
Abstract
Apolipoprotein C-III has been referred to as an important participant in the metabolism of triglyceride-rich lipoproteins, leading to hypertriglyceridemia and thereafter cardiovascular disease. Accumulating evidence indicates that apolipoprotein C-III is a multifaceted protein which not only regulates triglyceride metabolism, but also participates in the atherosclerotic lesion formation and several other pathological processes involved in atherosclerosis. Based on data from experiments and clinical trials, some novel therapies such as antisense technology emerge.
Similar content being viewed by others

Background
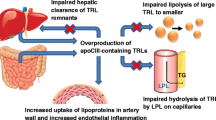
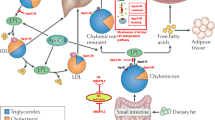
Triglyceride (TG) has been identified as an important risk factor for cardiovascular disease (CVD) for a long time, but randomized controlled trials of fenofibrate and omega-3 fatty acids to reduce TG failed to show any significant clinical benefits [1, 2]. Although genome-wide association study (GWAS) has positioned some single nucleotide polymorphisms (SNPs) associated with TG to be linked with CVD, these SNPs have pleiotropic relationships with other lipids [3] and therefore TG-related effects may be confounded. The longstanding controversy renders TG not to be a persuasive biomarker in CVD pathogenesis. Renewed interest in TG is brought by recent genetic observations that loss-of-function (LOF) mutations in the gene encoding apolipoprotein C-III (apoCIII) and relevant hypotriglyceremia are associated with decreased CVD risk (40–41 %) [4, 5]. ApoCIII, an apolipoprotein composed of 79 amino acids with a molecular weight of 8.8 kDa, resides on circulating HDL, low density lipoprotein (LDL) and triglyceride-rich lipoproteins (TRLs) such as chylomicrons (CM) and very low density lipoprotein (VLDL) [6]. ApoCIII promotes hypertriglyceridemia (HTG) via different mechanisms. First, apoCIII inhibits the activity of lipoprotein lipase (LPL) and disturbs lipids lipolysis [6]. Second, apoCIII interferes with the binding of apolipoprotein B (apoB) or apolipoprotein E (apoE) to hepatic receptors, thus leading to a delayed catabolism of TRL remnants [6]. Third, apoCIII favors the assembly and secretion of VLDL in the liver [7, 8]. ApoCIII and the lipoproteins that carry apoCIII acts as not only a key regulator in the TG metabolism, but also an independent predictor for CVD risk [9, 10]. The negative effects of apoCIII agree with gene analysis in which null mutation or missense mutation of apoCIII produces apoCIII deficiency and confers lifelong cardioprotection [4, 5, 11, 12]. These evidence supports the hypothesis that apoCIII may be instrumental to mediate TG-related harmful effects. This review puts emphases on the newly-discovered function of apoCIII beyond the well-known regulator of triglyceride metabolism. The novel method to inhibit apoCIII via antisense oligonucleotide (ASO) treatment and selective capture of apoCIII via zeolite nanoparticles will be mentioned too.
ApoCIII is directly involved in atherogenesis
Evidence from basic experiments
ApoCIII facilitates interaction of monocytes and endothelial cells
Interaction between monocytes and endothelial cells (ECs) is the initial step in the formation of atherosclerotic lesion [13]. ApoCIII itself and apoB lipoproteins containing apoCIII could activate pertussis toxin (PTX)-sensitive G protein pathway to activate phosphatidylcholine-specific phospholipase (PC-PLC). PC-PLC can catalyze PC to phospholylcholine and diacylglycerol (DAG) , and then DAG induces protein kinase C (PKC) α activation and leads to activation of nuclear factor-κB (NF-κB) [14], which increases β1-integrin expression in monocytes and enhances their adhesion to ECs. Besides, apoCIII also activates PKC β, leading to NF-κB activation and vascular cell adhesion molecule-1 (VCAM-1) expression in ECs, which contributes to EC dysfunction and monocyte recruitment [15] (Fig. 1). All of these findings can be demonstrated not only under static conditions but also under laminar flow. Since apoCIII can directly affect the interaction between monocytes and ECs, more attention is given to the direct atherogenic effects of apoCIII. A recent study by Zheng et al. extends the experiments from venous endothelium to coronary artery endothelium and provides in vivo evidence for the independent atherogenic effects of apoCIII and atheroprotective effects of statins because of decreasing apoCIII [16]. The study has demonstrated that apoCIII can promote NF-κB and subsequent VCAM-1 expression, leading to EC activation and monocytes adhesion, all of which can be abolished by statin effectively [16]. Although these findings seem to have built a complete line of evidence to explain the effects of apoCIII on vascular inflammation, many questions, for example, how apoCIII activates PTX-sensitive G protein , how apoCIII activates PKC β, are there any other mediators used by apoCIII to activate PKC α beyond PTX senstive G protein, are there any other molecules can also be affected by statins to reduce vascular inflammation, still require further exploration.

