal thyroid function, no history of drinking, no malignancy, no viral hepatitis, no drug-induced liver disease, no autoimmune liver disease, and all other specific diseases that cause fatty liver.
NAFLD patients were divided into mild, moderate, and severe NAFLD groups based on abdominal ultrasound testing. The standard of ultrasound fatty liver classification is mainly based on the shape of the liver, the echo of the liver parenchyma, the degree of attenuation behind the liver, and the visualization of the intrahepatic duct. Generally, patients with mild fatty liver have a normal liver shape and a sharp angle. In patients with moderate fatty liver, the liver is slightly larger, and the angle becomes blunt. In patients with severe fatty liver, the liver is obviously enlarged, and the angle is round and blunt. The liver parenchyma echoes, the patients with mild fatty liver are fine and thicker, and the echoes of patients with severe fatty liver are obviously thicker. The attenuation at the rear of the liver changes from normal to obvious, and the intrahepatic ducts change from normal to unclear. Different grades can distinguish mild, moderate and severe fatty liver. The condition was diagnosed by the ultrasound physician. Healthy adults who were also having a health check-up at the same time had samples randomly collected. This study was performed in accordance with ethical standards and was approved by the Ethics Committee of Shandong University (No: [2017] S048).
Materials and reagents
C57/BL6 mice were purchased from the Experimental Animal Center of Shandong University. The high-fat diet (No: D12492) and basal diet (No: D12450B) were both products of Research Diets (New Brunswick, USA). The human hepatic cell line LO-2 (also named HL-7702) was purchased from Procell Life Science & Technology Co., Ltd. (Wuhan; China). The human TWIST1, TWIST2 and PPARγ ELISA kits were Jianglai Biology (Shanghai, China) products. RPMI-1640 medium, fetal bovine serum (FBS) and 0.25% trypsin-0.02% EDTA were all purchased from GIBCO (Invitrogen, California, USA). The rabbit anti-mouse TWIST1 mAb was purchased from Sigma (St. Louis, MO, USA). Rabbit anti-rabbit TWIST2 pAb and rabbit anti-mouse PPARγ mAb were purchased from Abcam (Cambridge, MA, USA). The rabbit anti-mouse β-actin mAb and secondary antibodies, including horseradish peroxidase-conjugated anti-mouse IgG for TWIST1, PPARγ, and β-actin and horseradish peroxidase-conjugated anti-rabbit IgG for anti-TWIST2, were purchased from ZSGB-BIO (Beijing, China). Oleic acid (OA) and all other general reagents used in this study were purchased from Sigma (St. Louis, MO, USA).
Serum determination of TWIST1, TWIST2 and PPARγ in patients
The levels of TWIST1, TWIST2 and PPARγ in the serum of patients were analyzed by ELISA per the manufacturer’s instructions. Generally, standard or serum (each 50 μL) was added to the plate wells; HRP-conjugated antibody (100 μL) was added to the plate to mix with standard or serum at 37 °C for 1 h. After being washed 5–6 times, the plates were treated with 50 μL each of substrates A and B and incubated at 37 °C for 15 min, and then 50 μL of terminating buffer was added to stop the reaction. Finally, the OD value was checked under an enzyme-linked immunoassay analyzer at 450 nm.
Induction of NAFLD in C57/BL6 mice
Sixty-four male C57/BL6 mice (6 weeks of age) were included in the study and housed 6 per cage. The mice were housed in a specific environment: 25 °C, 55% relative humidity, cycle of 12 h light and 12 h dark. They were free to eat and drink tap water ad libitum. After a week, they were randomly assigned to two groups, the control group (basal diet, 4% fat, with 20 g% lard) and the high-fat diet (HFD) group (HFD, 60% fat, with 225 g% lard). They were fed a control diet or HFD for 4, 8, 12, or 16 weeks, and their body weights were recorded every week. After the mice were euthanized, liver tissues were collected and snap frozen. Hematoxylin and eosin (H&E) staining and Oil red O staining were conducted on liver sections. The additional procedures were carried out according to the ‘Principles of Laboratory Animal Care’ established by the National Institutes of Health. This study was performed in accordance with ethical standards and was approved by the Ethics Committee of Shandong University (No: [2017] S048).
