
Abstract
Drug resistance represents a significant obstacle in cancer treatment, underscoring the need for the discovery of novel therapeutic targets. Ubiquitin-specific proteases (USPs), a subclass of deubiquitinating enzymes, play a pivotal role in protein deubiquitination. As scientific research advances, USPs have been recognized as key regulators of drug resistance across a spectrum of treatment modalities, including chemotherapy, targeted therapy, immunotherapy, and radiotherapy. This comprehensive review examines the complex relationship between USPs and drug resistance mechanisms, focusing on specific treatment strategies and highlighting the influence of USPs on DNA damage repair, apoptosis, characteristics of cancer stem cells, immune evasion, and other crucial biological functions. Additionally, the review highlights the potential clinical significance of USP inhibitors as a means to counter drug resistance in cancer treatment. By inhibiting particular USP, cancer cells can become more susceptible to a variety of anti-cancer drugs. The integration of USP inhibitors with current anti-cancer therapies offers a promising strategy to circumvent drug resistance. Therefore, this review emphasizes the importance of USPs as viable therapeutic targets and offers insight into fruitful directions for future research and drug development. Targeting USPs presents an effective method to combat drug resistance across various cancer types, leading to enhanced treatment strategies and better patient outcomes.
Similar content being viewed by others

Introduction
Cancer poses a significant public health challenge globally, being the first or second leading cause of death in 112 countries [1, 2]. Normal cell growth is controlled by stringent regulatory mechanisms; however, alterations in specific cell components can lead to dysfunction in these mechanisms, resulting in cancer [3]. Cancer treatments are tailored based on the type and stage of cancer, as well as the patient's overall health, including surgical resection, chemotherapy, radiation therapy, targeted therapy, and the burgeoning field of immunotherapy [4]. Despite advancements in molecular and tumor biology that have significantly transformed the cancer treatment landscape and substantially enhanced therapeutic outcomes over the last decades, resistance to treatment continues to be a formidable challenge, particularly in patients with advanced or metastatic disease [5, 6]. Drug resistance, a primary cause of reduced treatment efficacy, is influenced by varied mechanisms [7]. Previous study outlines the key factors of drug resistance, proposing a conceptual framework that encompasses tumor heterogeneity, physical barriers, tumor burden and growth kinetics, undruggable cancer drivers, the immune system and the microenvironment, along with the many consequences of applying therapeutic pressures [8]. Although researches are ongoing to find new drugs and combinations to address drug resistance, the complex molecular mechanisms behind drug resistance remain largely elusive [9]. The identification of new drug resistance biomarkers and a deeper understanding of drug resistance mechanisms are crucial endeavors that will significantly advance personalized precision medicine for cancer treatment [10].
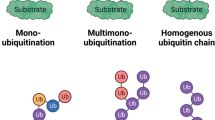
Post-translational modification (PTM) of proteins is a critical mechanism for modulating protein structure and function in both physiological and pathological conditions, encompassing ubiquitination, phosphorylation, methylation, acetylation, glycosylation, SUMOylation, among others [11]. Ubiquitination, a prevalent form of PTM, involves an ATP-dependent process that attaches ubiquitin to specific proteins. This attachment, involving the 76-amino acid peptide, ubiquitin, initiates protein degradation by the 26S proteasome complex [12]. In recent years, it has become increasingly evident that ubiquitination plays a pivotal role in controlling a broad range of cellular processes beyond protein degradation via the ubiquitin-proteasome system (UPS) (Fig. 1A). Ubiquitin modification acts as a versatile signaling mechanism, regulating protein stability, translocation, signaling activation/inactivation, and even influencing the organization of cellular structures such as organelle membranes and chromatin [13]. The dynamic and precise control of these diverse processes is achieved through the concerted action of a hierarchical enzymatic cascade involving E1 activating enzymes, E2 conjugating enzymes, and E3 ligases. The ubiquitination sequence begins with ubiquitin-activating enzyme E1 binding and activating ubiquitin, followed by the transfer of activated ubiquitin to ubiquitin-conjugating enzyme E2. Then ubiquitin ligase E3 recognizes the substrate and facilitates the transfer of ubiquitin from E2, leading to substrate degradation [14]. E3 ligases, known for their substrate specificity, are crucial in the ubiquitination pathway. The human genome contains approximately 1000 E3 ligases, categorized into RING-between-RING (RBR) family E3s, homology to E6AP C terminus (HECT) domain-containing E3s, and extremely fascinating novel gene (RING) finger domain-containing E3s [15]. These enzymes work together to recognize specific target proteins, transfer ubiquitin molecules, and generate distinct ubiquitin codes, including (i) mono-ubiquitination, where a single ubiquitin molecule is connected; (ii) poly-ubiquitination, forming polyubiquitin chains; (iii) multi-ubiquitination or poly-mono-ubiquitination, with multiple ubiquitin molecules bound [16] (Fig. 1B). In polyubiquitination, ubiquitin is often joined through seven Lysine residues (K6, K11, K27, K29, K33, K48, and K63) and the initial methionine (M1) [17]. These different types of ubiquitin modifications confer specific functional consequences, directing proteins to degradation, influencing protein localization and trafficking, and modulating the activation or inactivation of signaling pathways (Fig. 1C). Moreover, emerging studies have revealed the involvement of ubiquitination in shaping organelle dynamics, regulating membrane fusion events, modulating chromatin structure and DNA repair processes [17]. These findings highlight the multifaceted and intricate roles of ubiquitin in cellular physiology, underscoring its significance as a crucial PTM.

Overview of the ubiquitination and deubiquitination process and their functional implications. A The mechanism of the ubiquitin proteasome system. B Different types of ubiquitination: monoubiquitination, multiubiquitination, and polyubiquitination. C Different substrate fates result from diverse mechanisms of polyubiquitination through the M1 methionine residue or through the seven distinct lysine residues of ubiquitin, K6, K11, K27, K29, K33, K48, and K63. D The specific features of USPs involved in biological processes
Like other PTMs, ubiquitination is reversible. Deubiquitinating enzymes (DUBs), a type of peptidase, can accurately cleave the C-terminal isopeptide bond of ubiquitin and detach the substrate protein from ubiquitin, thus reversing the ubiquitination process, a phenomenon known as deubiquitination [18]. Ubiquitination and deubiquitination together constitute the complex UPS, which regulates the balance of misfolded proteins in eukaryotic cells. To date, approximately 100 DUB species have been identified in humans, divided into seven subfamilies: ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), ovarian tumor proteases (OTUs), Machado-Joseph disease protein proteases (MJDs), Jab1/Mov34/MPN+ proteases (JAMMs), Zinc Finger ubiquitin-specific proteases (ZUP/ZUFSPs), and motif interacting with ubiquitins (MIUs)-containing novel DUB family members (MINDYs) [19]. The USP family, with over 50 members, is the largest and most diverse, accounting for about 60% of DUBs. USPs are a class of cysteine-dependent proteases, an analogous mechanism of action of the cysteine protease papain, which features three highly conserved subdomains resembling the fingers, thumb, and palm of the right hand [19, 20]. USPs are characterized by the presence of a conserved catalytic domain known as the USP domain, which exhibits protease activity and enables the cleavage of ubiquitin from target proteins. In addition to the USP domain, various USPs possess additional domains or motifs, such as ubiquitin-like (UBL) domain, zinc finger ubiquitin-binding (ZnF-UBP) domain, and domains specific to USP (DUSP), ubiquitin-interacting motifs (UIM) and ubiquitin-associated (UBA), among others [19]. These additional domains influence substrate recognition, protein-protein interactions, and subcellular localization, further augmenting the functional repertoire of USPs. Notably, USPs exhibit diverse substrate specificities, allowing them to target specific ubiquitinated proteins or substrates and regulate distinct signaling pathways and cellular functions [19]. Furthermore, USPs display differential cellular localization, with some USPs predominantly localized in the nucleus, while others are primarily cytoplasmic. This subcellular distribution of USPs contributes to their spatial regulation of protein deubiquitination events [20] (Fig. 1D). USPs control a range of cell processes that are significant in the setting of cancer, including the cell cycle, DNA damage repair mechanisms, chromatin remodeling, and several signaling pathways [17, 18].