Pleiotropic effects of apoCIII. PKC indicates protein kinase C; NF-κB, nuclear factor-κB; VCAM-1, vascular cell adhesion molecule-1; EC, endothelial cells; ROS, reactive oxygen species; SMC, smooth muscle cells; HDL, high density lipoprotein; SR-BI, scavenger receptor class B type I
ApoCIII promotes smooth muscle cell proliferation
Smooth muscle cells (SMCs) have been identified as an crucial participant in the progression of atherosclerotic lesion and neointimal formation. A recently published article demonstrates that apoCIII can promote SMC proliferation via Akt signalling pathway mediated by reactive oxygen species in vitro, leading to aggravated restenosis and atherogenesis [17] (Fig. 1). The researchers built reliable HTG models through human apoCIII transgenic (apoCIIItg) mice and observed significant atherosclerotic lesion development in apoCIIItg mice with LDL receptor deficient background. Furthermore, they crossed apoCIIItg or apoCIII−/−mice with glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1-deficient (GPIHBP1−/−) mice, which exhibited extreme HTG because of impaired LPL anchoring to ECs, and harvested authentic TRL with or without apoCIII (TRL+ apoCIII or TRL–apoCIII) from the crossed mice to further distinguish the effect between TRL and apoCIII without impairing TRL properties. It was shown that TRL+ apoCIII promoted SMC proliferation while TRL–apoCIII not. The study adds more weight to the hypothesis that apoCIII can aggravate atherogenesis directly. However, no significant difference exists between apoCIII−/−and WT mice, which seems contradicted with findings from human researches that apoCIII LOF mutation provides cardioprotection [4, 5]. This may be caused by species difference that deletion of mouse apoCIII is not the same as apoCIII LOF mutation in human. Overall, more experiments are still needed to address the role of human apoCIII on human atherogenesis and restenosis.
Evidence from clinical studies
ApoCIII alters platelet activity
Platelets and the coagulation system are importantly involved with atherogenesis and atherothrombosis [18]. Results from a prospective study concerning 663 subjects with angiographically proven CVD suggest that basal serum concentration of apoCIII ≥ 10.5 mg/dl represents an independent risk factor of overall and cardiovascular mortality in a 5-year follow-up [19] (Table 1). Besides, the sub-analysis from this study reveals an independent association between elevated apoCIII and enhanced thrombin generation [19]. Moreover, another retrospective study about 933 unrelated patients enrolled in Verona heart Study has found that increased concentration of apoCIII is associated with elevated thrombin level in the plasma even after adjustment for other risk factors and apoCIII presents similar correlation with the presence of Factor II (FII) G20210A gene variant, the most important genetic determinant of FII and hereditary risk factor for venous thrombosis [20] (Table 1). It’s noteworthy that no significant relationship has been observed between thrombin activity and TRLs [20]. Although no causal mechanism could be inferred from these analysis because of the intrinsically flaw of cross-sectional design, they indicate that apoCIII could alter the homeostatic balance in a procoagulant way and may promote atherothrombotic complications.
ApoCIII modifies risk factors of atherosclerosis
Evidence from clinical studies
HDL dysfunction
HDL has been reported to exert various atheroprotective properties, including anti-apoptotic effects, anti-inflammatory effects and cholesterol efflux capacity [21]. All of these can be altered by pathological disorders and HDL with defects in these properties is referred to as dysfunctional HDL, which in turn contributes to the disorders, especially CVD.