Intraperitoneal glucose tolerance test (IPGTT) and intraperitoneal insulin tolerance test (IPITT)
Before the mice were sacrificed at the 16th week, IPGTT and IPITT were conducted in a portion of the mice to determine the ability of mice to respond to glucose and insulin. After fasting overnight for IPGTT or six hours for IPITT, 12 mice were injected with 50% glucose (2.0 g/kg, i.p.), and 12 mice were injected with insulin (0.65 U/kg, i.p). Blood glucose levels in the mice were analyzed by means of a glucose test strip (Bayer, Germany) after blood samples were taken via the caudal vein at 0, 30, 60, 90, 120, 150 and 180 min after glucose or insulin injection. The area under the curve (AUC) was compared among different groups to analyze the IPGTT and IPITT.
Establishment of the LO-2 hepatocyte steatosis model
LO-2 hepatocytes were cultured in RPMI 1640 supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin. The cells were cultured in a humidified chamber at 37 °C and 5% CO2. For steatosis induction, RPMI-1640 supplemented with 10% FBS and OA (50 μg/mL) was added to the cells when they reached approximately 60% confluence. The culture medium and OA were replaced every 24 h for 24, 48, 72, and 96 h.
Hematoxylin-eosin (H&E), oil red O staining of frozen liver sections and LO-2 hepatocytes
After the mice were sacrificed and the livers were removed and snap frozen, H&E staining was performed on frozen sections. The general steps are as follows: Methanol fixative for 60 s; Hematoxylin 2 min; Washed with phosphate buffered saline (PBS) for 30 s; Hydrochloric acid alcohol washed for 3 s; Washed with PBS for 30 s; Lithium carbonate 10 s; Washed with PBS for 30 s; 90% alcohol for 10 s; Eosin for 5 s; 90% alcohol for 5 s; 95% alcohol for 20 s, twice; 100% alcohol for 20 s; 100% alcohol for 20 s; Xylene for 30 s, twice. The resulting slides were visualized with an inverted phase contrast microscope.
For Oil red O staining, LO-2 cells were grown on cover slips, and after removal of cell culture medium, cells were washed three times with PBS and fixed with 4% tissue cell fixative solution (Solarbio Science) at room temperature for 10 min. Frozen slices for hepatic tissue were similarly fixed. After washing three times with PBS, the cells or frozen slices were stained with freshly diluted Oil red O solution (0.1% Oil red O dissolved in 60% isopropyl alcohol and 40% distilled water) for 15–30 min. Cells or frozen slices were then washed with 60% isopropyl alcohol for 1 min and washed with PBS three times. Images were obtained using an inverted phase contrast microscope. Oil red O staining was quantified by an absorbance assay. Briefly, the Oil red O stain was solubilized with isopropyl alcohol and the optical density at 510 nm was measured by spectrophotometry (Multiskan Go, Thermo Scientific). All experiments were conducted in triplicate.
Protein extraction and western blotting analysis
Total protein was extracted using radioimmunoprecipitation assay (RIPA) lysis buffer containing protease inhibitor cocktail (Thermo Scientific, USA) and phosphatase inhibitor cocktail (Thermo Scientific, USA), and the protein concentration was analyzed by Pierce™ BCA protein assay as previously shown [14, 15]. The protein (40 μg for each sample) was resolved on SDS-PAGE gels and transferred to a Hybond-P PVDF membrane. The membranes were blocked with 5% milk or 5% bovine serum albumin (BSA) for 1 h. The membranes were incubated with primary antibodies (anti-TWIST1, 1:1000; anti-TWIST2, 1:1000; anti-PPARγ, 1:1000 and anti-β-actin, 1:2500) at 4 °C overnight. After washing three times (20 min each) with TBST, the secondary antibodies were incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse (1:5000) antibodies for 1 h at room temperature. Blots were washed three times (20 min each), and image development was performed with electrochemiluminescence (ECL) reagent. All experiments were performed in triplicate.
