
Abstract
Background
Dietary intervention is the cornerstone of non-alcoholic steatohepatitis (NASH) treatment. However, histological evidence of its efficacy is limited and its impact on hepatic pathways involved in NASH is underreported. The efficacy of the angiotensin receptor type 1 blocker losartan is controversial because of varying results in a few animal and human studies. We evaluated the effect of dietary intervention versus losartan on NASH and associated systemic metabolic features in a representative mouse model.
Methods
Male C57BL/6 J mice with high fat-high sucrose diet (HF-HSD) induced NASH, obesity, insulin resistance and hypercholesterolemia were subjected to dietary intervention (switch from HF-HSD to normal chow diet (NCD)) (n = 9), continuation HF-HSD together with losartan (30 mg/kg/day) (n = 9) or continuation HF-HSD only (n = 9) for 8 weeks. 9 mice received NCD during the entire experiment (20 weeks). We assessed the systemic metabolic effects and performed a detailed hepatic histological and molecular profiling. A P-value of < 0.05, using the group with continuation of HF-HSD only as control, was considered as statistically significant.
Results
Dietary intervention normalized obesity, insulin resistance, and hypercholesterolemia (for all P < 0.001), and remarkably, completely reversed all histological features of pre-existent NASH (for all P < 0.001), including fibrosis measured by quantification of collagen proportional area (P < 0.01). At the hepatic molecular level, dietary intervention targeted fibrogenesis with a normalization of collagen type I alpha 1, transforming growth factor β1, tissue inhibitor of metalloproteinase 1 mRNA levels (for all P < 0.01), lipid metabolism with a normalization of fatty acid translocase/CD36, fatty acid transport protein 5, fatty acid synthase mRNA levels (P < 0.05) and markers related to mitochondrial function with a normalization of hepatic ATP content (P < 0.05) together with sirtuin1 and uncoupling protein 2 mRNA levels (for both P < 0.001). Dietary intervention abolished p62 accumulation (P < 0.01), suggesting a restoration of autophagic flux. Losartan did not significantly affect obesity, insulin resistance, hypercholesterolemia or any histological NASH feature.
Conclusions
Dietary intervention, and not losartan, completely restores the metabolic phenotype in a representative mouse model with pre-existent NASH, obesity, insulin resistance and hypercholesterolemia.
Similar content being viewed by others


Background
Non-alcoholic fatty liver disease (NAFLD) is the most prevalent liver disease in the Western world, affecting 20–30% of the adult population [1]. Due to its strong pathophysiological and epidemiological association with obesity, insulin resistance, dyslipidemia and hypertension, NAFLD is considered as the hepatic manifestation of the metabolic syndrome [2]. NAFLD is the collective noun for a spectrum of histological abnormalities ranging from isolated steatosis to nonalcoholic steatohepatitis (NASH). The latter is characterized by steatosis, hepatocyte ballooning and inflammation with or without fibrosis [3]. NASH can progress to cirrhosis and related complications, including hepatocellular carcinoma [4]. As a consequence, NASH is the most rapidly rising indication for liver transplantation [5, 6].
This societal and medical relevance of NASH has inspired numerous animal studies and human trials in order to find an effective treatment. Dietary modification and physical exercise remain the cornerstones of treatment, although available evidence that these measures can reverse NASH is rather limited [7–11]. When applied together, they may dose-dependently improve the histological features of NASH in a subset of patients, as shown in two clinical trials [8, 11]. Data on the histological effects of diet only interventions on established NASH are even more scarce [10]. However, knowledge of the therapeutic potential of dietary intervention is important, not in the least for patients who cannot increase their level of physical exercise due to physical limitations or lack of time. Furthermore, the mechanisms behind the possible beneficial effects of dietary intervention remain largely underexplored [12].
In addition, not a single pharmacological agent has been approved for the treatment of NASH so far [7, 9, 13]. Research efforts rather improved the insight in NASH pathophysiology, including the role of the renin-angiotensin system (RAS). In the liver, angiotensin II, the main effector of the RAS, is thought to play a role in the development and progression of NASH, possibly by the generation of oxidative stress and inflammatory cytokines leading to steatosis, inflammation and fibrosis [14]. Therefore, attenuating the effects of angiotensin II by administering losartan, an angiotensin II type 1 receptor blocker, could be beneficial for NASH and other components of the metabolic syndrome [14]. However, in vivo evidence regarding the efficacy of losartan is only based on a few studies in non-physiological animal models and two human pilot trials with each a limited number of patients [14–17]. The lack of efficacy of losartan in other studies, further fueled the controversy [18, 19].
Previously, we developed a NASH mouse model induced by the administration of a ‘Western’ high fat-high sucrose diet (HF-HSD), with close resemblance to the hepatic and systemic metabolic profile of typical NASH patients [20]. In the current study, we assessed the therapeutic efficacy of dietary intervention (based on a switch from HF-HSD to normal chow diet) and losartan in this mouse model with pre-existent NASH, insulin resistance, obesity and hypercholesterolemia.