Accumulating evidence suggests that apoCIII correlates tightly with HDL dysfunction. A prospective case-control study has identified two types of HDL: HDL with apoCIII and HDL without apoCIII, and the two HDL subgroups are oppositely related to CVD risk. The adjusted relative risk per standard deviation for HDL-C with apoCIII is 1.18 (1.03–1.34) and for HDL-C without apoCIII is 0.66 (0.53–0.83) [22] (Table 1). In a comparative study, apolipoprotein A-I containing HDL particles with apoCIII is significantly higher in obese subjects than normal weight (10 % vs. 4 %, P = 0.009) and apoCIII concentration in these HDL particles is 1.5–2 times higher in obese subjects (P ≤ 0.004), both of which indicated increased risk for CVD [23] (Table 1). In the Chin-Shan Community Cardiovascular Cohort study in Taiwan, HDL apoCIII to VLDL apoCIII ratio is considered as a more reliable marker to predict CVD than conventional apolipoproteins or lipid factors (odds ratio: 2.04; 95 % CI: 1.46–2.84; P < 0.0001) [24] (Table 1).
Besides, the concentration of apoCIII in HDL is significantly higher in CVD patients than in non-CVD patients according to the results from enzyme-linked immunosorbent assay method [25] (Table 1) and mass spectrometry [26]. ApoCIII increase in HDL from CVD patients can activate mitogen-activated protein kinase (MAPK) signalling pathway via phosphorylation of p38 in ECs and thus increase the expression of pro-apoptotic protein tBID, altering HDL from anti-apoptotic to pro-apoptotic [26] (Fig. 1).
The HDL dysfunction related to apoCIII is not only seen in CVD patients, but also seen in patients with chronic kidney disease (CKD). Holzer et al. found that apoCIII was highly abundant in HDL isolated from hemodialysis patients and correlated inversely with HDL cholesterol efflux capacity [27]. However, the study did not analyze the relationship between apoCIII and other changed HDL components which may also affect HDL cholesterol efflux capacity in CKD. Therefore, it’s unreliable to justify that enrichment of HDL with apoCIII would impair HDL cholesterol efflux capacity independent of other HDL components.
These findings indicate that apoCIII increases in HDL may exert some disruptive impacts on HDL functional properties and provide a novel mechanism for HDL dysfunction.
Evidence from basic experiments
Diabetes mellitus
Patients with diabetes mellitus often display abnormal lipid profiles and are susceptible to CVD. Elevated glucose can upregulate apoCIII expression in the transcriptional level via the activation of transcription factors carbohydrate response element-binding protein and hepatocyte nuclear factor-4α and thus plasma apoCIII concentration presents a positive correlation with fasting plasma glucose as well as glucose excursion after oral glucose load in obese humans [28]. However, the relationship between insulin and apoCIII still remains controversial.
In vitro experiments have shown that apoCIII interacted with scavenger-receptor BI (SR-BI) and then upregulated β1 integrin expression, leading to the hyperactivation of β cell Ca2+ channels through the coactivation of PKA and Src kinase pathways [29] (Fig. 1). This finding has been supported by another study carried on a special animal model diabetes-prone BB rat, which develops type 1 diabetes mellitus (T1DM) at around the age of 60 days spontaneously [30]. Of note, antisense treatment to decrease the level of endogenous apoCIII can delay the onset of T1DM [30], which suggests that apoCIII is an essential participant in the development of T1DM.
As for T2DM, it’s well-known that it is featured by two major defects: insulin resistance and β cell failure. A cross-sectional study about 1422 T2DM subjects found that apoCIII correlated positively with coronary artery calcification, a biomarker of subclinical atherosclerosis [31]. Juntti-Berggren et al. found that islet insulin resistance could promote local apoCIII production [32]. Besides, they transplanted apoCIII knockout islet and islet from ob/ob or B6 mice into the anterior chamber of the eye in an ob/ob mouse or a HFD-treated B6 mice, both of which were characterized by high systemic serum levels of apoCIII and impaired glucose tolerance [32]. By this way a delicate system was created where the islets rich and deficient in apoCIII coexisted in one animal model . The elevation of intraislet apoCIII production increased local inflammation, deranged Ca2+ handling in β cells and thus β cell apoptosis [32]. From this perspective, ApoCIII serves as a bridge linking insulin resistance and β cell failure in T2DM. Besides, systemically decreasing apoCIII in vivo via ASO treatment can improve glucose tolerance in T2DM [32].
In contrast to the studies above, one study investigated effects of apoCIII on intact rat pancreatic islets in the presence of proinflammatory cytokines ( interferon-γ + interleukin 1β) to find that apoCIII reduced β cell apoptosis by stimulating phosphorylation of Akt [33]. The disagreement may be caused by the differences about experiment factors such as models and intervention methods. Of note, it’s inappropriate to draw a conclusion from one study that apoCIII may exert some protective function on islet cells.