RNA sequencing analysis
RNA sequencing analysis was performed using the Illumina HiSeq 4000 platform (KangChen Bio-tech, Shanghai, China). Briefly, cultured LO-2 cells were treated with DMSO (the control group) and OA (50 μg/mL, 48 h). After treatment, cells were lysed with TRIzol reagent. RNA was extracted as normal protocol. RNA (1–2 μg) was used to establish the RNA library based on specific kits. It includes RNA fragmentation, primed 1st strand cDNA synthesis, dUTP-based 2nd strand cDNA synthesis, adaptor ligation and PCR amplification. The library was qualified using an Agilent 2100, quantified by qPCR, and then subjected to sequencing based on Illumina X-ten/NovaSeq. Three biological replicates were prepared in each group. Genes were considered differentially expressed if the expression difference between two groups showed a twofold change or greater (log2FC > 1.0, fold change). Gene Ontology (GO) enrichment analysis was performed on the http://geneontology.org website, and the enrichment scores for molecular function (MF), biological process (BP) and cellular component (CC) were analyzed. Pathway analysis for differentially expressed genes was further conducted based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, whose website is https://www.genome.jp/kegg/.
Statistical analysis
The data were analyzed using SPSS 24.0 software (mean ± SD). The correlations between TWIST1, TWIST2, PPARγ, body mass index (BMI), fasting glucose, fasting insulin, or homeostasis model assessment (HOMA) and fatty liver severity were analyzed by ANOVA. For western blotting, we used Image J to determine the relative gray values of each band by comparison with β-actin. A t test was used to compare the differences between two groups, and ANOVA was used to compare the differences between more than two groups. P < 0.05 was considered an indicator of significant difference.
Results
Analysis of TWIST1, TWIST2, and PPARγ protein levels in human serum samples
The serum samples were collected at the clinical laboratory. A total of 406 human serum samples were collected in this study. The samples were divided into four groups based on abdominal ultrasound testing, with a healthy control group (n = 99), mild NAFLD group (n = 102), moderate NAFLD group (n = 107), and severe NAFLD group (n = 98). The Materials and Methods section describes the criteria for determining whether a fatty liver is mild, moderate, or severe. Sex, age, height, and body weight were recorded for each individual, and the basic clinical characteristics of these individuals are shown in Table 1. They were further divided into four groups based on BMI (kg/m2), with thin group (BMI<18.5 kg/m2, n = 4), normal group (BMI = 18.5–23.9 kg/m2, n = 84), overweight group (BMI = 24–27.9 kg/m2, n = 137), and obesity group (BMI ≥ 28 kg/m2, n = 181).
As shown in Table 2, fasting blood glucose and insulin levels were determined by an automatic analyzer. Both blood glucose and blood insulin increased significantly with the severity of fatty liver disease (P < 0.05). Spearman rank correlation analysis showed that a positive correlation was found between the severity of fatty liver disease and BMI (r = 0.761, P < 0.001) and HOMA value (r = 0.607, P < 0.001). Then, TWIST1, TWIST2 and PPARγ levels were measured in the serum of patients using ELISA kits. For TWIST1, the levels in the mild and severe fatty liver groups decreased significantly (△P < 0.05, compared with healthy control) and increased significantly in the moderate fatty liver group (△P < 0.05, compared with healthy control). The levels of TWIST2 in all of the fatty liver groups decreased significantly (△P < 0.05, compared with healthy control). For PPARγ, compared with the control group, there were no significant changes in the mild fatty liver, moderate fatty liver, and severe fatty liver groups (P > 0.05).
Characterization of the in vivo model of NAFLD
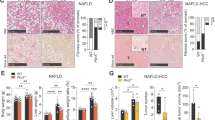
After 16 weeks on a high-fat diet, the livers in the HFD group were enlarged and had a white hue (Fig. 1a). H&E staining (Fig. 1b) and Oil red O staining (Fig. 1c) showed increased lipid and triglyceride contents in the HFD group indicative of steatosis. The mice in the HFD group had a much higher body weight that increased significantly from 6 weeks of age (*P < 0.05, Fig. 1d). Both the IPGTT (Fig. 1e) and IPITT (Fig. 1f) of the HFD group showed a decreased insulin response. TWIST1, TWIST2 and PPARγ protein levels in liver tissues all changed with feeding time (Fig. 1g). Both TWIST1 (the arrow showed the true band) and TWIST2 expressions in HFD tended to be decreased in the detection, and statistical significance showed only in TWIST2 at 3 months after the start of the HFD (Fig. 1h, i). There were no significant change in PPARγ expression (Fig. 1j).