In recent years, there has been growing interest in USPs as potential targets for inhibiting tumor formation and cancer progression. So far, over 40 USPs have been connected, either directly or indirectly, to pertinent cancer processes and anti-cancer therapies. The link between USPs and cancer drug resistance is increasingly being substantiated [21]. USPs contribute to drug resistance by catalyzing specific substrate proteins, promoting DNA damage, inducing cancer stem cells (CSCs) characteristics, interfering with cell apoptosis, and regulating transcription factors and key signaling pathways [22]. Gene editing and pharmacological inhibitors targeting USPs could mitigate drug resistance and render cancer cells more vulnerable to anticancer therapies. Current trials investigating the anti-cancer efficacy of USP inhibitors underscore the therapeutic potential of targeting USP-mediated deubiquitination in cancer patients [23, 24]. This review systematically concludes, for the first time, the intricate mechanisms of USP-mediated anticancer resistance across varied treatment modalities, such as chemotherapy, molecular targeted therapy, immunotherapy, and specific radiotherapy. It also explores current potential small molecule USP inhibitors and effective strategies for combining these inhibitors with other anti-cancer means, to modulate drug resistance, aiming to offer innovative approaches and insights for enhancing future cancer treatments.
Chemotherapy resistance mediated by USPs
Platinum
After its approval in 1978, cisplatin became a cornerstone in clinical practice as a foundational platinum anticancer drug. A decade later, carboplatin emerged as the second platinum-based drug to be clinically utilized. Then, in 2002, oxaliplatin also successfully entered in Europe and the United States [25]. Despite the advent of precision medicine and immunotherapy, platinum-based treatments, especially cisplatin, remain a mainstay in the treatment of many cancers, serving as the gold standard [26].
Cisplatin
USPs and DNA damage response (DDR) in cisplatin resistance
The DDR is a highly conserved mechanism that protects cells against DNA damage caused by external and internal factors. It consists of a network of multiple signaling pathways designed to detect and relay damage signals, facilitate damage identification and repair, and ensure the continuation of the normal cell cycle. Unrestricted cyclic DNA replication contributes to unlimited growth and reproduction of cancer cells, and cisplatin exerts its cytotoxic effect by inducing DNA damage and disrupting the protective DDR mechanisms [27]. γH2AX, the phosphorylated form of histone H2AX at Ser139, marks an early cellular response to DNA double-strand breaks (DSBs), initiating and activating the DDR system [28]. In lung cancer patients with cisplatin resistance, the upregulation of USP51 diminishes γH2AX formation and increases checkpoint kinase 1 (CHK1) phosphorylation, thereby ensuring an effective cell cycle [29]. USP22, a crucial regulator that enhances H2AX phosphorylation through its deubiquitinating activity, has been shown to contribute to robust DDR mechanisms in lung adenocarcinoma [30]. Notably, USP22 enhances the repair of DSBs by interacting with the partner and localizer of BRCA2 (PALB2), facilitating the recruitment of the PALB2-BRCA2-Rad51 complex during DDR, ultimately leading to cisplatin resistance [31].
USP7, a typical researched USP member, plays a pivotal role in regulating several key components of DDR pathways, including the MRN-MDC1 complex [32], CHK1 [33], Rad18 [34], RNF168 [35], CDC25A, and p53 [36]. Through its interactions with these proteins, USP7 influences the recruitment of downstream factors involved in DNA damage, modulates the overall functionality of DDR, and confers cellular resistance against genotoxic insults. In the research conducted by Liu et al., USP7 was shown to interact with SAMHD1, a crucial dNTP hydrolase, deubiquitinating it at K421 [37]. This action stabilizes SAMHD1, activating DDR by facilitating further interaction between USP7 and the C-terminal binding protein-interacting protein (CtIP), a key initiator of DSB repair, thus leading to cisplatin resistance [37]. Another significant member, USP1, is regulated at the translational level in cisplatin-resistant non-small cell lung cancer (NSCLC) cell lines [38]. USP1, in a complex with USP1 associated factor 1 (UAF1), removes monoubiquitin from target proteins, FANCD2 and PCNA, which are essential for DDR and chromatin recruitment [39, 40]. Moreover, USP1 can prevent K48-linked polyubiquitination of MAST1, whose overexpression is correlated with increased cisplatin resistance [41, 42]. The loss of USP1 enhances cisplatin-induced DNA damage, evidenced by larger γH2AX foci formation, and diminishes MAST1-mediated activation of phosphorylated MEK/ERK [43].
Zinc finger E-box binding homeobox 1 (ZEB1) is a key promoter of cisplatin resistance. While ZEB1's role in epithelial-mesenchymal transformation (EMT) and dedifferentiation is well-documented, recent findings also highlight its involvement in enhancing DNA repair and clearance of DSBs [44]. ZEB1 acts as a DNA repair regulator by directly interacting with USP7, thereby augmenting USP7's deubiquitinating activity on CHK1 [45]. Additionally, USP51 can interact with ZEB1, and the reduction of USP51 levels increases ZEB1 ubiquitination, significantly lowering cisplatin resistance in lung cancer cells [46]. On the contrary, overexpression of USP17, a potential downstream target of ZEB1, renders cancer cells more susceptible to cisplatin-induced DNA damage [47] (Fig. 2A).

USPs regulate platinum drugs resistance. A USPs contribute to cisplatin resistance by regulating DNA damage response, inhibiting apoptosis and enhancing epithelial to mesenchymal transition. B Detailed mechanisms of USPs involve in oxaliplatin resistance
USPs and cell apoptosis in cisplatin resistance
During the DDR, cells may initiate apoptosis to eliminate those with irreparable damage, thereby preventing the proliferation of cells harboring severe errors. Dysregulated apoptosis or evasion of apoptosis constitutes a pivotal mechanism by which cancer cells develop cisplatin resistance [48, 49]. Elevated expression of USP8 in cisplatin-resistant ovarian cancer (OC) cells has been documented. USP8 silencing markedly diminishes the levels of FLIPL, Claspin, and survivin, critical regulators of anti-apoptotic pathways [50]. Additionally, USP14's interaction with BCL6, a transcriptional repressor and proto-oncogene, plays a role in anti-apoptotic processes. Inhibition of USP14 effectively mitigates cisplatin resistance in OC cells, enhancing the proteasomal degradation of BCL6 [51]. Furthermore, reduced expression of USP46 contributes to cisplatin resistance by suppressing the apoptotic mediators Caspase3, Bax, and poly‑ADP ribose polymerase (PARP), while concurrently activating BCL2. And this process is potentially under the regulation of PUM2, a Pumilio RNA-binding protein family member [52].
USP39's association with the augmented migratory and invasive capacities of esophageal squamous cell carcinoma (ESCC) cells fosters tumor progression and metastasis. USP39 overexpression impedes PARP and Caspase3 activation, diminishing the apoptotic rate in ESCC cells treated with cisplatin [53]. Beyond ESCC, additional research indicates USP39's regulatory influence on cisplatin-induced apoptosis in colon cancer cells, a process contingent upon the tumor suppressor protein p53. USP39 knockdown escalates p53 levels, enhancing apoptosis and promoting G2/M arrest [54]. Moreover, USP35 stabilizes BIRC3, an apoptosis inhibitor protein (IAP) family member, by averting Lys48-mediated polyubiquitination, impacting PARP and Caspase3 expression in NSCLC cells [55]. Notably, acetylation of USP31 at Lys1264 fosters cervical cancer cell survival and resistance to cisplatin-induced apoptosis. The deacetylase sirtuin 1 (SIRT1) counteracts USP31's oncogenic traits and bolsters cisplatin-induced apoptosis through deacetylation [56] (Fig. 2A).
USPs, EMT, and stemness in cisplatin resistance
EMT is a complex biological process transforming epithelial cells into mesenchymal-like cells [57]. CSCs are a subset of tumor cells characterized by pronounced self-renewal capabilities [58]. EMT-induced stemness facilitates the migration of cancer cells from the primary tumor, promotes distant metastasis, and enhances resistance to platinum-based therapies [59]. In triple-negative breast cancer (TNBC) cells, a positive correlation exists between USP22 expression and cisplatin resistance. USP22 overexpression significantly boosts the extracellular acidification rate and spheroid formation while upregulating expression of stemness genes and EMT markers. These unique cellular effects are mediated through USP22's interaction with c-Myc which enhances c-Myc deubiquitination and reduces intracellular glycolysis [60]. In lung adenocarcinoma, USP22 inhibition decreases ALDH1A3 expression, heightens the sensitivity of tumor cells to cisplatin, particularly CD133+ cancer-initiating cells, and attenuates their stem cell-like properties [61].
