
Abstract
Background
The current research aimed to expound the genes and pathways that are involved in coronary artery disease (CAD) and ischaemic stroke (IS) and the related mechanisms.
Methods
Two array CAD datasets of (GSE66360 and GSE97320) and an array IS dataset (GSE22255) were downloaded. Differentially expressed genes (DEGs) were identified using the limma package. The online tool Database for Annotation, Visualization and Integrated Discovery (DAVID) (version 6.8; david.abcc.ncifcrf.gov) was used to annotate the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) enrichment analyses of the DEGs. A protein-protein interaction (PPI) network was constructed by Cytoscape software, and then Molecular Complex Detection (MCODE) analysis was used to screen for hub genes. The hub genes were also confirmed by RT-qPCR and unconditional logistic regression analysis in our CAD and IS patients.
Results
A total of 20 common DEGs (all upregulated) were identified between the CAD/IS and control groups. Eleven molecular functions, 3 cellular components, and 49 biological processes were confirmed by GO enrichment analysis, and the 20 common upregulated DEGs were enriched in 21 KEGG pathways. A PPI network including 24 nodes and 68 edges was constructed with the STRING online tool. After MCODE analysis, the top 5 high degree genes, including Jun proto-oncogene (JUN, degree = 9), C-X-C motif chemokine ligand 8 (CXCL8, degree = 9), tumour necrosis factor (TNF, degree = 9), suppressor of cytokine signalling 3 (SOCS3, degree = 8) and TNF alpha induced protein 3 (TNFAIP3, degree = 8) were noted. RT-qPCR results demonstrated that the expression levels of CXCL8 were increased in IS patients than in normal participants and the expression levels of SOCS3, TNF and TNFAIP were higher in CAD/IS patients than in normal participants. Meanwhile, unconditional logistic regression analysis revealed that the incidence of CAD or IS was positively correlated with the CXCL8, SOCS3, TNF and TNFAIP3.
Conclusions
The CXCL8, TNF, SOCS3 and TNFAIP3 associated with inflammation may serve as biomarkers for the diagnosis of CAD or IS. The possible mechanisms may involve the Toll-like receptor, TNF, NF-kappa B, cytokine-cytokine receptor interactions and the NOD-like receptor signalling pathways.
Similar content being viewed by others

Background
Coronary artery disease (CAD) and ischaemic stroke (IS) are prominent causes of disability, mortality, morbidity, functional deterioration and healthcare expenses and account for approximately 30% of all deaths worldwide [1,2,3,4]. Twins and family studies have proven that both CAD and IS are highly heritable [5, 6], and hereditary elements are thought to account for approximately 30–60% of CAD and IS cases [7]. Atherosclerosis is generally regarded as the pathological foundation of CAD [8] and IS [9]. In addition, there is some evidence of several shared genetic characteristics of both diseases [10]. Both diseases are risk factors for one another [11, 12], and they are considered to be therapeutic targets for clinical research and for evaluating the risk of major adverse cardiac events (MACEs). A recent study showed that CAD and IS result from various factors and can be influenced by genomic background, lifestyle, environmental factors and alterations of plasma lipid levels as well as their interactions with each other [13]. To some extent, there is a consensus on the effectiveness of the early prevention of CAD and IS.
As a novel and practical approach for identifying CAD and IS susceptibility genes, a microarray analysis may be helpful for the early diagnosis of CAD and IS [14, 15]. However, the sensitivity and reproducibility of microarray results may be limited [16, 17]. Thus, a comprehensive analysis may be useful to improve the reliability and integrity of the conclusions. Through this method, we can achieve a more accurate approach of identifying susceptibility genes for CAD and IS and further explore their potential biological functions. The Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) [18] is an international public database for next-generation sequence functional genomic datasets and high-throughput microarray data submitted by researchers worldwide. In this study, we downloaded two CAD datasets (GSE66360 and GSE97320) and one IS dataset (GSE22255) to identify differentially expressed genes (DEGs) in patients suffering from CAD or IS and healthy controls. The purpose of the present research was to confirm new biomarkers for the early diagnosis of CAD and IS.