Characterization of an in vivo model of fatty liver disease and changes in TWIST1, TWIST2 and PPARγ protein expression in the liver (n = 8). a Comparison of the sizes of control and high-fat diet (HFD) group mice and their livers. b H&E staining of livers from control and HFD mice.c Oil red O staining of livers from control and HFD group mice. d Changes in body weight over time. e Intraperitoneal glucose tolerance test (IPGTT). f Intraperitoneal insulin tolerance test (IPITT). g Relative protein levels of TWIST1, TWIST2 and PPARγ in the livers of control and HFD group mice. The arrow shows that the upper band is the true Twist1. H/I/J) Semiquantitative assays of TWIST1 (h), TWIST2 (i), and PPARγ (j) in the livers of control and HFD group mice
Characterization of the in vitro model of hepatocyte steatosis
LO-2 cells were treated with OA (50 μg/mL) for 24, 48, 72, and 96 h to induce steatosis. Obvious red-stained lipid droplets could be seen with Oil red O staining compared with the control (Fig. 2a, b), showing that the LO-2 cell steatosis model was established.

The LO-2 steatosis model shows increased lipid uptake and decreased TWIST2 expression. a Oil red O staining in the control and oleic acid (50 μg/mL) groups. b OD values for oil red O staining in the control and oleic acid groups. c TWIST1, TWIST2 and PPARγ expression in the control and oleic acid treatment groups in cultured LO-2 cells. β-Actin was used as a loading control. The arrow shows that the upper band is the true Twist1. d/e/f: Semiquantitative assays of TWIST1 (d), TWIST2 (e), and PPARγ (f)
As shown in Fig. 2c, both TWIST1 (the arrow showed the true band) and PPARγ protein expression gradually increased with time (24, 48, 72, and 96 h) under OA treatment in LO-2 cells. However, similar increases could be found in control cells without OA exposure. The increased cell density may be the possible cause of TWSIT1 and PPARγ changes in LO-2 control cells under no external stimulation. Statistical analysis based on semi-quantitative analysis using Image J software showed that Both TWIST1 (Fig. 2d, the arrow showed the true band) and TWIST2 (Fig. 2e) in OA treatment tended to be decreased, and a significant difference could be found in TWIST2 at both 48 and 72 h. No significant change in PPARγ (Fig. 2f) expression between the control and OA (50 μg/mL) treatment groups (P > 0.05).
RNA sequencing analysis and RT-PCR both showed that TWIST2 was decreased significantly by OA treatment
RNA sequencing of LO-2 cells treated with OA for 48 h showed that 1884 genes were upregulated and 1975 genes were downregulated in the OA treatment group compared with the control group (Fig. 3a). Cluster analysis of the top 20 upregulated and downregulated genes is shown in Fig. 3b. TWIST1 and PPARγ mRNA levels showed no significant difference (P > 0.05) between the groups in the RNA sequencing results, but TWIST2 was downregulated significantly with OA treatment (P < 0.05) (Fig. 3c). Further GO enrichment analysis of the RNA-seq data revealed 542 differentially expressed (DE) upregulated genes and their associated biological processes (BPs) (Fig. 3d) and 514 DE downregulated genes and associated BPs (Fig. 3e). The details of the top 20 upregulated and downregulated genes under oleic acid treatment by RNA sequencing are shown in Table 3.