TWIST and Snail, crucial EMT transcription factors, diminish the chemotherapy sensitivity of cancer cells [62]. USP29-mediated TWIST1 deubiquitination induces cisplatin resistance in TNBC, stabilizing TWIST1 and promoting EMT and CSC activities. CDK1, a USP29 activator, facilitates this process through USP29 phosphorylation, enhancing the TWIST1-driven malignant phenotype [63]. Additionally, USP1, phosphorylated by DDR kinases ATM and ATR, initiates Snail deubiquitination, fostering cisplatin resistance, metastatic potential, and stemness in OC cells [64]. Subsequent studies reveal that, USP45, recruited by MYH9 and MYH10, deubiquitinates Snail in serous ovarian cancer (SOC) [65]. Furthermore, USP27X and Snail expressions are positively linked in breast and pancreatic cancers. During EMT, TGFβ-induced USP27X upregulation stabilizes Snail1 expression in epithelial cells and cancer-associated fibroblasts (CAFs), reducing cisplatin sensitivity [66]. Given TGFβ's role in EMT induction, the regulation of SMAD2, a critical TGFβ pathway component, by USPs is notable [67]. USP32 overexpression in gastric cancer (GC) enhances SMAD2 deubiquitination, correlating with advanced tumor stages, increased cisplatin resistance, and poorer survival [68]. Moreover, in cisplatin-resistant laryngeal squamous cell carcinoma (LSCC) cells, USP34’s interaction with SOX2, a key CSC- and EMT-related transcription factor, decreases SOX2 polyubiquitination and augments LSCC cell sensitivity to cisplatin [69] (Fig. 2A).
Oxaliplatin
Oxaliplatin is a fundamental component of FOLFOX, the standardized first-line treatment regimen for gastrointestinal cancers, which also includes 5-fluorouracil (5-Fu) and leucovorin [70]. Recent studies have underscored the pivotal roles of long non-coding RNAs (lncRNAs) in oxaliplatin resistance [71]. The influence of USPs in modulating lncRNAs, and their ensuing effects on oxaliplatin resistance, should not be overlooked.
A recently discovered lncRNA, lnc-RP11-536 K7.3, has been found to be associated with oxaliplatin resistance and indicates a poor prognosis in colorectal cancer (CRC) patients. Functionally, lnc-RP11-536 K7.3 interacts with SOX2, initiating the transcriptional activation of USP7 mRNA. This activation of USP7 facilitates the deubiquitination of hypoxia-inducible factor (HIF-1α), thereby bestowing resistance to oxaliplatin in cancer cells [72]. Conversely, another lncRNA, AC092894.1, was found to be significantly downregulated in oxaliplatin-resistant CRC cells. AC092894.1 serves as a scaffold molecule, enabling the deubiquitination of the androgen receptor (AR) by USP3, fostering the transcription of RASGRP3, and subsequently activating the MAPK signaling pathway, which augments oxaliplatin-induced apoptosis [73]. Moreover, the expression of lncRNA HULC, regulated by miR-6825-5p, miR-6845-5p, and miR-6886-3p, elevates the deubiquitination effect of USP22 on SIRT1, making hepatocellular carcinoma (HCC) cells resistant to oxaliplatin and inducing protective autophagy in HCC cells [74].
Given the clinical practice of oxaliplatin combined with 5-Fu, USP-mediated dual resistance to oxaliplatin and 5-Fu has been thoroughly investigated in numerous studies. Upregulation of USP35 promotes CRC cell proliferation and imparts resistance to both oxaliplatin and 5-Fu. Further investigations demonstrated that USP35 directly targets α-L-fucosidase 1 (FUCA1) for deubiquitination, and the USP35-FUCA1 axis elevates nucleotide excision repair (NER) components, culminating in platinum resistance [75]. In contrast, a decrease in USP38 expression was noted in clinical CRC samples, which significantly enhanced the sensitivity of CRC cells to oxaliplatin and 5-Fu. Notably, USP38 plays a crucial role in amplifying oxaliplatin and 5-Fu resistance by removing Lysine 63 ubiquitin chains from histone deacetylase 3 (HDAC3) in CRC cells, accompanied by an increase in H3K27 acetylation [76] (Fig. 2B).
Carboplatin
Carboplatin, which is structurally akin to cisplatin, exhibits lower toxicity and fewer side effects than cisplatin; however, resistance remains a challenge [77]. Studies have shown that USP39 protein is overexpressed in carboplatin-resistant OC samples. Mechanistic analyses indicate that USP39 promotes the phosphorylation of AKT, EGFR, and cyclin B1, while it deters the activation of PARP and Caspase-3, thereby enhancing cell proliferation, migration, and invasion, and curbing apoptosis [78]. Additionally, USP48 exhibits high expression in carboplatin-resistant OC cells, too. The reduction of USP48 markedly mitigates chemoresistance to carboplatin and curtails the metastasis of OC cells [79].
Doxorubicin (adriamycin, dox)
Dox, a member of the anthracycline class, is a prevalent anticancer agent employed in treating various cancers. It exerts its therapeutic effects by intercalating into DNA strands, inducing DNA damage and disrupting DNA replication [80, 81].
USPs, cell cycle, and cell apoptosis in Dox resistance
USP7 has been identified as a critical regulator of Dox resistance across several cancer types, including HCC [82], pancreatic ductal adenocarcinoma (PDAC) [83], and neuroblastoma (NB) [84]. In Dox-resistant HCC cells, the disruption of USP7 not only amplifies Dox-induced apoptosis but also impedes cell proliferation via the prolonged activation of the pro-apoptotic protein Bax [82]. In PDAC cells, inhibition of USP7 boosts sensitivity to Dox, correlating with a notable rise in Dox nuclear localization [83]. Additionally, the inhibition of USP7 intensifies the cytotoxic effects of Dox on NB cells, particularly those with an operational USP7-HDM2-p53 axis, increasing their susceptibility to Dox-induced p53-mediated apoptosis [84]. Also in NB, cell viability is influenced by USP14 expression. A synergistic antitumor response is observed when USP14 inhibition is paired with Dox treatment [85].
Bioinformatics analysis has revealed a notable positive correlation between USP37 expression and Dox resistance in BC. The combined approach of USP37 knockdown and Dox treatment significantly increases cleaved Caspase 3 and Bax levels while suppressing BCL2 expression, resulting in cell cycle arrest and enhanced apoptosis [86]. Furthermore, numerous studies have shown that β-transducin repeat-containing protein (β-Trcp)'s regulation of cell cycle depends on its capacity to target Cdc25A [87, 88]. β-Trcp as an E3 ligase engages in specific binding with USP47, and mutations in β-Trcp can impair this interaction. Crucially, disrupting USP47 leads to Cdc25A accumulation, which diminishes cell survival and elevates cellular sensitivity to Dox-induced apoptosis [89]. Additionally, USP8 has been identified as an inhibitor of Dox-induced cell cycle arrest and apoptosis by modulating various receptor tyrosine kinases (RTKs) in HCC, including EGFR, c-Met, p-AKT, p-STAT3, and p-Raf [90].
USPs, stemness, and metastasis in Dox resistance
The role of ATP-binding cassette (ABC) transporter-mediated drug efflux is critically examined in TNBC [91]. An increased expression of ABC transporters correlates with resistance to taxanes and anthracyclines, as these drugs, including Dox and paclitaxel, are substrates of p-glycoprotein (Pgp), encoded by the ABCB1 gene [92]. USP7, acting as a specific regulator of ABCB1, engages directly with ABCB1, reducing K48-linked polyubiquitination. Inhibition of USP7 significantly counters resistance to Dox and paclitaxel in TNBC cells, thus diminishing tumorigenesis and distant metastasis in an orthotopic BC mouse model [93]. Additionally, a rise in USP29 expression enhances resistance of NSCLC cells to Dox and paclitaxel by deubiquitinating Snail1 via USP29 [94]. Co-IP assays confirmed that USP45 binds directly to MYC, selectively removing K48-linked ubiquitin chains from MYC, thereby intensifying Dox resistance in cancer cells. The USP45/MYC axis elevates the expression of MYC-targeted downstream factors and CSC-associated proteins, leading to an increase in tumorsphere formation and CD133+ cell populations [95]. Conversely, a notable decrease in USP16 expression was observed in HCC cells. USP16 levels are associated with the carboxyl-terminal truncated form of the Hepatitis B virus X protein (Ct-HBx)-induced upregulation of CSC markers, colony formation, and augmented resistance of HCC cells to Dox [96].