Lp-PLA2
Although whether lipoprotein-associated phospholipase A2(Lp-PLA2) is an independent risk factor for atherosclerosis remains controversial, Lp-PLA2 activity and mass shows a strong, positive association with atherosclerosis [34]. Results from in vitro experiments and animal models revealed that apoCIII could upregulate Lp-PLA2 expression via MAPK and NF-kB pathways in a dose- and time-dependent manner and thus induce inflammation [35]. In addition, in vitro experiments indicated that the downstream Lp-PLA2 could also increase apoCIII expression in the liver in turn [35]. However, the detailed mechanisms in the circle still await further investigation.
Future prospects
Species-specific antisense suppression of apoCIII has produced robust reduction in serum apoCIII and TG and demonstrated good tolerance in various preclinical models, including human apoCIII transgenic mice, rats, mouse, nonhuman primates [36]. In the phase I clinical study in healthy human volunteers, administration of human apoCIII ASO (volanesorsen, formally ISIS-APOCIIIRx) brought about a profound dose- and time-dependent reduction of apoCIII and TG without remarkable adverse effects [36]. The phase 2 study evaluated the pharmacodynamic effects of volanesorsen in adult patients with severe or uncontrolled HTG [37]. When volanesorsen was administrated as a single agent, plasma apoCIII was reduced on apoB100 and apoAI containing lipoproteins as well as lipoprotein (a) uniformly [38] in a dose-dependent fashion (apoCIII: 40 %-100 mg, 63.8 %-200 mg, 79.6 %-300 mg). TG decreased in concordance with apoCIII (TG: 31.3 %-100 mg, 57.7 %-200 mg, 70.9 %-300 mg) and showed a strong, positive correlation with apoCIII, while HDL-C presented a dose-dependent increase (HDL-C: 36.6 %-100 mg, 36.2 %-200 mg, 45.7 %-300 mg). Similar changes occurred when volanesorsen was given as an add-on to stable fibrate therapy. No serious treatment-associated safety issues were identified. The apparent clinical benefits brought by volanesorsen presented not only in patients with HTG but also in those with familial chylomicronemia syndrome (FCS). In FCS patients, absence of LPL caused insufficient TRLs removal and resulted in chylomicronemia (TG > 880 mg/dl). ASO treatment exhibited substantial efficacy to lower TG less than 500 mg/dl independent of LPL [39]. Short-term efficacy and apparent safety of volanesorsen brings light to further long-term evaluation. Ongoing phase III trials about volanesorsen include the APPROACH (the APPROACH study: A Study of ISIS-APOCIIIRx in Patients with Famial Chylomicronemia Syndrome, NCT02211209), the COMPASS (the COMPASS study: A Study of Volanesorsen in Patietns with Hypertriglyceridemia, NCT02300233), and the BROADEN (the BROADEN study: A Study of Volanesorsen in Patients with Partial Lipodystrophy, NCT02527343). These studies aim to evaluate the efficacy and safety of 300 mg volanesorsen versus placebo administered subcutaneously once a week for 52 ,26, 52 weeks respectively in patients with corresponding diseases. The primary outcome is measured by the percent change in fasting TG from baseline to 13 weeks. ApoCIII ASO treatment links genetic insights to molecular mechanisms and keeps abreast with the ‘precision medicine’.
In addition to the promising ASO treatment, zeolite nanoparticles, whose negative charge help it to interact electrostatically with the positively charged amino acid residues of apoCIII , selectively capture plasma apoCIII and may be a potential therapy to reduce plasma apoCIII in the future [40].
Conclusions
Considerable evidence indicates that apoCIII can affect the development of CVD not only by regulating triglyceride metabolism but also through its direct atherogenic effects. Novel understanding of apoCIII in atherogenesis makes it an attractive therapy target and some innovative approaches to modifying it emerge. Although full illustration of apoCIII function still awaits further exploration, to lower the apoCIII levels in circulation via specific intervention seems to be an intriguing therapy for cardiovascular protection.