RNA sequencing analysis of transcriptional changes and GO biological process analysis in cultured LO-2 cells treated with OA (50 μg/mL, 48 h). a After sequencing analysis, the mRNA transcriptional changes between the control and test groups were analyzed. Shown here is a scatter map of mRNA transcriptional changes. b The top 20 upregulated and downregulated genes in the OA treatment group were selected and are shown in a heat map. c RT-PCR was conducted to determine TWIST1, TWIST2 and PPARγ mRNA changes in response to OA treatment as described in the methods section. The results are shown in the bar graphs. d GO biological process analysis was conducted, and the classifications of the upregulated genes are shown. e The classifications of the downregulated genes for GO biological processes are shown
Genes in the PPAR signaling pathway were enriched significantly based on RNA sequencing
Compared with the control, the DE genes in OA-treated LO-2 cells (50 μg/mL, 48 h) were involved in 62 pathways, with 42 upregulated (top 10 shown in Fig. 4a) and 20 downregulated (top 10 shown in Fig. 4b). In the top 10 upregulated pathways of DE genes, the IL-17 signaling pathway [hsa04657], TNF signaling pathway [hsa04668], and PPAR signaling pathway [hsa03320] were listed. Four genes in the PPAR signaling pathway were upregulated in OA (50 μg/mL, 48 h)-treated cells (P = 0.0018). These four genes are acyl-CoA synthetase long chain family member 4 (ACSL4), angiopoietin like 4 (ANGPTL4), carnitine palmitoyltransferase 1A (CPT1A), and perilipin 4 (PLIN4). RT-PCR verified their up regulation (Fig. 4c). A schematic model for the PPAR function is proposed, as shown in Fig. 4d. During adipogenesis, PPAR can be induced by unsaturated fatty acids or 9-cis-retinoic acid and can upregulate ACSL4, ANGPTL4, CPT1A, and PLIN, which can regulate lipid metabolism or adipocyte differentiation.

PPAR signaling pathway genes were enriched significantly in LO-2 cells by OA treatment (50 μg/mL, 48 h). a KEGG pathway analysis based on the KEGG website. The top 10 upregulated pathways of differentially expressed (DE) genes are shown here. b The top 10 downregulated pathways of DE genes based on the KEGG website shown here. c The mRNA levels of four genes in the PPAR signaling pathway, namely, ACSL4, ANGPTL4, CPT1A, and PLIN4, showed increased transcription based on RT-PCR. d A schematic model of the PPARγ pathway in lipid metabolism and adipocyte differentiation was drawn, indicating the mechanism of the PPAR signaling pathway in NAFLD
Discussion
The present study focuses on the role of TWIST1, TWIST2 and PPARγ in nonalcoholic steatohepatitis progression. To do this, the serum protein levels of TWIST1, TWIST2, and PPARγ in the serum of NAFLD patients were examined with varying stages of disease, and significant changes in the TWIST proteins were noted. In vitro and in vivo models of the disease were also studied, and TWIST2 seemed to be affected the most in these models. The results using each of these models provide additional evidence that the TWIST2 transcription factor may play a role in NAFLD.
TWIST2 seems to be particularly important in NAFLD because the results and the publication of a recent article showed that TWIST2 was involved in the development of NAFLD [16]. TWIST2 is a basic helix-loop-helix (bHLH) transcription factor that is highly related to TWIST1 and appears to have some redundant and nonredundant functions [17]. TWIST transcription factors bind as homodimers or heterodimers to E-box consensus sites and are transcriptional activators or repressors depending on the cellular context [17, 18]. TWIST proteins have been extensively studied for their oncogenic properties and their role in the epithelial-to-mesenchymal transition (EMT), but their involvement in other processes is less well known [19]. twist2-null mice have many abnormalities in fat and glucose metabolism and are presumed to die soon after birth because of the upregulation of cytokines in an NF-κB-dependent manner [20]. A decrease in TWIST2 protein in the in vitro and in vivo models of NAFLD was found. Interestingly, a decrease in TWIST2 protein levels in the serum of NAFLD patients at all stages of the disease was also noticed. This suggests that in NAFLD, a reduction in the level of TWIST2 protein may be an early event that increases inflammation. It also suggests that TWIST2 protein levels in serum may be a biomarker of NAFLD. It has recently been shown that TWIST1 mRNA and protein can be found in exosomes secreted from hepatic stellate cells (HSCs) and that the levels of TWIST1 mRNA and protein decrease in models of hepatic injury and fibrosis [21]. It is likely that TWIST2 is also secreted from the liver compartment and that changes in TWIST2 protein content can be indicative of a developing disease process.
TWIST1 serum protein levels varied greatly with the severity of disease, with significantly lower levels in the mild fatty liver group and significantly higher levels in the moderate fatty liver group. The level of TWIST1 protein in the severe fatty liver group was the same as that in the control group. This pattern of TWIST1 serum protein may indicate complex regulation of the TWIST1 gene during the development of NAFLD. TWIST1 was considered an anti-inflammatory marker, as low mRNA and protein expression of TWIST1 was associated with increased expression of proinflammatory cytokines and decreased insulin sensitivity in humans [22]. Lower levels of TWIST1 in the early stages of NAFLD may lead to increased inflammation and disease progression.