Materials and methods
CAD and IS microarray data sets
Two CAD datasets (GSE66360 and GSE97320) and another IS dataset pf IS (GSE22255) were obtained from the GPL570 Affymetrix Human Genome U133 Plus 2.0 array. The GSE22255 dataset included 20 normal samples and 20 IS samples. An integrated analysis of 53 normal samples and 52 CAD samples from the two CAD datasets was performed. The original files in CEL format were transformed into an expression value matrix using the Affy package in R with the RMA method to normalize the expression values and the SVA method to remove batch differences [19]. Then, the bioconductor package was used to transform the probe ID into a gene symbol [20]. When multiple probes corresponded to one common gene, the average value was taken as its expression value.
Differentially expressed gene (DEG) identification
The DEGs between patients suffering from CAD or IS and healthy participants were identified using the limma package [21]. The threshold values were P < 0.05 and |log fold change (FC)| > 1. To visualize the shared DEGs between the CAD datasets and the IS dataset, an online tool (bioinformatics.psb.ugent.be/webtools/Venn) was used to draw a Venn diagram.
Functional enrichment analysis
The online tool Database for Annotation, Visualization and Integrated Discovery (DAVID) (version 6.8; david.abcc.ncifcrf.gov) was used to annotate the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway [22] and Gene Ontology (GO) enrichment analyses [23] of the common differentially expressed genes. P < 0.05 was defined as the threshold for significant enrichment for KEGG and GO analyses.
PPI interaction network construction and module analysis
A PPI interaction network of common DEGs was constructed with the Search Tool for the Retrieval of Interacting Genes database (version 11.0; www.string-db.org) [24], and a combined score of > 0.9 was defined as the cut-off value. Cytoscape 3.7.1 (www.cytoscape.org) was applied to visualize the PPI network [25]. Degrees were used to verify the significance of protein nodes in the PPI network. As one of the core components of the PPI network, the network module may have specific biological functions. The Cytoscape software (version 3.61) Molecular Complex Detection (MCODE) plugin was used to identify the most common and largest module clusters with the following parameters: EASE ≤0.05, count ≥2 and MCODE score > 6 [26].
Sample verification and diagnostic criteria
A total of 420 unrelated participants (202 IS patients and 218 CAD patients) were recruited from the First Affiliated Hospital of Guangxi Medical University from Jan. 1, 2015 to Dec. 31, 2016. CAD was defined as significantly coronary artery stenosis (≥ 50%) in at least anyone of the three main coronary vessels or their main branches (branch diameter ≥ 2 mm) [27]. All patients with IS received a brain magnetic resonance imaging (MRI) scan and strict neurological examination. The diagnostic criteria for IS were derived from the International Classification of Diseases (9th Revision). All subjects with a history of type 1 diabetes, neoplasm, autoimmune disorder, abnormal renal or liver function, haemopathy or thyroid dysfunction were excluded. The patients with CAD had no history of IS, and the patients with IS had no history of CAD.
A total of 203 healthy controls matched by ethnic group (Han Chinese), age, and gender were also recruited. All subjects were healthy, and none of them had a history of CAD, myocardial infarction, IS or type 2 diabetes mellitus (T2DM), as determined by history-taking, questionnaires, or critical clinical examination. All participants were randomly recruited from the Physical Examination Center of the First Affiliated Hospital, Guangxi Medical University in the same period. Before the beginning of the study, all participants signed a written informed consent form. The research proposal was approved by the Ethics Committee of the First Affiliated Hospital, Guangxi Medical University (No: Lunshen-2011-KY-Guoji-001; Mar. 7, 2011).