Abbreviations
- apoB:
-
apolipoprotein B
- apoCIII:
-
apolipoprotein C-III
- apoE:
-
apolipoprotein E
- ASO:
-
Antisense oligonucleotide
- CKD:
-
Chronic kidney disease
- CM:
-
Chylomicron
- CVD:
-
Cardiovascular disease
- DAG:
-
Diacylglycerol
- EC:
-
Endothelial cell
- FCS:
-
Familial chylomicronemia syndrome
- GPIHBP1:
-
Glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1
- GWAS:
-
Genome-wide association study
- HTG:
-
Hypertriglyceridemia
- LDL:
-
Low density lipoprotein
- LOF:
-
Loss-of-function
- LPL:
-
Lipoprotein lipase
- Lp-PLA2:
-
Lipoprotein-associated phospholipase A2
- NF-κB:
-
Nuclear factor-κB
- PC-PLC:
-
Phosphatidylcholine-specific phospholipase
- PKC:
-
Protein kinase C
- PTX:
-
Pertussis toxin
- SMC:
-
Smooth muscle cell
- SNP:
-
Single nucleotide polymorphism
- SR-BI:
-
Scavenger-receptor BI
- T1DM:
-
Type 1 diabetes mellitus
- TG:
-
Triglyceride
- TRL:
-
Triglyceride-rich lipoproteins
- VCAM-1:
-
Vascular cell adhesion molecule-1
- VLDL:
-
Very low density lipoprotein
References
ACCORD Study Group, Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–74.
ORIGIN Trial Investigators, Bosch J, Gerstein HC, Dagenais GR, et al. n-3 fatty acids and cardiovascular outcomes in patients with dysglycemia. N Engl J Med. 2012;367:309–18.
Do R, Willer CJ, Schmidt EM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–52.
TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute, Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31.
Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, et al. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41.
Ooi EMM, Barrett PHR, Chan DC, et al. Apolipoprotein C-III: understanding an emerging cardiovascular risk factor. Clin Sci (Lond). 2008;114:611–24.
Sundaram M, Zhong S, Bou Khalil M, et al. Expression of apolipoprotein CIII in McA-RH7777 cells enhances VLDL assembly and secretion under lipid-rich conditions. J Lipid Res. 2010;51:150–61.
Yao Z. Human apolipoprotein C-III: a new intrahepatic protein factor promoting assembly and secretion of very low density lipoproteins. Cardiovasc Hematol Disord Drug Targets. 2012;12:133–40.
Mendivil CO, Rimm EB, Furtado J, et al. Low-density lipoproteins containing apolipoprotein C-III and the risk of coronary heart disease. Circulation. 2011;124:2065–72.
Scheffer PG, Teerlink T, Dekker JM, et al. Increased plasma apolipoprotein C-III concentration independently predicts cardiovascular mortality: the Hoorn study. Clin Chem. 2008;54:1325–30.
Valleix S, Verona G, Jourde-Chiche N, et al. D25V apolipoprotein C-III variant causes dominant hereditary systemic amyloidosis and confers cardiovascular protective lipoprotein profile. Nat Commun. 2016;7:10353.
Pollin TI, Damcott CM, Shen H, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–5.
Randolph GJ. Mechanisms That Regulate Macrophage Burden in Atherosclerosis. Circ Res. 2014;114:1757–71.
Kawakami A, Aikawa M, Nitta N, et al. Apolipoprotein CIII-Induced THP-1 Cell Adhesion to Endothelial Cells Involves Pertussis Toxin-Sensitive G Protein- and Protein Kinase C-Mediated Nuclear Factor- κB Activation. Arterioscler Thromb Vasc Biol. 2007;27:219–25.
Kawakami A, Aikawa M, Alcaide P, et al. Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation. 2006;114:681–7.
Zheng C, Azcutia V, Aikawa E, et al. Statins suppress apolipoprotein CIII-induced vascular endothelial cell activation and monocyte adhesion. Eur Heart J. 2013;34:615–24.
Li H, Han Y, Qi R, et al. Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: the effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc Res. 2015;107:579–89.
Borissoff JI, Spronk HM, ten Cate H. The hemostatic system as a modulator of atherosclerosis. N Engl J Med. 2011;364:1746–60.
Olivieri O, Martinelli N, Girelli D, et al. Apolipoprotein C-III predicts cardiovascular mortality in severe coronary artery disease and is associated with an enhanced plasma thrombin generation. J Thromb Haemost. 2010;8:463–71.
Olivieri O, Martinelli N, Baroni M, et al. Factor II activity is similarly increased in patients with elevated apolipoprotein CIII and in carriers of the factor II 20210A allele. J Am Heart Assoc. 2013;2:e000440.
Rosenson RS, Brewer Jr HB, Ansell B, et al. Translation of high- density lipoprotein function into clinical practice: current prospects and future challenges. Circulation. 2013;128:1256–67.