Cell adhesion is integral to the EMT process, and cell adhesion-mediated drug resistance (CAM-DR) is identified as a pivotal mechanism in drug resistance in multiple myeloma (MM) [97]. USP14 is implicated in CAM-DR in MM, where it fosters Dox resistance by inhibiting apoptosis and altering the Wnt signaling pathway [98].
Paclitaxel
Paclitaxel, a member of the taxane class, influences various cellular oncogenic processes, including mitosis, apoptosis, angiogenesis, inflammatory response, CSC formation, and reactive oxygen species (ROS) production [99, 100]. Notably, as paclitaxel is often used in conjunction with cisplatin or Dox, certain USP-mediated resistance mechanisms previously mentioned may be relevant to paclitaxel resistance as well [63, 93, 94].
USPs, cell mitosis, and cell apoptosis in paclitaxel resistance
PLK1 is pivotal in regulating mitosis and orchestrating G2/M cell cycle transition [101, 102]. Recent findings disclose a direct interaction between USP7 and PLK1, with both showing overexpression in paclitaxel-resistant cancer cells. The dual knockdown of USP7 and PLK1 markedly enhances the susceptibility of paclitaxel-resistant cells to apoptosis by influencing chromosome alignment during mitosis [103]. Following this research, targeting USP7 prompts the formation of multiple spindle poles, triggering mitotic catastrophe and apoptosis in lung, prostate, and cervical cancer cells. Synergistic anticancer outcomes are achieved by combining USP7 and PLK1 inhibitors, chiefly through the suppression of MDR/ABCB1 expression [104].
USP33 overexpression impedes paclitaxel-triggered apoptosis in resistant prostate cancer cells. It interacts with DUSP1, preventing its Lys48-linked polyubiquitination and the subsequent activation of JNK [105]. Intriguingly, Skp1-CUL1-F-box (SCF) E3 ubiquitin ligase system targets procaspase-3, modulating the apoptotic threshold to shield cells from apoptosis [106]. A notable decrease in USP15 expression has been identified in paclitaxel-resistant OC samples. Restoring USP15 expression in paclitaxel-treated cells enhances procaspase-3 deubiquitination, detaches it from the SCF complex, and induces apoptosis, thereby counteracting OC cell resistance to paclitaxel [107] (Fig. 3A).

USPs regulate the resistance to paclitaxel, 5-Fu and temozolomide (TMZ). A USPs affect paclitaxel resistance by altering cell mitosis, cell apoptosis and reactive oxygen species production. B USP22's deubiquitination of SIRT1 and BMI1 supports cancer stem cells formation, aiding in 5-Fu efflux and resistance. C USPs participate in regulating TMZ resistance in glioma cells
USPs, ROS, and oxidative stress (OS) in paclitaxel resistance
An imbalance between ROS production and antioxidant defense mechanisms triggers OS responses. Paclitaxel promotes ROS generation, which in turn increases OS levels, inducing DNA damage and mutations that contribute to genomic instability and the development of drug-resistant clones [99]. Previously discussed, USP29 upregulation in response to OS stabilizes Snail1 expression, enhancing stemness and resistance to paclitaxel and Dox in NSCLC cells [94]. Crucially, USP2a overexpression in prostate cancer cells reduces ROS production and stabilizes mitochondrial membranes, granting resistance to OS induced by prooxidants like cisplatin, Dox, and paclitaxel [108]. This protective mechanism of USP2a involves regulating c-Myc through miR-34b/c to boost intracellular antioxidant glutathione (GSH) levels, thereby mitigating the oxidative cascade initiated by these chemotherapy agents [108].
NF-E2-related factor 2 (Nrf2) is a transcription factor that preserves cellular redox balance by upregulating genes associated with antioxidant response elements (AREs) [109,110,111]. Zhang et al.'s research demonstrated that USP15 deubiquitinates Keap1, enhancing its E3 ligase activity and prolonging Nrf2 ubiquitination, thus suppressing the Nrf2-dependent antioxidant response. A decrease in USP15 expression elevates Nrf2 levels via a Keap1-dependent pathway, leading to increased paclitaxel resistance [112].
CAFs promote cancer cell growth and drug resistance by releasing various bioactive compounds, including exosomes [113,114,115]. An intricate study revealed that cisplatin and paclitaxel activate USP7, prompting CAFs to emit exosomal miR-522. USP7 then reduces ALOX15 expression by deubiquitinating and stabilizing heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1), diminishing lipid-ROS accumulation and decreasing ferroptosis, ultimately reducing chemotherapy sensitivity in GC cells [113] (Fig. 3A).
5‑Fu
5-Fu is a pyrimidine analog classified as an antimetabolite, frequently used alongside other chemotherapy agents. Its primary anticancer action is the noncompetitive inhibition of thymidylate synthase (TS), essential for RNA and DNA synthesis [116].
USPs and stemness in 5-Fu resistance
Emerging research indicates that enhanced stemness characteristics mediate 5-Fu resistance in cancer cells. The aforementioned USP16 and USP38 in HCC and CRC also influence 5-Fu resistance by modulating stemness and the expression of related stem cell markers [94, 96]. In recurrent and chemoresistant CRC cells, USP22 expression is elevated, with miR-305p identified as an upstream regulator [117]. Inhibiting USP22 expression can adversely affect the Wnt/β-catenin pathway, thus reducing CRC stemness and the cells' resistance to 5-Fu [118]. Furthermore, BMI1, part of the polycomb group (PcG) proteins crucial for stem cell renewal [119], is targeted alongside cisplatin to synergistically suppress growth in head and neck squamous cell carcinoma (HNSCC) cells [120]. It is posited that increased USP22 expression contributes to 5-Fu resistance in HCC cells by elevating BMI1 expression. In a mouse model injected with a 5-Fu-resistant HCC cell line, targeting USP22 led to a significant tumor size reduction post 5-Fu treatment [121] (Fig. 3B).
USPs and SIRT1 in 5-Fu resistance
SIRT1, a class III histone deacetylase, serves as an acetylation mediator within the USP22 and SAGA coactivator complex [122, 123]. Studies have demonstrated that USP22 directly interacts with SIRT1, activating the AKT/GSK-3β/multidrug resistance-associated protein 1 (MRP1) pathway, thereby enhancing 5-Fu efflux and reducing 5-Fu-induced apoptosis in HCC cells [124]. Furthermore, a positive feedback loop exists between c-MYC and SIRT1, where USP22 increases SIRT1 stability through MYC mediation, concurrently decreasing p53 levels [125]. Through SIRT1 deubiquitination, USP22 potentially triggers autophagy, diminishing HCC cell sensitivity to chemotherapeutic agents, including 5-Fu [74] (Fig. 3B).
Temozolomide (TMZ)
TMZ, an orally administered chemotherapy drug, is predominantly used to treat glioblastoma (GBM), an extremely aggressive brain cancer. As an alkylating agent, TMZ induces DNA damage and inhibits cell division [126, 127]. Recent findings indicate that USP4, upregulated in TMZ-resistant GBM cells, inhibits apoptosis in a p53-dependent manner, and this resistance is further amplified by p53-specific inhibitors [103].
Glioma stem cells (GSCs) are a unique population among GBM characterized by their remarkable self-renewal ability and the acquisition of TMZ resistance [128]. USP 6 N-terminal-like protein (USP6NL) is a GTPase-activating protein that plays a regulatory role in EGFR endocytosis [129]. In GBM-resistant cells, the expression levels of USP6NL, as well as CSC markers (CD44 and CD133), transcription factors (Nanog and SOX2), and the efflux transporter ABCG2, were significantly upregulated. Notably, USP6NL was found to interact with EGFR and deubiquitinate it to enhance TMZ-induced autophagy [130]. Additionally, USP36 interacts with and upregulates ALKBH5, an m6A demethylase. Depleting USP36 diminishes GSC self-renewal and increases their sensitivity to TMZ in vitro and in vivo [131]. Significantly, TRAF4, a scaffold protein with E3 ligase activity, binds to Caveolin-1 (CAV1) to inhibit ZNRF1-mediated ubiquitination and facilitate USP7-mediated deubiquitination, thus enhancing CAV1 stability, promoting stemness, and increasing GBM cell resistance to TMZ [132] (Fig. 3C).