Quantitative real-time PCR
RT-qPCR was used to validate the four significantly dysregulated mRNAs identified by the microarray results in the 603 subjects. Total RNA was extracted from peripheral blood mononuclear cells (PBMCs) that were separated from blood samples using TRIzol reagent and reverse transcribed into cDNA using the PrimeScript RT reagent kit (Takara Bio, Japan) according to the manufacturer’s instructions. The resulting cDNA was used as a template for RT-qPCR. Supplementary Table 2 shows the sequences of the specific primers designed by Sangon Biotech (Shanghai, China) and used to detect the 5 hub genes. Quantitative RT-PCR was performed using Taq PCR Master Mix Kit (Takara) on an ABI Prism 7500 sequence-detection system (Applied Biosystems, USA) using RT Reaction Mix in a total volume of 20 μL with conditions of 95 °C pre-denaturation for 30 s, 95 °C for 30 s, and 60 °C for 30 s for 40 cycles.
Diagnostic criteria
In our Clinical Science Experiment Center, 0.56–1.70 mmol/L serum triglyceride (TG), 3.10–5.17 mmol/L total cholesterol (TC), 0.80–1.05 g/L apolipoprotein (Apo) B, 2.70–3.10 mmol/L low-density lipoprotein cholesterol (LDL-C), 1.20–1.60 g/L ApoA1, 1.16–1.42 mmol/L high-density lipoprotein cholesterol (HDL-C) and a ApoA1/ApoB ratio of 1.00–2.50 were defined as normal values. The diagnostic criteria of hyperlipidaemia [28], hypertension [29], obesity, normal weight, and overweight [30] were referred to in previous studies. Participants who had been previously diagnosed with diabetes and participants with 2-h postprandial plasma glucose ≥11.1 mmol/L or fasting plasma glucose ≥7.0 mmol/L were defined as diabetic patients [31].
Statistical analyses
All data were x (Version 22.0). The values are presented as the mean ± SD. The chi-square test was used to calculate the differences in the rates between patients and controls. Independent samples t test was used to analyse differences in general characteristics between patients and controls. Unconditional logistic regression was used to evaluate the relationship between genes and clinical variables and the incidence of CAD or IS. The pheatmap and ggplot2 packages (https://cran.r-project.org/) were used to draw the volcano plot and heat map.
Results
Identification of DEGs in GSE97320, GSE66360 and GSE22255
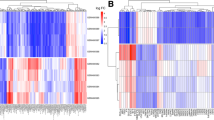
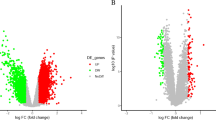
After data normalization and removed of batch differences, a total of 643 genes, including 178 downregulated genes and 465 upregulated genes, were defined as DEGs between the patients with CAD and healthy controls according to the following criteria: |logFC| > 1 and P < 0.05. A total of 29 DEGs, including 2 downregulated genes and 27 upregulated genes were identified between IS patients and healthy controls, and 20 common upregulated DEGs between the CAD patients and controls and between IS patients and controls were identified (Table 1 and Fig. 1). Analysis of heatmap clustering and the volcano plot showed that the identified DEGs can easily distinguish patients with CAD or IS from healthy controls (Figs. 2 and 3).

Venn map showing the intersection of DEGs between CAD vs normal and IS vs normal

Cluster heat maps of DEGs. Red represents CAD/IS group and green represents control group. a: Top 50 up-regulated DEGs between CAD and control; b: Top 27 up-regulated and 2 down- regulated DEGs between IS and control

Volcano plots of DEGs. Up-regulated genes are marked with red dots, and down-regulated genes are marked with green dots. a: CAD vs. control; b: IS vs. control
KEGG pathway and GO functional enrichment analysis
The online tool DAVID was used to predict the potential biological functions of the DEGs. A total of 21 KEGG pathways, including the Toll-like receptor signalling pathway (TNF, JUN, CXCL8, and IL1B); the NF-kappa B signalling pathway (TNF, CXCL8, IL1B, and TNFAIP3); the TNF signalling pathway (TNF, SOCS3, JUN, IL1B, and TNFAIP3), 11 molecular functions, 3 cellular components, and 49 biological processes were enriched in the present study, and GO:0006915~apoptotic process (IL1B and TNFAIP3), GO:0042346~positive regulation of NF-kB import into nucleus (TNF and IL1B), GO:0045429~positive regulation of nitric oxide biosynthetic process (TNF and IL1B), GO:0006954~inflammatory response (TNF, CXCL8, IL1B, and TNFAIP3), GO:0050995~negative regulation of lipid catabolic process (TNF and IL1B), GO:0034116~positive regulation of heterotypic cell-cell adhesion (TNF and IL1B), GO:0048661~positive regulation of smooth muscle cell proliferation (TNF and JUN), GO:0010803~regulation of tumour necrosis factor-mediated signalling pathway (TNF and TNFAIP3), GO:0001525~angiogenesis (JUN and CXCL8) and GO:0043122~regulation of I-kappaB kinase/NF-kB signalling (TNF and IL1B) were selected for further analysis, as presented in Fig. 4. More detailed information is presented in Supplementary Table 1.