Methods
Animals, diet and experimental set-up
Male C57BL/6 J mice (The Jackson Laboratory, Bar Harbor, Maine, USA) were housed under a 14 h light- 10 h dark cycle at 21–23 °C and had ad libitum access to water during the entire experiment. Mice were fed a ‘Western’ high fat-high sucrose diet (HF-HSD) with 44.6% of kcal derived from fat (of which 61% saturated fatty acids) and 40.6% of kcal derived from carbohydrates (primarily sucrose 340 g/kg diet) (TD.08811, 45%kcal Fat Diet, Harlan Laboratories Inc., Madison, WI, USA) or a normal chow diet (NCD) (V1534-000 ssniff R/M-H, ssniff Spezialdiäten GmbH, Soest, Germany). Losartan (30 mg/kg bodyweight/day) (Cayman Chemical Company, Ann Arbor, MI, USA) was administered via drinking water. Food intake per cage (n = 3–4 mice per cage) was measured when renewing the pellets weekly. The drinking water with losartan was renewed daily.
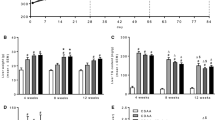
We applied a curative set-up, wherein 27 mice were fed a HF-HSD for 12 weeks (started at age of 6 weeks) to induce the phenotype with NASH, obesity, insulin resistance and hypercholesterolemia, as we published previously [20, 21]. Subsequently, the mice were randomized to the different intervention groups (Fig. 1): 1) HF-HSD group: continuation HF-HSD for the remaining 8 weeks (n = 9); 2) HF-HSD-LOS group: continuation HF-HSD together with losartan for the remaining 8 weeks (n = 9) and 3) Return to NCD group: switch from HF-HSD to NCD (ad libitum) for the remaining 8 weeks of the experiment (n = 9). 9 mice received NCD during the entire experiment (20 weeks) (NCD group). Mean food intake during these last 8 weeks of the experiment was 3.40 g/mouse/day (≈10.37 kcal/mouse/day) for the NCD group, 2.85 g/mouse/day (≈ 13.39 kcal/mouse/day) for the HF-HSD group, 2.74 g/mouse/day (≈ 12.87 kcal/mouse/day) for the HF-HSD-LOS group and 2.88 g/mouse/day (≈ 8.78 kcal/mouse/day) for the Return to NCD group. All procedures were approved by the animal welfare committee of the University of Leuven (protocol number: P088/2011).

Experimental design. Losartan was given in a dose of 30 mg/kg/day via drinking water
In vivo phenotyping
Body weight was monitored weekly on the same day. Two weeks before sacrifice, intra-peritoneal glucose tolerance test (IPGTT) was performed in 6 h fasted mice. Tail vein glucose levels were measured with a Bayer Contour® glucometer immediately before (time point 0 min) and 15, 30, 60, 90 and 150 min after glucose administration (1 g glucose/kg bodyweight). Insulin resistance was estimated using the Homeostasis Model of Insulin Resistance (HOMA-IR) index: (fasting insulin (ng/ml) X fasting glucose (mg/dl))/405 [22].
Sacrifice
Sacrifice was performed as described previously [20]. After a 6 h fasting period, mice were anesthetized with sodium pentobarbital (i.p. injection, 50 mg/kg body weight) and killed by blood sampling via cardiac puncture. Plasma was obtained by centrifugation of blood (6000 rpm for 5 min at 4 °C) that was collected in heparinized syringes. Tissues were either snap frozen in liquid nitrogen and together with the plasma stored at -80 °C till further biochemical and molecular analyses or preserved for histological analysis.
Biochemical analyses
Alanine aminotransferase (ALT) and total cholesterol were measured on 200 μl plasma, obtained through cardiac puncture at sacrifice, via automated procedures. Plasma insulin was measured with a mouse Insulin Elisa Kit (Crystal Chem Inc., Downers Grove, IL, USA). ATP content was measured in liver tissue after homogenization in RIPA lysis buffer (1× PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM sodium orthovanadate (Na3VO4), 200 mM phenylmethanesulfonylfluoride (PMSF) in isopropanol, protease inhibitor cocktail), using the CellTiter-Glo Luminescent Cell Viability Assay (Promega Corp., Madison, WI, USA) according to the manufacturer’s instructions. 4-Hydroxynonenal (HNE), a byproduct of lipid peroxidation and thus a marker of oxidative stress, was measured following the manufacturer’s protocol (OxiSelect HNE-His Adduct ELISA Kit, Cell Biolabs Inc., San Diego, CA, USA).