Molecular targeted drug resistance mediated by USPs
PARP inhibitors
PARP is a critical component of the DDR system, recognizing and binding to DNA single-strand breaks (SSBs), thereby facilitating SSBs repair. Repair of DSBs primarily occurs through two pathways: nonhomologous end-joining (NHEJ) and homologous recombination (HR) [133]. When genes, typically BRCA, responsible for HR at DSBs are mutated, DSB repair is impeded, increasing reliance on PARP-mediated SSBs repair. At the same time, if a PARP inhibitor impedes PARP activity at SSBs, DNA damage cannot be rectified through either SSB or DSB repair mechanisms, leading to cancer cell death. This elucidates why PARP inhibitors are particularly effective in tumor patients with BRCA mutations. Furthermore, PARP inhibitors can be synergistically combined with chemotherapy or radiotherapy to enhance DNA damage in cancer cells [134].
USPs and BRCA in olaparib resistance
BRCA1 is pivotal in facilitating HR and is recruited to DSBs through a series of signaling events [135]. Receptor-associated protein 80 (RAP80) plays an essential role in this recruitment, acting through a scaffolding protein to form a complex with BRCA1, thereby promoting the DDR [136]. Recent research indicates that ATM phosphorylation of USP13, following DNA damage, enables USP13 to deubiquitinate RAP80. This action renders OC cells resistant to olaparib by removing K63-linked ubiquitin chains from RAP80 [137]. Additionally, USP15, recruited to DSBs by MDC1, deubiquitinates BARD1 [138, 139], a BRCA1 binding partner, facilitating the interaction between BARD1 and HP1γ at DSBs, thus enhancing olaparib resistance in cancer cells [140].
CtIP as the key initiator of DDR, also collaborates with BRCA1 to influence olaparib resistance [141]. USP52 can directly deubiquitinate CtIP and facilitate its phosphorylation at Thr-847 [142]. Moreover, in BRCA1-deficient cells, USP1 expression is elevated, leading to its interaction with the essential cell cycle protein PCNA at the replication fork. This interaction prevents PCNA's ubiquitin-mediated degradation by E3 ligase RAD18. In the absence of USP1, persistent loading of the translesion synthesis (TLS) polymerase and the build-up of ubiquitinated PCNA induce replication fork instability, significantly increasing the susceptibility of cancer cells to olaparib [143] (Fig. 4A).

USPs regulate molecular targeted drug resistance. A USPs deubiquitinate key nuclear proteins, enhancing DNA damage repair and leading to resistance to PARP inhibitors. Inhibitors targeting USP7 improve the therapeutic efficacy of PARP inhibitors in treating cancer. B USPs are involved in imatinib (IM) resistance in chronic myeloid leukemia (CML) and gastrointestinal stromal tumors
USP7 and CCDC6 in Olaparib resistance
USP7 plays a significant role in mediating cancer cell resistance to PARP inhibitors [93, 144,145,146,147,148]. In pancreatic cancer, USP7 deubiquitinates fructose-1,6-bisphosphatase 1 (FBP1) at K206, hindering its nuclear translocation. By preventing FBP1's association with DNA (cytosine-5)-methyltransferase 1 (DNMT1), USP7 inhibits PARP1 entrapment in chromatin, contributing to olaparib resistance [144].
The interaction between USP7 and CCDC6 is crucial in conferring resistance to PARP inhibitors. CCDC6, an ATM substrate, can dephosphorylate γH2AX at S139, maintaining stable DNA damage checkpoints [145]. Studies have identified a positive association between USP7 and CCDC6 expression levels. While the E3 ligase FBXW7 targets CCDC6 for ubiquitination and destabilization, leading to mitotic arrest, USP7's deubiquitination of CCDC6 counters this effect, enhancing its stability and influencing CCDC6 turnover [146]. Inhibiting USP7 promotes CCDC6 degradation, diminishes γH2AX levels, and markedly increases cell sensitivity to PARP inhibitors in various cancer types, including NSCLC [146], prostate cancer [145], lung neuroendocrine cancer [147], bladder cancer [93], and SOC [148] (Fig. 4A).
Protein kinase inhibitors
Tyrosine kinase inhibitors (TKIs)
USPs mediate Imatinib (IM) resistance in chronic myeloid leukemia (CML)
IM, a quintessential TKI, is primarily used for treating CML and gastrointestinal stromal tumors (GISTs). CML is a clonal disorder of pluripotent hematopoietic cells, characterized by the presence of a gene, BCR-ABL, that encodes a constitutively active tyrosine kinase fusion protein [149]. IM specifically targets this BCR-ABL protein, significantly inhibiting CML progression [150].
In CML cell lines and peripheral blood mononuclear cells (PBMCs) from CML patients, a reduction in USP15 expression was noted. This decrease in USP15 is due to the upregulation of STAT5A and the direct activation of miR-202-5p, which specifically targets and downregulates USP15 mRNA, causing inhibitory deubiquitination of Caspase6 and apoptosis [151]. Furthermore, research has verified an increase in USP6 expression in IM-resistant CML cells [152]. Elevated USP6 levels facilitate the deubiquitination of glutaminase-1 (GLS1), enhancing the conversion of glutamine to glutamate and ammonia, thus impeding IM-induced apoptosis [153]. This pivotal deubiquitination step can be targeted for inhibition by miR-146a-5p contained in exosomes from human umbilical cord mesenchymal stem cells (hucMSCs) [152]. Additionally, USP47 is overexpressed in primary CML cells, where it deubiquitinates Y-box binding protein 1 (YB-1). Targeting USP47 presents a promising strategy to counter IM resistance and effectively eradicate leukemia stem/progenitor cells in CML [154] (Fig. 4B).
USPs mediate IM resistance in GISTs
GISTs are the predominant malignant mesenchymal tumors of gastrointestinal tract. The c-KIT protein, a common tyrosine kinase in GISTs, is the primary target of IM, particularly the hyperactive mutant form of the c-KIT protein [155]. IM has proven effective in controlling the disease in 70-85% of patients with advanced c-KIT-positive GISTs [156].
The regulation of autophagy-related protein 5 (ATG5) is vital for autophagic activity and IM resistance [157]. USP13 has been shown to deubiquitinate ATG5, thereby enhancing autophagy and increasing IM resistance in GIST cells, a process dependent on serine/threonine-protein kinase PAK1 [158]. The stabilization of USP13 mRNA is facilitated by N6-methyladenosine methyltransferase-like 3 (METTL3) with the aid of the m6A reader IGF2BP2 [158].
Tumor-derived exosomes also play a significant role in mediating IM resistance [159, 160]. Aligning with previous findings, exosomes from hucMSCs and USP6 contribute to IM resistance in CML [152]. Recent studies have indicated that exosomes from IM-resistant GIST cells can confer resistance to IM-sensitive cells, facilitated by Ras-related protein 35 (Rab35). In this context, the transcription factor ETV1 upregulates USP32 expression, which then interacts with Rab35, reducing its K48-ubiquitination and maintaining its stability, thus promoting the resistant mechanism [161] (Fig. 4B).
Multiple targeted RTK inhibitors
Sorafenib, a multi-kinase inhibitor, is a recommended treatment for patients with advanced HCC [162]. USP22 plays a role in mediating sorafenib resistance in HCC cells through a complex series of mechanisms. Under normoxic conditions, HIF1α is degraded by the UPS system. However, under hypoxic conditions, HIF1α is stabilized and forms a complex with HIF1β, triggering the transcription of downstream genes [163, 164]. USP22 can enhance hypoxia-induced HCC stemness and glycolysis by deubiquitinating and stabilizing HIF1α. Moreover, USP22 can be transcriptionally upregulated by HIF1α, creating a positive feedback loop that amplifies stemness characteristics and reduces the sensitivity of HCC cells to sorafenib [165]. Notably, a self-activated cascade-responsive nanoplatform, galactose-decorated lipopolyplex (Gal-SLP), has been developed for targeted HCC therapy, facilitating the co-delivery of sorafenib and shUSP22 to achieve a synergistic effect. Sorafenib, encapsulated within Gal-SLPs, initiates a ROS cascade, enabling the rapid release of shUSP22, which inhibits downstream SIRT1/AKT/MRP1 and ABCC1 pathways, increases intracellular sorafenib accumulation, and disrupts glycolysis in HCC cells. This approach demonstrates significant antitumor efficacy and excellent biosafety in a patient-derived xenograft (PDX) model [166, 167].