Functional annotation for DEGs. a GO enrichment analysis of DEGs; b KEGG pathways analysis of DEGs
PPI network construction and module analysis for DEGs
A PPI network including 24 nodes and 68 edges was constructed with the STRING online tool. After MCODE analysis, the top 5 high degree genes, including C-X-C motif chemokine ligand 8 (CXCL8, degree = 9), Jun proto-oncogene (JUN, degree = 9), tumour necrosis factor (TNF, degree = 9), suppressor of cytokine signalling 3 (SOCS3, degree = 8), and TNF alpha induced protein 3 (TNFAIP3, degree = 8), were identified in the present study (Fig. 5).

PPI network construction and identification of hub genes. a PPI network of the selected DEGs. The edge shows the interaction between two genes. Significant modules identified from the PPI network using the MCODE with a score > 6.0. b Moldule-1 with MCODE = 9
Validation by RT-qPCR
The RT-qPCR results revealed that the expression levels of CXCL8 were increased in IS patients than in normal participants and the expression levels of SOCS3, TNF and TNFAIP3 genes were higher in CAD/IS patients than in normal participants. Meanwhile, there was no difference in the expression of JUN between CAD/IS patients and the control group. The RT-qPCR results in our study were in accordance with the results of the microarray analysis (Fig. 6). The primer sequences for the abovementioned genes are shown in Supplementary Table 2.

Relative expression levels of five hub genes identified from the microarray data were verified by RT-qPCR. *P < 0.05
Biochemical characteristics and unconditional logistic regression analysis
As mentioned in Table 2, the female to male ratio, age, serum ApoB levels, the proportion of drinkers, height and diastolic blood pressure were similar between the controls and patients. The proportion of smokers, weight, systolic blood pressure, glucose, body mass index (BMI), pulse pressure, and serum LDL-C, TG and TC levels were significantly lower and serum ApoA1, HDL-C levels and the ApoA1/ApoB ratio were significantly higher in controls than in both CAD and IS patients.
Unconditional logistic regression analysis revealed that the overexpression of CXCL8, SOCS3, TNF and TNFAIP3, hyperlipidaemia, smoking and diabetes were considered independent risk factors for the incidence of CAD or IS; the incidence of IS was also positively correlated with hypertension, and the incidence of CAD was negatively correlated with alcohol consumption (Fig. 7).