RNA isolation and quantitative RT-PCR
At sacrifice, tissue samples were collected for RNA isolation and stored at -80 °C until analysis. RNA was obtained by a two-step procedure. First, the RNA was isolated using the Trizol method according to the manufacturer’s instructions (Invitrogen Life Technologies, Ghent, Belgium) and was further purified with the RNeasy Kit (Qiagen, Chatsworth, CA, USA). One microgram of cellular RNA was reverse transcribed into cDNA using SuperScript II reverse transcriptase and random hexamer primers (Invitrogen Life Technologies, Ghent, Belgium). The PCR reaction was carried out in a mixture that contained appropriate sense- and anti-sense primers and a TaqMan MGB probe in TaqMan Universal PCR Master Mixture (Applied Biosystems, Foster City, CA, USA) (Col1a1: Mm00801666_g1, TGF-β1: Mm01178820_m1, Timp1: Mm00441818_m1, FAT/CD36: Mm01135198_m1, FATP5: Mm00447768_m1, SREBP-1c: Mm00550338_m1, FAS: Mm00662319_m1, ACC1: Mm01304257_m1, PPAR-α: Mm00440939_m1, CPT1A: Mm01231183_m1, MCAD: Mm01323360_g1, PGC1-α: Mm01208835_m1, SIRT1: Mm00490758_m1, UCP2: Mm00627599_m1, TNF-α: Mm00443260_g1, p62: Mm00448091_m1 and Atg3: Mm00471287_m1. Beta-2-microglobulin was used as housekeeping gene (B2M: Mm00437762_m1). Real-time PCR amplification and data analysis were performed using the A7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Each sample was assayed in duplicate in a MicroAmp optical 96-well plate. The ΔΔCt-method was used to determine relative gene expression levels.
Western blot
Liver samples were homogenized with the Precellys 24 tissue homogenizer (Bertin Technologies, distributed by VWR, Leuven, Belgium) in a buffer containing 20 mM Tris-HCl pH 7.6, 10% glycerol, 1% Nonidet P-40, 2 μg/ml aprotinin, 5 μg/ml leupeptin, 0.5 μg/ml pepstatin, 10 mM sodium orthovanadate, 34 μg/ml phenylmethylsulfonyl fluoride, 10 mM sodium pyruvate, 100 mM sodium fluoride and 10 mM EDTA. Coomassie Protein Assay Reagent (Pierce Biotechnology Inc., Rockford, IL, USA) in combination with a standard curve of bovine serum albumin was used to determine the corresponding protein content of the homogenates.
Western blots for autophagic markers were performed as described previously [23]. Primary antibodies were purchased from Abcam, Cambridge, UK (microtubule-associated protein-1 light chain 3 (LC3) and actin) or Novus Biologicals, Littleton, CO, USA (p62). Secondary antibodies were obtained from DakoCytomation, Glostrup, Denmark (horseradish peroxidase-conjugated goat-anti-rabbit or goat-anti-mouse antibodies). Actin expression was used to control for equal loading. Relative protein levels were normalized to the mean of the ratio in the NCD mice.
Histological analyses
Histological analyses were performed as described previously [20]. Liver samples were routinely fixed in buffered formalin (4%) and embedded in paraffin. Serial 4 μm thick sections were stained with hematoxylin and eosin (H&E) and picrosirius red to assess fibrosis. All liver biopsies were analysed by an expert liver pathologist, blinded to the dietary condition or pharmacological intervention. Steatosis, activity and fibrosis were semi-quantitatively scored according to the NASH-Clinical Research Network criteria [24]. The amount of steatosis (percentage of hepatocytes containing fat droplets) was scored as: 0 (<5%), 1 (5–33%), 2 (>33–66%) and 3 (>66%). Hepatocyte ballooning was classified as: 0 (none), 1 (few) or 2 (many cells/prominent ballooning). Foci of lobular inflammation were scored as 0 (no foci), 1 (<2 foci per 200× field), 2 (2–4 foci per 200× field) and 3 (>4 foci per 200× field). Fibrosis was scored as stage F0 (no fibrosis), stage F1a (mild, zone 3, perisinusoidal fibrosis), stage F1b (moderate, zone 3, perisinusoidal fibrosis), stage F1c (portal/periportal fibrosis), stage F2 (perisinusoidal and portal/periportal fibrosis), stage F3 (bridging fibrosis) and stage F4 (cirrhosis). Diagnosis of NASH was based on accepted histological criteria [25, 26]. Severity of the disease was assessed using the NAS (nonalcoholic fatty liver disease activity score) as the unweighted sum of scores of steatosis, hepatocyte ballooning and lobular inflammation [24]. Percentage of fibrosis was quantitated by morphometry from digitalized sirius red stained sections using the Aperio system after tuning the threshold of fibrosis detection under visual control. Result is expressed as collagen proportional area [27].
Statistical analyses
Statistical analyses were performed with GraphPad Prism version 6 (GraphPad Software, La Jolla, CA, USA) and JMP10.0.0 (SAS Institute Inc, Cary, NC, USA). Significant differences were estimated by one-way ANOVA and Tukey post-hoc analysis (parametric samples) or Kruskal-Wallis and Dunn’s multiple comparisons test (nonparametric samples). A P-value of < 0.05 was considered as statistically significant. Significance is represented by: *: P < 0.05, **: P < 0.01 and ***: P < 0.001. Error bars are standard error of mean (SEM).