In addition of USP22, USP29 also deubiquitinates HIF1α, contributing to sorafenib resistance in HCC cells by promoting the transcriptional activation of target genes, especially hexokinase 2 (HK2), a key enzyme in glycolytic pathway [168]. Furthermore, ENKUR, a crucial adaptor protein involved in the localization of a Ca2+-permeable ion channel in sperm [169], is noted for its inhibitory effects on tumor proliferation, metastasis, and sorafenib resistance in HCC. Detailed studies reveal that ENKUR can interact with β-catenin, inhibiting its nuclear translocation and subsequently reducing c-Jun and MYH9 levels. The decreased expression of MYH9 impairs the recruitment of USP7 and the deubiquitination of c-Myc, enhancing the sensitivity of HCC cells to sorafenib treatment [170].
Bruton's tyrosine kinase (BTK) inhibitors
Ibrutinib, a prototypical BTK inhibitor, is predominantly employed in treating blood cancers. It targets BTK, an essential component of the B-cell receptor (BCR) signaling pathway, thus impeding B-cell activation, proliferation, and survival [171]. In chronic lymphocytic leukemia (CLL), USP7 is overexpressed and interferes with HR pathways, leading to an accumulation of unrepaired DSBs. Inhibiting USP7 significantly enhances the sensitivity of ibrutinib-resistant CLL cells to clinically achievable doses of chemotherapeutic agents [172]. Additionally, USP14 is implicated in inhibiting tumor-specific apoptosis in ibrutinib-resistant Waldenström macroglobulinemia (WM) cells. The inhibition of USP14 results in the downregulation of BCR-associated elements, disruption of mitochondrial membrane integrity and endoplasmic reticulum stress mechanisms, culminating in increased apoptosis in resistant WM cells [173].
Receptor inhibitors
AR inhibitors
Prostate cancer progression is predominantly driven by AR signaling [174]. Consequently, androgen deprivation therapy (ADT), which diminishes circulating testosterone levels and blocks cellular AR signaling via surgical or chemical castration, remains the cornerstone of treatment for prostate cancer. Based on the response to ADT [175], prostate cancer is classified into hormone-sensitive prostate cancer (HSPC) and castration-resistant prostate cancer (CRPC). Although enzalutamide effectively inhibits AR signaling in CRPC treatment, some CRPC cells evolve to resist enzalutamide by upregulating AR or its splice variant AR-V7 [176].
USP22 is overexpressed in CRPC tumor samples, where it deubiquitinates AR/AR-V7, thereby increasing their accumulation [177]. The lncRNA PCBP1-AS1 amplifies USP22's deubiquitination effect. Inhibiting PCBP1-AS1 markedly restores the sensitivity of resistant cells to enzalutamide [178]. Similarly, USP14 deubiquitinates AR/AR-V7 and can outcompete the E3 ligase MDM2, preventing AR's ubiquitination by MDM2 [179]. Kinesin family member 15 (KIF15) facilitates the interaction between USP14 and AR/AR-V7, promoting enzalutamide resistance in prostate cancer cells [180]. Research has identified that nobiletin, a polymethoxylated flavonoid from citrus fruit peels, possesses significant anticancer properties. It induces G0/G1 phase arrest and heightens the sensitivity of AR-V7+ cells to enzalutamide by selectively inhibiting the interactions between AR-V7 and USP14/USP22 [181]. Additionally, glucose-regulated protein 75 (GRP75) hinders the degradation of sinusoidal eye homeobox homolog 1 (SIX1) by facilitating its deubiquitination by USP1. Inhibiting the GRP75-USP1-SIX1 protein complex formation in preclinical models has been shown to delay tumor progression and augment enzalutamide efficacy [182].
Estrogen receptor (ER) inhibitors/Endocrine therapy
ER is present in about 70% of breast cancers (BC) and is a pivotal therapeutic target [183]. Patients with ER+ BC benefit from anti-estrogen endocrine therapies, including tamoxifen, an ER antagonist; fulvestrant, an ER modulator; and letrozole, an aromatase inhibitor [184]. Elevated USP22 levels can deubiquitinate ERα, enhancing its transactivation to cis-regulatory elements of ERα target genes, thereby increasing BC cell resistance to tamoxifen [185]. USP15, identified as a novel factor in protecting against ERα degradation, when knocked down, enhances K48-linked ERα ubiquitination, significantly boosting the efficacy of tamoxifen against BC cells [186]. Furthermore, as a crucial component of the PI3K pathway, AKT phosphorylates USP35 at Ser613. The activated USP35 interacts with ERα, boosting its transcriptional activity, which diminishes the effectiveness of tamoxifen and fulvestrant treatments [187].
EGFR inhibitors
EGFR-RTK activation plays an important role in the progression of NSCLC. To address this, a series of EGFR-TKI inhibitors, including gefitinib, erlotinib, afatinib, and osimertinib, have been developed specifically for NSCLC patients harboring EGFR mutations [188]. USP8 has emerged as a novel target to counteract gefitinib resistance, with its inhibition leading to the downregulation of multiple RTKs and the induction of cell death in gefitinib-resistant NSCLC cells, while sparing normal cells [189]. In addition to USP8, USP13 inhibits the ubiquitin-mediated degradation of EGFR by the Cbl family of E3 ubiquitin ligases, thereby selectively stabilizing mutant EGFR through a peptidase-independent mechanism [190]. Concurrently, USP22 deubiquitinates EGFR on late endosomes, enhancing its recycling and the sustained activation of various downstream signaling pathways upon EGF stimulation [191]. Moreover, microRNA-124a is identified as a tumor suppressor that targets USP14, reducing stemness and increasing the sensitivity of NSCLC cells to gefitinib [192]. Nonetheless, the precise mechanisms underlying these interactions remain largely unexplored and necessitate further investigation.
HER2/ERBB2 inhibitors
HER2-targeted therapies are developed to counteract the overexpression or amplification of HER2 protein in cancers, particularly BC. Trastuzumab, a monoclonal antibody targeting the HER2 receptor, stands out as the most prevalent HER2-targeted treatment. Additional therapies, such as pertuzumab, ado-trastuzumab emtansine (T-DM1), and lapatinib, impede the HER2 pathway through various mechanisms [193]. In the study by Shamshad et al., USP27X was found to be overexpressed in HER2+ resistant BC cells, where it deubiquitinates the CCND1 protein. The ablation of USP27X markedly reduces CCND1 levels and enhances the sensitivity of BC cells to lapatinib [194]. Persistent HER2 protein expression represents a critical resistance mechanism against HER2-targeted therapies. USP2 has been identified as a key regulator of HER2 stability, binding to internalized HER2 to avert its lysosomal degradation. Targeting USP2 reduces HER2 levels by promoting its ubiquitination and degradation [195], offering a potential strategy to overcome resistance in HER2-targeted BC therapies.
Proteasome inhibitors
Bortezomib (BTZ), a seminal proteasome inhibitor (PI), is extensively employed in the treatment of MM, where it notably impedes NF-κB activation and augments IκBα stability [196]. USP7's role involves deubiquitinating NEK2, thereby stabilizing its expression. Elevated NEK2 levels lead to the binding and phosphorylation of PP1α, initiating the canonical NF-κB pathway and engendering BTZ resistance in MM cells [197]. The ablation of USP7 markedly diminishes colony formation and mitigates BTZ resistance in MM cells by fortifying IκBα expression and obstructing the NF-κB pathway [198,199,200].
Research consistently shows that autophagy inhibition can significantly slow MM cell growth and induce apoptosis [201]. USP12 emerges as a critical regulator in this context, interacting with and deubiquitinating the autophagy mediator, high mobility group box-1 (HMGB1). The knockdown of USP12 decreases HMGB1 levels, curtails autophagy, and consequently boosts MM cell susceptibility to BTZ [202].
Table 1 encapsulates the described drug resistance mechanisms in cancers, as mediated by the deubiquitination activities of USPs.
Immunotherapy resistance mediated by USPs
Cancer immunotherapy seeks to mobilize the human immune system, utilizing the body's innate ability to eliminate cancer cells [203]. Despite the approval of targeted antibodies against key immune checkpoints, such as programmed death protein-1 (PD-1), programmed death-ligand 1 (PD-L1), and cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) for various cancers, a significant subset of patients encounters resistance and treatment failure [204]. Emerging researches suggest that modulating USP-mediated deubiquitination of proteins in antitumor immune responses may offer a strategy to circumvent immunotherapy resistance [205].