Increased protein expression of PPAR is a general property of steatotic livers. The contribution of PPAR is to the maintenance of a steatotic phenotype in liver cells [23]. PPARγ is capable of activating the expression of genes involved in TG accumulation in hepatocytes and promoting the generation of fatty liver [24]. While no change was seen in PPARγ protein levels in the patient serum or in the in vivo and in vitro models, the RNA sequence analysis of OA-treated cells suggested that the PPAR signaling pathway was involved in the hepatic steatosis process. It is believed that PPAR may regulate downstream lipid metabolism or adipocyte differentiation through ACSL4, ANGPTL4, CPT1A, and PLIN4. Further investigation into the mechanism of how PPAR contributes to NAFLD is warranted.
Chronic inflammation has also been implicated in the development and progression of NAFLD [25, 26]. TWIST1 and TWIST2 have been shown to reduce inflammation by inhibiting the NF-kB pathway [18, 20]. As mentioned, twist-2-deficient mice produce high levels of cytokines [20]. An interesting finding of the GO analysis of the RNA sequencing data revealed an enhanced IL-17 signaling pathway in OA-treated cells. IL-17 signaling has become of greater significance in NAFLD in recent years [27]. An enhancement in the TNF signaling pathway was also found, another important inflammatory pathway implicated in NAFLD [28,29,30]. It has been reported that a significantly higher concentration of TNF-alpha is associated with both the type of inflammatory cells and with increased vascularization in various tissue types [31].
Study strength and limitations
The present findings prompt TWIST2 as a potential new biomarker for the early detection of NAFLD. Meanwhile, the involvement of the PPAR signaling pathway hints further perspective in hepatocytes steatosis. However, there are some limitations in this study. Although the function of PPAR is involved in steatosis, the mechanism of how PPAR contributes to NAFLD is unclear. It is necessary to determine the PPAR gene function and to study the phenotype if we block the PPAR signaling pathway. Also, the inconsistence of TWIST1 gene expression at protein level between in vitro, in vivo and in patient samples is needed to be fully examined. The mechanism of downregulation of the TWIST2 gene expression in 3rd month is also needed to be understood. Together this paper demonstrates the importance of the TWIST2 gene in the clinical prognosis and roles of PPAR and TWIST1 in NAFLD development needed fully investigated.
Conclusions
In summary, the present study shows that the TWIST2 and PPAR signaling pathways are important in NAFLD and sheds light on a potential mechanism of steatosis. The present findings establish TWIST2 as a new biomarker for the early detection of NAFLD. A simple blood test for TWIST2, a potential biomarker, may allow one to detect the early stages of NAFLD. Future studies on the PPAR signaling pathway will be urgent to clarify the roles of these genes in NAFLD.
Availability of data and materials
The data will be available on request.
Abbreviations
- TWIST1:
-
Twist family bHLH transcription factor 1
- TWIST2:
-
Twist family bHLH transcription factor 2
- PPARγ:
-
Peroxisome proliferator activated receptor gamma
- NAFLD:
-
Nonalcoholic fatty liver disease
- ELISA:
-
Enzyme-linked immunosorbent assay
- IL-17:
-
Interleukin (IL)-17
- TNF:
-
Tumor necrosis factor
- IR:
-
Insulin resistance
- NASH:
-
Nonalcoholic steatohepatitis
- VLDL:
-
very Low density lipoprotein
- EMT:
-
Epithelial-to-mesenchymal transition
- IPGTT:
-
Intraperitoneal glucose tolerance test
- IPITT:
-
Intraperitoneal insulin tolerance test
- H&E:
-
Hematoxylin and eosin
- PBS:
-
Phosphate-buffered saline;
- RIPA:
-
Radioimmunoprecipitation assay
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- BMI:
-
Body mass index
- HOMA:
-
Homeostasis model assessment
- DE:
-
Differentially expressed
- ACSL4:
-
Acyl-CoA synthetase long chain family member 4
- ANGPTL4:
-
Angiopoietin like 4
- CPT1A:
-
Carnitine palmitoyltransferase 1A
- PLIN4:
-
Perilipin 4
- HSCs:
-
Hepatic stellate cells
References
Povero D, Feldstein AE. Novel molecular mechanisms in the development of non-alcoholic Steatohepatitis. Diabetes Metab J. 2016;40(1):1–11. https://doi.org/10.4093/dmj.2016.40.1.1.