Results
Dietary intervention normalizes obesity, insulin resistance and hypercholesterolemia
Mice in which the HF-HSD was switched to NCD (Return to NCD), displayed a normalization of body weight (Fig. 2a) and epididymal fat pad weight (Fig. 2b). In addition, dietary intervention normalized glucose levels during IPGTT (Fig. 2c), HOMA-IR (Fig. 2d) and plasma total cholesterol levels (Fig. 2e).

Systemic metabolic effects of dietary intervention (Return to NCD) and losartan (HF-HSD-LOS). a Progression of body weight (n = 9 per group). b Epididymal fat pad weight (n = 9 per group). c Intra-peritoneal glucose tolerance test (6 h fasting, 1 g/kg glucose) with associated area under the curve values, determined 2 weeks before sacrifice (n = 9 for HF-HSD and HF-HSD-LOS mice and n = 6 for NCD and Return to NCD mice). d HOMA-IR (fasting glucose (mg/dl) X fasting insulin (ng/ml)/405) at sacrifice (n = 8 per group). e Fasting plasma total cholesterol (n = 9 per group). Asterisks indicate significant difference versus HF-HSD mice. Data are presented as mean. Error bars are SEM
On the contrary, losartan did not have an effect on established obesity. Body weight (Fig. 2a) and epididymal fat pad weight (Fig. 2b) were not different between the HF-HSD and HF-HSD-LOS group. Losartan did not affect glucose homeostasis. We observed no significant differences in glucose levels during IPGTT (Fig. 2c) or HOMA-IR (Fig. 2d) between HF-HSD and HF-HSD-LOS mice. Plasma total cholesterol levels were not influenced by losartan (Fig. 2e).
Dietary intervention, in contrast with losartan, completely reverses pre-existent NASH and fibrosis
Remarkably, switch from HF-HSD to NCD in mice with NASH, completely reversed steatosis, hepatocyte ballooning, inflammation and fibrosis (Fig. 3a). Histological scores of Return to NCD mice were identical to that of mice that were fed a NCD during the entire experiment (Fig. 3b and c). In correspondence, morphometric assessment showed a significant decrease of collagen proportional area in Return to NCD mice (Fig. 3d). In addition, liver weight and plasma alanine transaminase (ALT) levels (as a marker of liver damage) were normalized by the dietary intervention (Fig. 3e and f).

Effects on liver histology of dietary intervention (Return to NCD) and losartan (HF-HSD-LOS). a Representative hematoxylin and eosin (H&E) and picrosirius red stained liver histology images per dietary group. b Steatosis, hepatocyte ballooning, inflammation scores and NAFLD activity score (NAS). c Fibrosis score. d Morphometric analysis of collagen proportional area. e Liver weight. f Plasma ALT. Asterisks indicate significant difference versus HF-HSD mice. N = 9 per group for all assays. Data are presented as mean. Error bars are SEM
Losartan did not improve nor halt the progression of pre-existent NASH (Fig. 3a). Steatosis, hepatocyte ballooning, inflammation and fibrosis scores did not differ between HFD-HSD and HF-HSD-LOS mice (Fig. 3b and c). Collagen proportional area, liver weight and plasma ALT levels were not significantly affected by losartan (Fig. 3d-f).
Dietary intervention targets several pathways involved in NASH
In agreement with the histological findings, dietary intervention (Return to NCD) normalized hepatic mRNA levels of genes involved in fibrogenesis (Col1a1, TGF-β1, Timp1) (Fig. 4a). Furthermore, dietary intervention targets lipid metabolism. mRNA levels of genes involved in fatty acid uptake/transport (FAT/CD36 and FATP5) (Fig. 4b) and lipogenesis (FAS) (Fig. 4c) were normalized in Return to NCD mice.

Effect of dietary intervention (Return to NCD) and losartan (HF-HSD-LOS) on fibrogenesis and hepatic lipid metabolism. a mRNA levels of genes involved in fibrogenesis (Collagen type I alpha 1 (Col1a1), transforming growth factor β1 (TGF-β1) and tissue inhibitor of metalloproteinase 1 (Timp1)). b mRNA levels of genes involved in fatty acid transport (Fatty acid translocase/cluster of differentiation 36 (FAT/CD36), fatty acid transporter member 5 (FATP5)). c mRNA levels of genes involved in lipogenesis (Sterol regulatory element binding transcription factor 1 (SREBP1c), fatty acid synthase (FAS), acetyl-CoA carboxylase 1 (ACC1)). Asterisks indicate significant difference versus HF-HSD mice. N = 9 per group for all assays. Data are presented as mean. Error bars are SEM
At least on mRNA level, we observed no significant differences in fatty acid β-oxidation between the dietary groups at the moment of sacrifice (Fig. 5a). This probably can be explained by the decline of an initial increase of fatty acid β-oxidation in NASH, as we demonstrated previously, which also might be the case after dietary intervention [20]. Dietary intervention affects other parameters involved in mitochondrial function. mRNA levels of SIRT1 (key regulator of mitochondrial function), TNF-α (inducer of mitochondrial dysfunction) and UCP2 (uncouples oxidation from ATP synthesis) were normalized in Return to NCD mice (Fig. 5b). In addition, hepatic ATP levels were restored after dietary intervention (Fig. 5c). Hepatic HNE content (marker of oxidative stress) was not significantly decreased in Return to NCD mice (Fig. 5d). This indicates that oxidative damage by itself is not sufficient to sustain the NASH phenotype in our set-up.