The relative risk factors for CAD and IS CAD coronary artery disease; IS ischemic stroke. *P < 0.05. **P < 0.01
Discussion
Currently, the diagnosis of CAD is based on ischaemia-related symptoms, detailed physical examination, electrocardiogram changes, elevated biomarkers of myocardial injury and coronary angiography [32, 33]. Meanwhile, the diagnosis of ischaemic stroke (IS) is also based on the patient’s symptoms, signs, strict neurological examination and MRI scans [34]. However, the early diagnosis of CAD and IS is still limited. As a novel and practical approach for identifying CAD and IS susceptibility genes, microarray analysis may be helpful for the early diagnosis of CAD and IS [14, 15]. However, the sensitivity and reproducibility of microarray results may be limited [16, 17]. Thus, it is important for us to identify several new biomarkers for the early diagnosis of CAD and IS through the integrated analysis of different datasets. Therefore, in the present research, we integrated and analysed two different CAD datasets and an IS dataset, identified 20 common DEGs to further and analysed their KEGG pathways, GO functional enrichment, and PPI networks and modules to define five significantly DEGs (CXCL8, TNF, SOCS3, TNFAIP3, and JUN). However, when we verified the above results in our experiment, we found that the expression of CXCL8, TNF, SOCS3, and TNFAIP3 was higher in patients with CAD or IS than in healthy controls and that there was no significant difference in the expression of JUN between CAD or IS patients and the control group.
A recent study showed that CAD and IS result from various factors and can be influenced by genomic background, lifestyle, environmental factors, alterations in plasma lipid levels and the interactions of these factors [13]. Atherosclerosis is generally regarded as the pathological foundation of CAD [8] and IS [9]. Actually, atherosclerosis is not only a lipid-driven disease, but also a type of chronic inflammatory process involving numerous inflammatory cells and mediators [35]. The toll-like receptor signalling pathway plays a crucial role in adaptive and innate immune responses and represents an important medium between inflammation and atherosclerosis [36]. Toll-like receptors are the most characteristic pattern recognition receptors in the innate immune system and are expressed in all types of leukocytes, such as B, T and DC lymphocytes and macrophages/monocytes. The involvement of Toll-like receptors in immune and inflammatory responses may play a crucial role in various aspects of the formation and development of atherosclerotic lesions, and this effect may be related to multiple biological processes, including foam cell formation, the induction of leukocyte recruitment, lipid uptake and proinflammatory cytokine release, which are all facilitated by Toll-like receptors [37]. In the present study, enrichment analysis of KEGG pathways suggested that the TNF, CXCL8 and IL1B genes may be involved in the Toll-like receptor signalling pathway. Thus, we speculated that these genes might exert their biological functions through the Toll-like receptor signalling pathway.
The TNF signalling pathway plays a crucial role in inflammatory and autoimmune diseases. TNF-α is one of the most important members of the TNF superfamily is mainly secreted by macrophages and participates in the regulation of a wide spectrum of biological processes, including cell proliferation, lipid metabolism, apoptosis and differentiation. All of the above biological processes can lead to chronic immunoinflammatory lesions that eventually result in atherosclerosis [38]. Previous studies have proven that the NF-kappa B (NF-kB) signalling pathway plays a key role in the inflammatory reaction, leading to the transcription of genes involved in endothelial inflammation and injury. TNF-α is a major inflammatory cytokine involved in activating the NF-kB signalling pathway to induce the production of more inflammation mediators and reactive oxygen species [39, 40]. At the same time, numerous scientific studies have shown that, in atherogenesis, increased levels of IL-1β and TNF-α, as the two leading mediators of the inflammatory response, in result from increased transcriptional activity of the NF-kB gene [41, 42]. A recent compelling study showed that quercetin may play an anti-inflammatory role in the treatment of stable coronary heart disease by reducing the transcriptional activity of the NF-kB gene [43]. Similar studies have also shown that NF-kB acts as a key regulator of various genes involved in inflammation and cell survival and is activated after cerebral ischaemia in microglia, neurons, astrocytes and infiltrating inflammatory cells [44]. These results show that the TNF and NF-kB signalling pathways may be involved in the development of CAD and IS. In the present study, enrichment analysis of KEGG pathways indicated that the TNF, SOCS3, JUN, and TNFAIP3 genes may be involved in the TNF signalling pathway and that TNF, CXCL8 and TNFAIP3 may be involved in the NF-kB signalling pathway. Meanwhile, cytokine-cytokine receptor interactions (TNF, CXCL8, and IL1B) and the NOD-like receptor signalling pathway (TNF, CXCL8, IL1B, TNFAIP3) were also identified in our study.