Bellentani S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017;37(Suppl 1):81–4. https://doi.org/10.1111/liv.13299.
Watanabe S, Hashimoto E, Ikejima K, Uto H, Ono M, Sumida Y, et al. Evidence-based clinical practice guidelines for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatol Res. 2015;45(4):363–77. https://doi.org/10.1111/hepr.12511.
Bashiri A, Nesan D, Tavallaee G, Sue-Chue-Lam I, Chien K, Maguire GF, et al. Cellular cholesterol accumulation modulates high fat high sucrose (HFHS) diet-induced ER stress and hepatic inflammasome activation in the development of non-alcoholic steatohepatitis. Biochim Biophys Acta. 2016;1861:594–605.
Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52(5):1836–46. https://doi.org/10.1002/hep.24001.
Takaki A, Kawai D, Yamamoto K. Molecular mechanisms and new treatment strategies for non-alcoholic steatohepatitis (NASH). Int J Mol Sci. 2014;15(5):7352–79. https://doi.org/10.3390/ijms15057352.
Okubo H, Kushiyama A, Sakoda H, Nakatsu Y, Iizuka M, Taki N, et al. Involvement of resistin-like molecule beta in the development of methionine-choline deficient diet-induced non-alcoholic steatohepatitis in mice. Sci Rep. 2016;6(1):20157. https://doi.org/10.1038/srep20157.
Petta S, Muratore C, Craxi A. Non-alcoholic fatty liver disease pathogenesis: the present and the future. Dig Liver Dis. 2009;41(9):615–25. https://doi.org/10.1016/j.dld.2009.01.004.
Gruben N, Shiri-Sverdlov R, Koonen DP, Hofker MH. Nonalcoholic fatty liver disease: a main driver of insulin resistance or a dangerous liaison? Biochim Biophys Acta. 2014;1842:2329–43.
Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2011;8(1):35–44. https://doi.org/10.1038/nrgastro.2010.191.
Kim JY, Lee C, Oh M, Im JA, Lee JW, Chu SH, et al. Relationship between non-alcoholic fatty liver disease, metabolic syndrome and insulin resistance in Korean adults: a cross-sectional study. Clin Chim Acta. 2016;458:12–7. https://doi.org/10.1016/j.cca.2016.03.018.
Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30(1):255–89. https://doi.org/10.1146/annurev-cellbio-101512-122326.
Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology. 2016;150(4):956–67. https://doi.org/10.1053/j.gastro.2015.12.037.
Ren R, Chen Z, Zhao X, Sun T, Zhang Y, Chen J, et al. A possible regulatory link between twist 1 and PPARgamma gene regulation in 3T3-L1 adipocytes. Lipids Health Dis. 2016;15(1):189. https://doi.org/10.1186/s12944-016-0361-x.
Lu S, Wang H, Ren R, Shi X, Zhang Y, Ma W. Reduced expression of twist 1 is protective against insulin resistance of adipocytes and involves mitochondrial dysfunction. Sci Rep. 2018;8(1):12590. https://doi.org/10.1038/s41598-018-30820-z.
Zhou L, Li Q, Chen A, Liu N, Chen N, Chen X, et al. via KLF15-activating Twist2 ameliorated hepatic steatosis by inhibiting inflammation and improving mitochondrial dysfunction NF-κB-FGF21 or SREBP1c-FGF21 pathway. FASEB J. 2019;33(12):14254–69. https://doi.org/10.1096/fj.201901347RR.
Franco HL, Casasnovas J, Rodriguez-Medina JR, Cadilla CL. Redundant or separate entities?--roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res. 2011;39(4):1177–86. https://doi.org/10.1093/nar/gkq890.
Merindol N, Riquet A, Szablewski V, Eliaou JF, Puisieux A, Bonnefoy N. The emerging role of twist proteins in hematopoietic cells and hematological malignancies. Blood Cancer J. 2014;4(4):e206. https://doi.org/10.1038/bcj.2014.22.
Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14(1):79–89. https://doi.org/10.1016/j.ccr.2008.06.005.
Sosic D, Richardson JA, Yu K, Ornitz DM, Olson EN. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell. 2003;112(2):169–80. https://doi.org/10.1016/S0092-8674(03)00002-3.
Chen L, Chen R, Kemper S, Charrier A, Brigstock DR. Suppression of fibrogenic signaling in hepatic stellate cells by Twist1-dependent microRNA-214 expression: role of exosomes in horizontal transfer of Twist1. Am J Physiol Gastrointest Liver Physiol. 2015;309(6):G491–9. https://doi.org/10.1152/ajpgi.00140.2015.
Lallukka S, Sevastianova K, Perttila J, Hakkarainen A, Orho-Melander M, Lundbom N, et al. Adipose tissue is inflamed in NAFLD due to obesity but not in NAFLD due to genetic variation in PNPLA3. Diabetologia. 2013;56(4):886–92. https://doi.org/10.1007/s00125-013-2829-9.
Moran-Salvador E, Lopez-Parra M, Garcia-Alonso V, Titos E, Martinez-Clemente M, Gonzalez-Periz A, et al. Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011;25(8):2538–50. https://doi.org/10.1096/fj.10-173716.
Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, et al. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278(36):34268–76. https://doi.org/10.1074/jbc.M300043200.
Farrell GC, van Rooyen D, Gan L, Chitturi S. NASH is an inflammatory disorder: pathogenic, prognostic and therapeutic implications. Gut Liver. 2012;6(2):149–71. https://doi.org/10.5009/gnl.2012.6.2.149.
Del Campo JA, Gallego P, Grande L. Role of inflammatory response in liver diseases: therapeutic strategies. World J Hepatol. 2018;10(1):1–7. https://doi.org/10.4254/wjh.v10.i1.1.
Beringer A, Miossec P. IL-17 and IL-17-producing cells and liver diseases, with focus on autoimmune liver diseases. Autoimmun Rev. 2018;17(12):1176–85. https://doi.org/10.1016/j.autrev.2018.06.008.
Braunersreuther V, Viviani GL, Mach F, Montecucco F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J Gastroenterol. 2012;18(8):727–35. https://doi.org/10.3748/wjg.v18.i8.727.
Kakino S, Ohki T, Nakayama H, Yuan X, Otabe S, Hashinaga T, et al. Pivotal role of TNF-alpha in the development and progression of nonalcoholic fatty liver disease in a murine model. Horm Metab Res. 2018;50:80–7.
Younossi ZM. Non-alcoholic fatty liver disease - a global public health perspective. J Hepatol. 2019;70(3):531–44. https://doi.org/10.1016/j.jhep.2018.10.033.
Chang CC, Wang SS, Hsieh HG, Lee WS, Chuang CL, Lin HC, et al. Rosuvastatin improves hepatopulmonary syndrome through inhibition of inflammatory angiogenesis of lung. Clin Sci (Lond). 2015;129(6):449–60. https://doi.org/10.1042/CS20140622.
Acknowledgments
RNA sequencing analysis was performed by KangChen Bio-tech, Shanghai, China.
Funding
This study was funded by the Cultivation Fund of National Natural Science Foundation of China in Shandong Provincial Qianfoshan Hospital (No. QYPY2020NSFC1004), the Key Technology Research and Development Program of Shandong (No. 2017G006024), National Natural Science Foundation of China (No. 81400843), and Natural Science Foundation of Shandong Province (No. ZR2014HP033).
Author information
Authors and Affiliations
Contributions
SML and WSM contributed to funding acquisition, study concept, study design, data interpretation and revision of the manuscript. YMZ and XXG performed most of the experiments and contributed to data collection, data analysis and original draft writing; YQL, MJH, and YYZ contributed to data analysis; BYZ, PG, and TS performed some part of the research. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was performed in accordance with ethical standards and was approved by the Ethics Committee of Shandong University (No: [2017] S048). Informed consent was obtained from all individual participants included in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Y., Ge, X., Li, Y. et al. TWIST2 and the PPAR signaling pathway are important in the progression of nonalcoholic steatohepatitis. Lipids Health Dis 20, 39 (2021). https://doi.org/10.1186/s12944-021-01458-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-021-01458-0