Effect of dietary intervention (Return to NCD) and losartan (HF-HSD-LOS) on mitochondrial parameters. a mRNA levels of genes involved in fatty acid β-oxidation: Peroxisome proliferator activated receptor α (PPAR-α), carnitine palmitoyltransferase 1A (CPT1A) and medium chain acyl-Coenzyme A dehydrogenase (MCAD). b mRNA levels of genes involved in regulation of mitochondrial function (Peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1-α), Sirtuin1 (SIRT1), uncoupling protein 2 (UCP2) and tumor necrosis factor-α (TNF-α)). c ATP content. d HNE concentration as marker of oxidative stress. Asterisks indicate significant difference versus HF-HSD mice. N = 9 per group for all assays. Data are presented as mean. Error bars are SEM
Hepatic protein levels of p62 (a protein directing ubiquitinated proteins to the autophagic machinery that is known to accumulate when autophagy is impaired or insufficient) were increased in HF-HSD mice (Fig. 6a). In the absence of an increase in p62 mRNA levels (Fig. 6b), the p62 protein accumulation thus suggests an impaired autophagic flux [23, 28]. Disturbed autophagy in HF-HSD mice is further supported by reduced mRNA levels of Atg3 (Fig. 6c). Dietary intervention abolished the p62 protein accumulation and returned Atg3 mRNA to normal levels, suggesting restoration of the autophagic flux (Fig. 6a-c). The LC3-II/LC3-I ratio, which is used as a marker of mature autophagosome formation, was not affected by HF-HSD or the dietary intervention (data not shown).

Effect of dietary intervention (Return to NCD) and losartan (HF-HSD-LOS) on hepatic autophagic parameters. a Protein levels of p62. b mRNA levels of p62. c mRNA levels of autophagocytosis associated protein 3 (Atg3). Asterisks indicate significant difference versus HF-HSD mice. N = 9 per group for all assays. Data are presented as mean. Error bars are SEM
Finally, losartan significantly decreased mRNA levels of FAT/CD36, FAS and UCP2, but did not affect any other of the aforementioned parameters (Figs. 4, 5 and 6).
Discussion
In this study, dietary intervention completely reversed all features of pre-existent NASH, together with obesity, insulin resistance and hypercholesterolemia in a representative mouse model. Losartan had no effect on these established metabolic changes.
The beneficial effect of life style intervention on NASH, focused on both dietary and exercise habits, has been demonstrated in only a few randomized controlled trials with histological endpoints [8, 10, 11]. In addition, human studies are limited in time and do not allow clear differentiation of the dietary effects relative to physical activity [7, 10]. Moreover, only 30 to 40% of patients achieve target weight loss even in trials implementing an intensive life style program including behavorial strategies, which reflects the variable patient adherence [8, 10, 11, 29]. Therefore, we opted to use a relevant NASH mouse model to explore the full therapeutic potential of dietary intervention, which additionally allowed us to study the hepatic mechanisms behind it. To our knowledge, our dietary intervention represents the most effective therapy for NASH (i.e., complete resolution) demonstrated in rodents. Dietary intervention also resolved fibrosis in our model. This is an important finding since fibrosis is the major determinant for progression to end-stage liver disease and thus represents a main target to combat in NASH [20, 30, 31]. In 3 recent studies, a comparable dietary intervention (switch from a high fat diet to a NCD) was applied in obese mice with NASH [12, 32, 33]. However, the beneficial effects on liver histology were less pronounced, likely due to the shorter length of the dietary intervention (3 weeks) in one study and the use of genetically modified mice (hyperphagic foz/foz (Alms1 mutant) and low-density lipoprotein receptor knock out mouse model) in the two other studies [12, 32, 33].
The fact that dietary intervention affects the precipitating insult and in addition has the ability to normalize several molecular pathways involved in NASH, probably explains its superior efficacy compared with pharmacological agents that mostly only target a single or limited number of relevant pathways. We showed that dietary intervention targets pathways involved in fibrogenesis, lipid metabolism, mitochondrial function and autophagy, together contributing to the beneficial effect on histological level. The time point of sacrifice was 8 weeks after return to NCD. The study of earlier time points may clarify the possibility that a burst of clean-up of accumulating lipids by fatty acid oxidation and possibly autophagy took place in earlier stages after the dietary switch.
In contrast with our animal study, life style interventions (including dietary counselling) yield rather disappointing results in daily patient care. The lack of a well-organized multidisciplinary therapeutic approach from physician’s side probably reinforces this phenomenon [34, 35]. Therefore, in order to exploit the maximal therapeutic potential of dietary intervention, improving patient adherence, possibly by cognitive behaviour therapy embedded in structured programs, should be a main goal in the care of NASH patients in the coming years [35]. The lack of effective pharmacological alternatives and the additional beneficial effect of life style measures on other components of the metabolic syndrome further support this strategy.