Extensive researches indicate the involvement of various USPs in the deubiquitination of PD1/PD-L1 proteins. For instance, USP8, upregulated in pancreatic cancer, can deubiquitinate PD-L1. Targeting USP8 reduces PD-L1’s level, stimulating cytotoxic T-cells, and bolstering the anti-tumor immune response, which enhances the efficacy of PD-L1-targeted immunotherapy [206]. However, a more nuanced study yielded contrary results, indicating that targeting USP8 elevates PD-L1 expression. This increase is primarily due to the intensification of K63 ubiquitination, facilitated by the E3 ligase TRAF6, which counteracts K48 ubiquitination, thereby averting PD-L1 degradation. In this context, USP8 inhibition initiates innate immune responses, boosts IFN type I signaling, and increases MHC-1 production through TRAF6-NF-κB signaling [207]. A similar dichotomy is observed with USP7's influence on PD-L1. In gastric tumors, USP7 suppression diminishes PD-L1 levels, increases the susceptibility of GC cells to T-cell-mediated destruction, and enhances the immune response [208]. Conversely, research by Dai et al. in lung cancer demonstrated that USP7 inhibition might actually intensify PD-L1 expression, associated with greater infiltration of M1 macrophages and IFN-γ+CD8+ T cells, culminating in a robust anti-tumor effect [209]. These disparate findings underscore the complex and context-dependent nature of USP7/USP8's impact on PD-L1. Nevertheless, combining USP8/USP7 inhibitors with PD-1/PD-L1 blockade appears to significantly bolster anti-tumor efficacy (Fig. 5A).

USPs regulate significant cancer immunotherapy resistance. A Targeting USP8, USP7, and USP22 affects PD-L1 protein stability, alters immune cells infiltration in tumor microenvironment, and enhances cancer cell sensitivity to immunotherapy. B USPs modulate critical IFN signaling pathways to affect cell pyroptosis, MHC-I receptor expression, and cytokine release
USP22 has been found to interact directly with the C terminus of PD-L1 protein, facilitating its deubiquitination. In a liver cancer mouse model, USP22 knockdown significantly enhanced the efficacy of combined PD-L1 targeted immunotherapy and cisplatin by boosting tumor immunogenicity [210]. In pancreatic cancer, USP22 knockout amplified the response to concurrent anti-PD1 and anti-CTLA4 therapy, notably by diminishing myeloid cell infiltration and encouraging T cell and NK cell presence, thus converting “cold” tumors into “hot” tumors [211] (Fig. 5A). Conversely, melanoma studies indicate that USP22 loss does not enhance immunotherapy effectiveness but rather induces resistance to T cell-mediated cytotoxicity. USP22's ability to deubiquitinate STAT1 and activate the JAK-STAT pathway is crucial; without USP22, STAT1 degradation escalates, inhibiting IFNγ from engaging with its receptors IFNGR1 and IFNGR2, and thus disrupting T cell-mediated cytotoxic signaling [212]. Moreover, IFN-γ activation leads to STAT1 phosphorylation, which triggers its nuclear migration and the subsequent activation of lncRNA TINCR transcription. TINCR then associates with DNMT1, promoting the methylation of miR-199a-5p loci and diminishing miR-199a-5p's suppressive effect on USP20, thereby stabilizing USP20 mRNA. Consequently, USP20 deubiquitinates PD-L1, increasing BC cell resistance to PD-L1 inhibitors [213]. Additionally, ERK phosphorylation of PD-1 at Thr234 enables USP5-mediated deubiquitination. Inhibiting USP5 in T cells reduces PD-1 levels, augments effector cytokine production, and decelerates tumor progression in mice, significantly enhancing the response to anti-CTLA-4 or trametinib therapy [214] (Fig. 5B).
Pyroptosis is a distinct form of programmed cell death, differing from apoptosis, characterized as a regulatory necrosis mechanism in inflammatory cells under stress or infection conditions [215]. Researches have shown that pyroptosis plays a crucial role in modulating immunotherapy responses [216, 217]. USP18, by interacting with IFNα receptors and STAT2, diminishes the binding of STAT2-mediated transcription complexes to IFN response elements, thus attenuating type I IFN signaling [218]. Inhibiting USP18 enhances the expression of canonical IFN-stimulated genes (ISGs) and activates a subset of non-traditional ISGs and NF-κB target genes, such as PLK2, leading to the induction of cancer pyroptosis [216]. Gasdermin family member Gasdermin E (GSDME), is activated by Caspase 3, transitioning apoptosis to pyroptosis [219]. USP48 facilitates pyroptosis by binding with GSDME, stripping its K48-linked ubiquitin, and thereby augmenting the functions of T cells and tumor-associated macrophages (TAMs) within the tumor microenvironment (TME), significantly boosting the efficacy of PD-1 inhibitors [218] (Fig. 5B).
Several seminal studies have significantly advanced the understanding of intricate roles of USPs in modulating immunotherapy responses. First, the TET2 DNA dioxygenase is monoubiquitylated at K1299, which augments its enzymatic function and promotes lymphocytes infiltration into tumors [220]. USP15, by removing this monoubiquitin, negatively impacts TET2 activity. Its absence in melanoma cells leads to enhanced IFNγ-induced chemokine production and lymphocytes recruitment, thereby augmenting the immunotherapy responsiveness [221]. Second, the advent of immunomodulatory medicines (IMiDs) like lenalidomide, thalidomide, and pomalidomide has transformed treatment approaches for MM [222]. IMiDs act by binding to cereblon (CRBN), a substrate receptor of the CUL4-RBX1-DDB1-CRBN (CRL4CRBN) E3 ligase complex, thereby recruiting neosubstrates as the drug target for ongoing degradation [223]. This study found that USP15 counteracts the CRL4CRBN-mediated ubiquitylation of these neosubstrates. Inhibiting USP15 promotes the degradation of these substrates, enhancing the sensitivity of IMiD-resistant MM cells to treatment, and offering a new avenue for CRBN-based PROTACs therapies [224]. Moreover, oncogenic KRAS activation fosters pro-tumorigenic microenvironment [225]. In KrasG12D-driven lung cancer, USP12 suppression, triggered by AKT-mTOR signaling, leads to inadequate deubiquitination of PPM1B, resulting in NF-κB signaling hyperactivation and the creation of an immune-suppressive milieu. This environment, characterized by increased macrophage presence, vascularization, and reduced T-cell activity, diminishes the efficacy of anti-PD-1 immunotherapy [226]. Furthermore, the essential amino acid tryptophan's depletion and the rise of kynurenine, catalyzed by IDO1, are crucial for immune evasion [227]. In CRC, USP14 directly deubiquitinates IDO1, shielding it from K48 ubiquitination by TRIM21. USP14 inhibition decreases IDO1 levels, disrupts CD8+ cell activation, alters CD4+ T cell differentiation into Treg cells, boosts the immune response against tumors, and increases the effectiveness of anti-PD-1 treatment [228].
Bioinformatics analysis has demonstrated a significant association between USP35 and an immunosuppressive TME, as indicated by the negative correlation between USP35 levels and CD8+ T cell infiltration in skin cutaneous melanoma [229, 230]. Patients exhibiting high USP35 expression show reduced benefits from immunotherapy compared to those with lower expression levels. A comparable predictive trend is noted for USP51 in GC patients, where increased USP51 expression correlates with decreased immunotherapy efficacy [231]. However, the specific mechanisms through which USP35 and USP51 affect immunotherapy success remain unclear and warrant further experimental investigation.
Table 2 summarizes the resistance mechanisms to cancer immunotherapy mediated by USPs.
Radiotherapy resistance mediated by USPs
Radiotherapy, a prevalent cancer treatment modality, employs radiation to induce DNA damage and inhibit cell replication in cancer cells [232]. A key strategy to counteract tumor radioresistance involves disrupting the protective DDR mechanisms. In NSCLC, USP14 modulates DSB repair in response to ionizing radiation (IR) by influencing both NHEJ and HR pathways. Inhibiting USP14 enhances NHEJ efficiency, facilitates the recruitment of essential NHEJ proteins to chromatin, and increases the formation of IR-induced BRCA1 foci [233]. Moreover, radiation triggers the phosphorylation of DGCR8 by the kinase ATM, enhancing DGCR8's deubiquitination by USP51. This enhances the assembly of activated DGCR8 and RNF168 at DSB sites via MDC1, promoting DSB repair and contributing to radioresistance in cancer cells [234].