In addition, several main biological processes that may be involved in chronic inflammatory lesions that eventually result in atherosclerosis were identified by GO functional enrichment analysis of DEGs, such as GO:0006915~apoptotic process (TNFAIP3), GO:0042346~positive regulation of NF-kB import into nucleus (TNF), GO:0045429~positive regulation of nitric oxide biosynthetic process (TNF), GO:0006954~inflammatory response (TNF, CXCL8 and TNFAIP3), GO:0050995~negative regulation of lipid catabolic process (TNF), GO:0048661~positive regulation of smooth muscle cell proliferation (TNF), GO:0010803~regulation of tumour necrosis factor-mediated signalling pathway (TNF and TNFAIP3), GO:0001525~angiogenesis (CXCL8), GO:0042517~positive regulation of tyrosine phosphorylation of Stat3 protein (SOCS3) and GO:0043122~regulation of I-kappaB kinase/NF-kB signalling (TNF). This information about the relevant pathways is not novel; however, the analytical approach is different from that of previous studies. Thus, the combination of previous and current research results revealed that TNF, CXCL8, SOCS3 and TNFAIP3 may be involved in chronic inflammatory lesions that eventually result in atherosclerosis, CAD or IS. Furthermore, RT-qPCR and unconditional logistic regression also validated the above results in our CAD and IS patients. We obtained results that were consistent with those of the microarray analysis, which might increase the credibility of the conclusions.
Conclusions
Two microarray CAD datasets and an IS dataset were integrated and analysed in the present study. Five hub genes (SOCS3, JUN, TNF, CXCL8, and TNFAIP3) were identified following GO functional enrichment analysis, KEGG pathway enrichment analysis, PPI network construction and MCODE analysis, but only four genes (SOCS3, TNF, CXCL8, and TNFAIP3) were verified by RT-qPCR in our CAD or IS patients. The CXCL8, TNF, SOCS3, TNFAIP3 genes, which are associated with inflammation, may serve as biomarkers for the diagnosis of CAD or IS. The mechanism may involve the TNF signalling pathway, the Toll-like receptor signalling pathway, the NF-kappa B signalling pathway, cytokine-cytokine receptor interactions and the NOD-like receptor signalling pathway.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Apo:
-
Apolipoprotein
- BMI:
-
Body mass index
- CAD:
-
Coronary artery disease
- CXCL8:
-
C-X-C motif chemokine ligand 8
- DAVID:
-
Database for Annotation, Visualization and Integrated Discovery
- DEGs:
-
Differentially expressed genes
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene Ontology
- HDL-C:
-
High-density lipoprotein cholesterol
- IS:
-
Ischemic stroke
- JUN:
-
Jun proto-oncogene
- KEGG:
-
Kyoto Encyclopedia of Genes and genomes
- LDL-C:
-
Low-density lipoprotein cholesterol
- logFC:
-
Log fold change
- MACEs:
-
Major adverse cardiac events
- MCODE:
-
Molecular Complex Detection
- MRI:
-
Magnetic resonance imaging
- NF-kB:
-
NF-kappa B
- PPI:
-
Protein-protein interaction
- RT-PCR: SOCS3:
-
Suppressor of cytokine signaling 3
- T2DM:
-
Type 2 diabetes mellitus
- TC:
-
Total cholesterol
- TG:
-
Triglyceride
- TNF:
-
Tumor necrosis factor
- TNFAIP3:
-
TNF alpha induced protein 3
References
Castelli WP. Cholesterol and lipids in the risk of coronary artery disease--the Framingham Heart Study. Can J Cardiol. 1988;4(Suppl A):5A–10A.
Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet. 2012;380(9859):2095–128.
Finegold JA, Asaria P, Francis DP. Mortality from ischaemic heart disease by country, region, and age: statistics from World Health Organisation and United Nations. Int J Cardiol. 2013;168(2):934–45.
Bonita R. Epidemiology of stroke. Lancet. 1992;339(8789):342–4.