However, more insight is needed in the role of specific dietary components in NASH treatment strategies. The evidence for the harmful effects of saturated fatty acid and sucrose intake is solid, whereas the preventive or even therapeutic capacity of specific dietary components (i.e., nutraceutical products) is less clear in the context of NASH [36, 37]. Animal studies using ingredient matched control diets or well-designed human interventional studies could provide further insight. This might potentiate the use of these molecules as therapeutics and explain the possible beneficial effects of the Mediterranean diet in NAFLD/NASH [36, 37]. Another issue that remains to be addressed is the influence of genetic variation in humans (in contrast with the inbred C57BL/6 J mouse strain we used for our experiments) on treatment outcome. Very little is known about the effect of genetics on the response to dietary or pharmacological treatment of NASH [38]. It was recently shown that genetic variation in PNPLA3 (adiponutrin) indeed confers sensitivity to weight loss-induced decrease of liver fat in humans [39]. Therefore, identifying (genetic) predictors for response to treatment could potentiate an individualized approach, stimulate patients to persevere the necessary dietary changes and increase their physical activity, and may thus further improve clinical outcome [38].
In parallel, we explored the therapeutic potential of losartan. Losartan seems an attractive candidate for the treatment of NASH, because it is a safe drug widely used for the treatment of hypertension (which is a part of the metabolic syndrome) and it targets angiotensin II that is thought to play an important role in NASH [14, 40]. However, the few available preliminary human and animal studies yielded equivocal results [14–19]. In addition, losartan has only been tested in normal or hyperphagic Otsuka Long-Evans Tokushima fatty (OLETF) rats, which were fed a non-physiological choline-deficient L-amino acid-defined diet [16, 18]. In our representative HF-HSD model, losartan did not have a beneficial effect on established NASH, obesity and insulin resistance. At the molecular level, losartan only significantly decreased FAT/CD36 and FAS mRNA together with UCP2 mRNA, but these effects appear insufficient to impact hepatic steatosis or ATP content respectively. We used a losartan dose comparable to the dose used in previous rodent studies on the metabolic and cardiovascular effects of losartan [41]. It may be that the duration of losartan administration in our experimental set-up was not long enough, although the same intervention period was largely sufficient to resolve NASH and associated systemic metabolic features by dietary intervention. However, in contrast with the dietary switch group, losartan had to counteract the effects of the continued HF-HSD, mimicking the human obesiogenic environment. It might be interesting to explore whether higher losartan doses, a longer administration time or losartan in combination with dietary intervention or another drug would be effective in NASH resolution. Possibly, a better effect could be achieved by using telmisartan, because of its combination of peroxisome proliferator-activated receptor (PPAR)-γ and angiotensin receptor blocking activity [42, 43]. Taken together, our pre-clinical data do not support the use of losartan to treat NASH.
Conclusions
In contrast with losartan, dietary intervention completely restores the metabolic phenotype, on both hepatic and systemic level, in mice with pre-existent NASH, obesity, insulin resistance and hypercholesterolemia. Our mouse model and set-up represent a useful tool to further study the effects of dietary and pharmacological interventions in NASH and the metabolic syndrome. In addition, our results emphasize the need to identify the pathophysiological and therapeutic barriers that preclude similar results in the current treatment of human NASH. This would facilitate the development of effective strategies to help patients to implement and maintain a healthy diet style in order to treat NASH and other components of the metabolic syndrome.
Abbreviations
- ACC1:
-
Acetyl-CoA carboxylase 1
- ALT:
-
Alanine transaminase
- Atg3:
-
Autophagocytosis associated protein 3
- ATP:
-
Adenosine triphosphate
- B2M:
-
Beta-2-microglobulin
- Col1a1:
-
Collagen type I alpha 1
- CPT1A:
-
Carnitine palmitoyltransferase 1A
- FAS:
-
Fatty acid synthase
- FAT/CD36:
-
Fatty acid translocase/cluster of differentiation 36
- FATP5:
-
Fatty acid transporter member 5
- H&E:
-
Hematoxylin and eosin
- HF-HSD:
-
High fat - high sucrose diet
- HNE:
-
4-hydroxynonenal
- HOMA-IR:
-
Homeostasis model of insulin resistance
- i.p.:
-
Intra-peritoneal
- IPGTT:
-
Intra-peritoneal glucose tolerance test
- LC3:
-
Microtubule-associated protein-1 light chain 3
- MCAD:
-
Medium chain acyl-Coenzyme A dehydrogenase
- NAFLD:
-
Non-alcoholic fatty liver disease
- NAS:
-
NAFLD activity score
- NASH:
-
Non-alcoholic steatohepatitis
- NCD:
-
Normal chow diet
- OLETF:
-
Otsuka Long-Evans Tokushima fatty
- PGC1-α:
-
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PNPLA3:
-
Patatin-like phospholipase domain-containing protein 3
- PPAR-α:
-
Peroxisome proliferator activated receptor alpha
- PPAR-γ:
-
Peroxisome proliferator-activated receptor gamma
- RAS:
-
Renin-angiotensin system
- SIRT1:
-
Sirtuin1
- SREBP1c:
-
Sterol regulatory element binding transcription factor 1
- TGF-β1:
-
Transforming growth factor beta 1
- Timp1:
-
Tissue inhibitor of metalloproteinase 1
- TNF-α:
-
Tumor necrosis factor-alpha
- UCP2:
-
Uncoupling protein 2
References
Bedogni G, Miglioli L, Masutti F, Tiribelli C, Marchesini G, Bellentani S. Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology. 2005;42:44–52.
Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N, Melchionda N, Rizzetto M. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–23.
Cassiman D, Jaeken J. NASH may be trash. Gut. 2008;57:141–4.
Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–46.
Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhising RA. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology. 2011;141:1249–53.
Wong RJ, Cheung R, Ahmed A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology. 2014;59:2188–95.
Musso G, Gambino R, Cassader M, Pagano G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology. 2010;52:79–104.
Promrat K, Kleiner D, Niemeier H, Jackvony E, Kearns M, Wands J, Fava J, Wing R. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51:121–9.
Ratziu V. Pharmacological agents for NASH. Nat Rev Gastroenterol Hepatol. 2013;10:676–85.
Thoma C, Day CP, Trenell MI. Lifestyle interventions for the treatment of non-alcoholic fatty liver disease in adults: a systematic review. J Hepatol. 2012;56:255–66.
Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, Friedman SL, Diago M, Romero-Gomez M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology. 2015;149:367–378 e365. quiz e314-365.
Rius B, Titos E, Moran-Salvador E, Lopez-Vicario C, Garcia-Alonso V, Gonzalez-Periz A, Arroyo V, Claria J. Resolvin D1 primes the resolution process initiated by calorie restriction in obesity-induced steatohepatitis. FASEB J. 2014;28:836–48.
Verbeek J, Cassiman D, Lannoo M, Laleman W, van der Merwe S, Verslype C, Van Steenbergen W, Nevens F. Treatment of non-alcoholic fatty liver disease: can we already face the epidemic? Acta Gastroenterol Belg. 2013;76:200–9.
Georgescu EF. Angiotensin receptor blockers in the treatment of NASH/NAFLD: could they be a first-class option? Adv Ther. 2008;25:1141–74.
Yokohama S, Yoneda M, Haneda M, Okamoto S, Okada M, Aso K, Hasegawa T, Tokusashi Y, Miyokawa N, Nakamura K. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology. 2004;40:1222–5.
Yoshiji H, Noguchi R, Ikenaka Y, Namisaki T, Kitade M, Kaji K, Shirai Y, Yoshii J, Yanase K, Yamazaki M, et al. Losartan, an angiotensin-II type 1 receptor blocker, attenuates the liver fibrosis development of non-alcoholic steatohepatitis in the rat. BMC Res Notes. 2009;2:70.
Georgescu EF, Georgescu M. Therapeutic options in non-alcoholic steatohepatitis (NASH). Are all agents alike? Results of a preliminary study. J Gastrointestin Liver Dis. 2007;16:39–46.
Ibanez P, Solis N, Pizarro M, Aguayo G, Duarte I, Miquel JF, Accatino L, Arrese M. Effect of losartan on early liver fibrosis development in a rat model of nonalcoholic steatohepatitis. J Gastroenterol Hepatol. 2007;22:846–51.
Hirata T, Tomita K, Kawai T, Yokoyama H, Shimada A, Kikuchi M, Hirose H, Ebinuma H, Irie J, Ojiro K, et al. Effect of Telmisartan or Losartan for Treatment of Nonalcoholic Fatty Liver Disease: Fatty Liver Protection Trial by Telmisartan or Losartan Study (FANTASY). Int J Endocrinol. 2013;2013:587140.
Verbeek J, Lannoo M, Pirinen E, Ryu D, Spincemaille P, Vander Elst I, Windmolders P, Thevissen K, Cammue BP, van Pelt J, et al. Roux-en-y gastric bypass attenuates hepatic mitochondrial dysfunction in mice with non-alcoholic steatohepatitis. Gut. 2015;64:673–83.
Verbeek J, Jacobs A, Spincemaille P, Cassiman D. Development of a Representative Mouse Model with Nonalcoholic Steatohepatitis. Curr Protoc Mouse Biol. 2016;6:201–10.
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9.
Vanhorebeek I, Gunst J, Derde S, Derese I, Boussemaere M, Guiza F, Martinet W, Timmermans JP, D’Hoore A, Wouters PJ, Van den Berghe G. Insufficient activation of autophagy allows cellular damage to accumulate in critically ill patients. J Clin Endocrinol Metab. 2011;96:E633–45.
Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21.
Sanyal AJ, Brunt EM, Kleiner DE, Kowdley KV, Chalasani N, Lavine JE, Ratziu V, McCullough A. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology. 2011;54:344–53.
Bedossa P, Poitou C, Veyrie N, Bouillot JL, Basdevant A, Paradis V, Tordjman J, Clement K. Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology. 2012;56:1751–9.