Jensen MK, Rimm EB, Furtado JD, et al. Apolipoprotein C-III as a potential modulator of the association between HDL-cholesterol and incident coronary heart disease. J Am Heart Assoc. 2012;1:e000232.
Talayero B, Wang L, Furtado J, et al. Obesity favors apolipoprotein E- and C-III-containing high density lipoprotein subfractions associated with risk of heart disease. J Lipid Res. 2014;55:2167–77.
Chang PY, Lee CM, Hsu HC, et al. Identification of the HDL-ApoCIII to VLDL-ApoCIII ratio as a predictor of coronary artery disease in the general population: The Chin-Shan Community Cardiovascular Cohort (CCCC) study in Taiwan. Lipids Health Dis. 2012;11:162.
Xiong X, Liu H, Hua L, et al. The association of HDL-apoCIII with coronary heart disease and the effect of statin treatment on it. Lipids Health Dis. 2015;14:127.
Riwanto M, Rohrer L, Roschitzki B, et al. Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary disease: role of high-density lipoprotein-proteome remodeling. Circulation. 2013;127(8):891–904.
Holzer M, Birner-Gruenberger R, Stojakovic T, et al. Uremia Alters HDL Composition and Function. J Am Soc Nephrol. 2011;22:1631–41.
Caron S, Verrijken A, Mertens I, et al. Transcriptional activation of apolipoprotein CIII expression by glucose may contribute to diabetic dyslipidemia. Arterioscler Thromb Vasc Biol. 2011;31:513–9.
Shi Y, Yang G, Yu J, Yu L, Westenbroek R, Catterall WA, Juntti-Berggren L, Berggren PO, Yang SN. Apolipoprotein CIII hyperactivates β cell CaV1 channels through SR-BI/β1 integrin-dependent coactivation of PKA and Src. Cell Mol Life Sci. 2014;71:1289–303.
Holmberg R, Refai E, Höög A, et al. Lowering apolipoprotein CIII delays onset of type 1 diabetes. Proc Natl Acad Sci U S A. 2011;108:10685–9.
Qamar A, Khetarpal SA, Khera AV, et al. Plasma apolipoprotein C-III levels, triglycerides, and coronary artery calcification in type 2 diabetics. Arterioscler Thromb Vasc Biol. 2015;35:1880–8.
Avall K, Ali Y, Leibiger IB, et al. Apolipoprotein CIII links islet insulin resistance to β-cell failure in diabetes. Proc Natl Acad Sci U S A. 2015;119:E2611–9.
Størling J, Juntti-Berggren L, Olivecrona G, et al. Apolipoprotein CIII Reduces Proinflammatory Cytokine-Induced Apoptosis in Rat Pancreatic Islets via the Akt Prosurvival Pathway. Endocrinology. 2011;152:3040–8.
Rosenson RS, Stafforini DM. Modulation of oxidative stress, inflammation, and atherosclerosis by lipoprotein-associated phospholipase A2. J Lipid Res. 2012;53:1767–82.
Han X, Wang T. Apolipoprotein CIII regulates lipoprotein-associated phospholipase A2 expression via the MAPK and NFkB pathways. Biology Ope. 2015;4:661–5.
Graham MJ, Lee RG, Bell III TA, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112:1479–90.
Gaudet D, Alexander VJ, Baker BF, et al. Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N Engl J Med. 2015;373:438–47.
Yang X, Lee SR, Choi YS, et al. Reduction in lipoprotein-associated apoC-III levels following volanesorsen therapy: activity, apolipoprotein C3 cardiovascular In disease: The Communities Phase 2 randomized trial results. J Lipid Res. 2016;57:706–13.
Gaudet D, Brisson D, Tremblay K, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med. 2014;371:2200–6.
Rahimi M, Ng EP, Bakhtiari K, et al. Zeolite Nanoparticles for Selective Sorption of Plasma Proteins. Sci Rep. 2015;5:17259.
Acknowledgements
Not applicable.
Funding
This project was supported by grants from National Natural Science Foundation of China (No.81370393 and No.81670426); supported by the Fundamental Research Funds for the Central Universities of Central South University (No.2016zzts545).
Availability of data and materials
Not applicable.
Authors’ contributions
DP conceived of the scope of the review and helped draft the manuscript. ML were involved in the accumulation of the relevant references and drafted the manuscript. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Luo, M., Peng, D. The emerging role of apolipoprotein C-III: beyond effects on triglyceride metabolism. Lipids Health Dis 15, 184 (2016). https://doi.org/10.1186/s12944-016-0352-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-016-0352-y