Roberts R, Stewart AF. The genetics of coronary artery disease. Curr Opin Cardiol. 2012;27(3):221–7.
Dichgans M. Genetics of ischaemic stroke. Lancet Neurol. 2007;6(2):149–61.
Marenberg ME, Risch N, ., Berkman LF, Floderus B, ., Faire U, De. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med 1994; 330(15): 1041–1046.
Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111(25):3481–8.
Holmstedt CA, Turan TN, Chimowitz MI. Atherosclerotic intracranial arterial stenosis: risk factors, diagnosis, and treatment. Lancet Neurol. 2013;12(11):1106–14.
Banerjee A, Lim CC, Silver LE, Welch SJ, Banning AP, Rothwell PM. Familial history of stroke is associated with acute coronary syndromes in women. Circ Cardiovasc Genet. 2011;4(1):9–15.
Kannel WB, Wolf PA, Verter J. Manifestations of coronary disease predisposing to stroke. The Framingham study. Jama. 1983;250(21):2942–6.
Sacco RL, Benjamin EJ, Broderick JP, Dyken M, Easton JD, Feinberg WM, Goldstein LB, Gorelick PB, Howard G, Kittner SJ, Manolio TA, Whisnant JP, et al. American Heart Association prevention conference. IV. Prevention and rehabilitation of stroke. Risk factors. Stroke. 1997;28(7):1507–17.
Zhang Q-H, Yin R-X, Chen W-X, Cao X-L, Wu J-Z. TRIB1 and TRPS1 variants, G × G and G × E interactions on serum lipid levels, the risk of coronary heart disease and ischemic stroke. Sci Rep. 2019;9(1):2376.
Sinnaeve PR, Donahue MP, Grass P, Seo D, Vonderscher J, Chibout SD, Kraus WE, Sketch M Jr, Nelson C, Ginsburg GS, Goldschmidt-Clermont PJ, Granger CB. Gene expression patterns in peripheral blood correlate with the extent of coronary artery disease. PLoS One. 2009;4(9):e7037.
Krug T, Gabriel JP, Taipa R, Fonseca BV, Domingues-Montanari S, Fernandez-Cadenas I, Manso H, Gouveia LO, Sobral J, Albergaria I, Gaspar G, Jimenez-Conde J, et al. TTC7B emerges as a novel risk factor for ischemic stroke through the convergence of several genome-wide approaches. J Cereb Blood Flow Metab. 2012;32(6):1061–72.
Ntzani EE, Ioannidis JP. Predictive ability of DNA microarrays for cancer outcomes and correlates: an empirical assessment. Lancet. 2003;362(9394):1439–44.
Ein-Dor L, Kela I, Getz G, Givol D, Domany E. Outcome signature genes in breast cancer: is there a unique set? Bioinformatics. 2005;21(2):171–8.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41(Database issue):D991–5.
Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–15.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–52.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2.
Franceschini N, Carty C, Buzkova P, Reiner AP, Garrett T, Lin Y, Vockler JS, Hindorff LA, Cole SA, Boerwinkle E, Lin DY, Bookman E, et al. Association of genetic variants and incident coronary heart disease in multiethnic cohorts: the PAGE study. Circ Cardiovasc Genet. 2011;4(6):661–72.
Ramazauskiene V. Diet and serum lipids: changes over socio-economic transition period in Lithuanian rural population. BMC Public Health. 2011;11(1):447.
Whitworth JA. 2003 World Health Organization (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J Hypertens. 2003;21(11):1983–92.
Zhou BF. Predictive values of body mass index and waist circumference for risk factors of certain related diseases in Chinese adults--study on optimal cut-off points of body mass index and waist circumference in Chinese adults. Biomed Environ Sci. 2002;15(1):83–96.
Alberti G, Zimmet PZ: Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus. Provisional report of a WHO Consultation. 1998.
Su J, Gao C, Wang R, Xiao C, Yang M. Genes associated with inflammation and the cell cycle may serve as biomarkers for the diagnosis and prognosis of acute myocardial infarction in a Chinese population. Mol Med Rep. 2018;18(2):1311–22.