Bedossa P, Dargere D, Paradis V. Sampling variability of liver fibrosis in chronic hepatitis C. Hepatology. 2003;38:1449–57.
Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–97.
Yki-Jarvinen H. Nutritional modulation of nonalcoholic fatty liver disease and insulin resistance: human data. Curr Opin Clin Nutr Metab Care. 2010;13:709–14.
Schuppan D, Pinzani M. Anti-fibrotic therapy: lost in translation? J Hepatol. 2012;56 Suppl 1:S66–74.
Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, Hultcrantz R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61:1547–54.
Larter CZ, Yeh MM, Haigh WG, Van Rooyen DM, Brooling J, Heydet D, Nolan CJ, Teoh NC, Farrell GC. Dietary modification dampens liver inflammation and fibrosis in obesity-related fatty liver disease. Obesity (Silver Spring). 2013;21:1189–99.
Lytle KA, Jump DB. Is Western Diet-Induced Nonalcoholic Steatohepatitis in Ldlr-/- Mice Reversible? PLoS One. 2016;11:e0146942.
Centis E, Marzocchi R, Suppini A, Dalle Grave R, Villanova N, Hickman IJ, Marchesini G. The role of lifestyle change in the prevention and treatment of NAFLD. Curr Pharm Des. 2013;19:5270–9.
Centis E, Moscatiello S, Bugianesi E, Bellentani S, Fracanzani AL, Calugi S, Petta S, Dalle Grave R, Marchesini G. Stage of change and motivation to healthier lifestyle in non-alcoholic fatty liver disease. J Hepatol. 2013;58:771–7.
Scicchitano P, Cameli M, Maiello M, Modesti P, Muiesan M, Novo S, Palmiero P, Saba P, Pedrinelli R, Ciccone M. Nutraceuticals and dyslipidaemia: Beyond the common therapeutics. J Funct Foods. 2014;6:11–32.
De Wit N, Afman L, Mensink M, Muller M. Phenotyping the effect of diet on non-alcoholic fatty liver disease. J Hepatol. 2012;57:1370–3.
Dongiovanni P, Anstee QM, Valenti L. Genetic predisposition in NAFLD and NASH: impact on severity of liver disease and response to treatment. Curr Pharm Des. 2013;19:5219–38.
Sevastianova K, Kotronen A, Gastaldelli A, Perttila J, Hakkarainen A, Lundbom J, Suojanen L, Orho-Melander M, Lundbom N, Ferrannini E, et al. Genetic variation in PNPLA3 (adiponutrin) confers sensitivity to weight loss-induced decrease in liver fat in humans. Am J Clin Nutr. 2011;94:104–11.
Matthew Morris E, Fletcher JA, Thyfault JP, Rector RS. The role of angiotensin II in nonalcoholic steatohepatitis. Mol Cell Endocrinol. 2013;378:29–40.
Johansson M, Wickman A, Skøtt O, Gan L, Bergstrom G. Blood pressure is the major driving force for plaque formation in aortic-constricted ApoE mice. J Hypertension. 2006;24:2001–8.
Nakagami H, Morishita R. Obesity and gastrointestinal hormones-dual effect of angiotensin II receptor blockade and a partial agonist of PPAR-gamma. Curr Vasc Pharmacol. 2011;9:162–6.
Nakagami H, Osako MK, Morishita R. Potential effect of angiotensin II receptor blockade in adipose tissue and bone. Curr Pharm Des. 2013;19:3049–53.
Acknowledgements
We thank Inge Derese (Laboratory of Intensive Care Medicine) for her technical help with the quantitative RT-PCR analyses and Western Blotting.
Funding
JV benefitted from a travel grant for a long stay abroad from the FWO Vlaanderen. PS is supported by IWT Vlaanderen and KT by Industrial Research Fund KU Leuven (IOF-M). GVdB, through the University of Leuven, receives long-term structural research financing via the Methusalem program, funded by the Flemish Government (METH/08/07) and holds a European Research Council Advanced Grant (AdvG-2012-321670) from the Ideas Programme of the EU FP7. FN and DC are fundamental clinical researchers for the FWO Vlaanderen.
Availability of data and material
The datasets during and/or analysed during the current study are available from the corresponding author on reasonable request.
Authors’ contributions
JV: concept and design, experiments and procedures, interpretation of data and writing the manuscript. PS, IV, PB: experiments and procedures, interpretation of data and critical revision of the manuscript. IVE, PW: experiments and procedures. GVdB, JvP, SvdM, FN, BC and KT: interpretation of data and critical revision of the manuscript. DC: concept and design, interpretation of data and critical revision of the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All procedures were approved by the animal welfare committee of the University of Leuven.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Verbeek, J., Spincemaille, P., Vanhorebeek, I. et al. Dietary intervention, but not losartan, completely reverses non-alcoholic steatohepatitis in obese and insulin resistant mice. Lipids Health Dis 16, 46 (2017). https://doi.org/10.1186/s12944-017-0432-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-017-0432-7