Li WJ, Yin RX, Cao XL, Chen WX, Huang F, Wu JZ. DOCK7-ANGPTL3 SNPs and their haplotypes with serum lipid levels and the risk of coronary artery disease and ischemic stroke. Lipids Health Dis. 2018;17(1):30.
Miao L, Yin RX, Huang F, Chen WX, Cao XL, Wu JZ. The effect of MVK-MMAB variants, their haplotypes and GxE interactions on serum lipid levels and the risk of coronary heart disease and ischemic stroke. Oncotarget. 2017;8(42):72801–17.
Li B, Li W, Li X, Zhou H. Inflammation: a novel therapeutic target/direction in atherosclerosis. Curr Pharm Des. 2017;23(8):1216–27.
Lin J, Kakkar V, Lu X. Essential roles of toll-like receptors in atherosclerosis. Curr Med Chem. 2016;23(5):431–54.
Cole JE, Georgiou E, Monaco C. The expression and functions of toll-like receptors in atherosclerosis. Mediators Inflamm. 2010;2010:393946.
Cabal-Hierro L, Lazo PS. Signal transduction by tumor necrosis factor receptors. Cell Signal. 2012;24(6):1297–305.
Read MA, Whitley MZ, Williams AJ, Collins T. NF-kappa B and I kappa B alpha: an inducible regulatory system in endothelial activation. J Exp Med. 1994;179(2):503–12.
Kumar S, Singhal V, Roshan R, Sharma A, Rembhotkar GW, Ghosh B. Piperine inhibits TNF-alpha induced adhesion of neutrophils to endothelial monolayer through suppression of NF-kappaB and IkappaB kinase activation. Eur J Pharmacol. 2007;575(1–3):177–86.
De Martin R, Hoeth M, Hofer-Warbinek R, Schmid JA. The transcription factor NF-kappa B and the regulation of vascular cell function. Arterioscler Thromb Vasc Biol. 2000;20(11):E83–8.
Brunetti ND, Correale M, Pellegrino PL, Munno I, Cuculo A, De Gennaro L, Gaglione A, Di Biase M. Early inflammatory cytokine response: a direct comparison between spontaneous coronary plaque destabilization vs angioplasty induced. Atherosclerosis. 2014;236(2):456–60.
Chekalina N, Burmak Y, Petrov Y, Borisova Z, Manusha Y, Kazakov Y, Kaidashev I. Quercetin reduces the transcriptional activity of NF-kB in stable coronary artery disease. Indian Heart J. 2018;70(5):593–7.
Ridder DA, Schwaninger M. NF-kappaB signaling in cerebral ischemia. Neuroscience. 2009;158(3):995–1006.
Acknowledgements
We thank all the participants of this study.
Funding
This study was supported by the National Natural Science Foundation of China (No: 81460169). There was no role of the funding body in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
P.-F.Z. and F.-J.L. conceived the study, participated in the design, performed the statistical analyses, and drafted the manuscript. R.-X.Y. conceived the study, participated in the design, carried out the epidemiological survey, collected the samples, and helped to draft the manuscript. L.-Z.C., H.L., R.-J.N., Y.W. and P.-J.L. carried out the epidemiological survey and collected the samples. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study design was approved by the Ethics Committee of the First Affiliated hospital, Guangxi Medical University (No: Lunshen-2011-KY-Guoji-001; March 7, 2011). Informed consent was obtained from all participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1
: Table S1 KEGG pathways and GO function enrichment analyses of twenty common upregulated-DEGs. Table S2 PCR primers for quantitative real-time PCR
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zheng, PF., Liao, FJ., Yin, RX. et al. Genes associated with inflammation may serve as biomarkers for the diagnosis of coronary artery disease and ischaemic stroke. Lipids Health Dis 19, 37 (2020). https://doi.org/10.1186/s12944-020-01217-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-020-01217-7