Histone methylation and acetylation by various enzymes, are crucial in DDR and radioresistance [235]. USP7 facilitates the deubiquitination of histone demethylase PHF8, elevating cyclin A2 levels, which attracts more BLM and KU70 to DSBs, thereby enhancing cellular resistance to radiation [236]. Additionally, USP38 associates with histone deacetylase HDAC1, removes its K63-linked ubiquitin chains, and bolsters the deacetylase activity of HDAC1 on histone H3K56. The absence of USP38 diminishes NHEJ efficiency and heightens cell vulnerability to IR [237].
The CHK family plays a crucial role in regulating cell cycle and mitosis, significantly impacting radiotherapy resistance. In BC cells, USP7 collaborates with LINC02582 to deubiquitinate and stabilize CHK1, targeting miR-200c and enhancing radioresistance [238]. Similarly, USP39 maintains CHK2 stability through deubiquitination. However, its depletion leads to increasing radiation resistance, accompanied with CHK2 dysfunction, impairing the G2/M checkpoint activation after DNA damage and reducing apoptosis [239].
Radiotherapy is a primary treatment modality for GBM, yet resistance to it is common in GBM patients [240]. USP1, highly expressed in GBM and particularly in cells positive for GSC-enrichment markers (CD133 or CD15), modulates the stability of ID1 and CHEK1, which are critical for DDR and stem cell maintenance. Inhibiting USP1 enhances GBM cell radiosensitivity and curtails GSC clonogenic growth and survival [241]. Moreover, USP44's interaction with histone H2B is disrupted by lincRA1, which binds to H2B and maintains H2Bub1 levels, impeding USP44's binding, inhibiting autophagy, and fostering radioresistance in GBM [242]. Additionally, the UCH domain of USP3 interacts with the N-terminus of Claspin, stabilizing it against ubiquitination and consequently activating ATR-CHK1 signaling, which contributes to the radioresistance in GBM cells [243].
In addition to the above studies, USPs also play pivotal roles in various pathways, modulating the activity of essential proteins in radioresistance. For instance, USP9X influences apoptosis by targeting MCL-1 [244, 245] or regulates TGFβ signaling via KDM4C [246], while USP7 and USP24 target p53 [247, 248], USP13 targets PTEN [249], USP53 interacts with DNA damage binding protein 2 (DDB2) [250], and USP28 modulates HIF-1α [251]. Due to space constraints, an in-depth discussion of these mechanisms is beyond the scope of this review. For reference, Table 3 succinctly summarizes these mechanisms.
Overcome anti-cancer drug resistance by USP inhibitors
Recent advances in USP inhibitors as therapeutic agents have demonstrated significant anti-cancer potential, with extensive reviews covering their development and clinical applications [14, 252, 253]. This section highlights USP inhibitors crucial for overcoming drug resistance in cancer treatment (Table 4).
USP7 inhibitors
Among USP inhibitors, USP7 inhibitors are the most varied and thoroughly researched. The thiophenyl compound P22077, a notable USP7 inhibitor, induces apoptosis by targeting USP7 and enhancing intracellular ROS production [260]. It stabilizes p53 and degrades HDM2, augmenting the cytotoxic effects of Dox and etoposide on NB cells [84]. In HCC and PDAC, P22077 lessens the cells' sensitivity to Dox [82, 83]. Additionally, P22077 disrupts the USP7-CHK1 interaction, aiding in overcoming cytarabine resistance in AML [254]. The combination of P22077 with the PLK1 inhibitor volasertib shows synergistic efficacy in paclitaxel-resistant lung cancer [104]. Interestingly, P22077 not only targets USP7 but also addresses IM resistance in CML by inhibiting USP47, enhancing the effectiveness against TKI-resistant CML cells and reducing Lin−Sca1+c-Kit+ CML stem/progenitor cell numbers in CML models [154].
Through high-throughput screening, scientists identified another novel USP7 inhibitor, the thiophenyl compound P5091, which induces apoptosis in BTZ-resistant MM cells. When combined with lenalidomide, dexamethasone, or SAHA (an HDAC inhibitor), P5091 demonstrates synergistic therapeutic effects [177]. In MM cells, the concurrent use of the NEK2 inhibitor INH1 and P5091 markedly impedes cell growth and overcomes NEK2-related and inherent BTZ resistance by modulating the NF-κB and PP1α/AKT pathways [175]. Moreover, the hypoxia-selective epigenetic agent RRx-001 triggers MM cell apoptosis through Caspase activation, increased ROS release, and reduced global methylation, exhibiting synergistic anti-MM effects with P5091 in overcoming BTZ resistance [255]. P5091 also enhances the sensitivity of lung neuroendocrine tumor cells to PARP inhibitors by diminishing CCDC6 levels and hampering HR repair, showing combined efficacy against lung neuroendocrine and CRPC [145, 147]. As for immunotherapy, P5091 escalates PD-L1 expression, while it blocks PD-1 and reprograms TAMs in TME, facilitating an effective antitumor response in Lewis lung carcinoma [209].
GNE-6776, another prominent USP7 inhibitor, exhibits significant inhibitory activity against the USP7 catalytic domain [261], markedly increasing apoptosis in chemoresistant TNBC cells [93]. HBX19818, which covalently binds to USP7's active site, enhances the sensitivity of chemoresistant and p53-deficient CLL cells to chemotherapy [172]. Notably, RAPT Therapeutics, Inc. has developed a unique USP7 inhibitor, compound 41 [262], which re-sensitizes MYCN-amplified chemoresistant tumors to cisplatin and etoposide by reducing N-MYC levels and increasing cleaved Caspase 3 [256].
USP1 inhibitors
Given the functional role of USP1 as part of the USP1/UAF1 complex, extensive researches have been conducted to develop inhibitors targeting this complex. In 2011, the first USP1/UAF1 inhibitor was identified through a high-throughput screening using Ub-Rho110 [40]. After that, pimozide and GW7647, identified as the most effective compounds, demonstrate noncompetitive and reversible inhibition of USP1/UAF1. In NSCLC cells, they increase the monoubiquitylation of PCNA and FANCD2 [40]. The combination of pimozide and the MAST1 inhibitor lestaurtinib markedly decreases MAST1 expression and the phosphorylation of MEK1 and ERK in cancer cells, enhancing their sensitivity to cisplatin [43]. In a model of rituximab/chemotherapy-resistant diffuse large B-cell lymphoma, pimozide synergizes with etoposide, destabilizing MAX, thereby inhibiting cell proliferation and inducing apoptosis, autophagy, and cell cycle arrest [257]. However, the interaction of pimozide and GW7647 with other proteins, independent of their DUB activity, may restrict their application in certain contexts.
The discovery of C527 and its more potent derivatives in 2013 marked a significant advancement, although their selectivity remains limited [263]. SJB3-019A, a derivative of C527, diminishes MM cell viability and mitigates resistance to BTZ. Its combinatory application with the HDAC inhibitor ACY-1215, BTZ, lenalidomide, or pomalidomide shows synergistic cytotoxic effects on MM cells [258]. Additionally, a new compound, ML323, surpasses GW7647 in terms of potency. With excellent selectivity against human DUBs, deSUMOylases, deneddylases, and unrelated proteases, ML323 boosts cytotoxicity in cisplatin-resistant NSCLC by blocking PCNA and FANCD2 deubiquitination [38, 264]. Moreover, ML323 selectively targets a subgroup of BRCA1-deficient cells that have developed resistance to PARP inhibitors through replication fork stabilization [143].
USP13 inhibitors
USP13 inhibitors play a critical role in modulating DNA repair mechanisms. USP13 can deubiquitinate DNA topoisomerase 2 binding protein 1 (TopBP1), influencing DNA chain breakage and repair processes. Depleting USP13 enhances cellular sensitivity to replication stress inducers such as hydroxyurea (HU), camptothecin (CPT), ultraviolet (UV) radiation, and 5-Fu [265]. An imaging-based screening method led to the identification of spautin-1, a potent autophagy inhibitor that targets both USP10 and USP13 [266]. Spautin-1 disrupts RAP80-BRCA1 complex formation, impeding the DDR and enhancing the sensitivity of OC cells to olaparib. Combining spautin-1 with olaparib offers a superior synergistic therapeutic effect compared to olaparib alone [137]. In addition, spautin-1, when used with the EGFR inhibitor afatinib, significantly reduces the viability of NSCLC cells [197]. In a GIST cell-derived mouse xenograft model, spautin-1 triggers ATG5 degradation, and its use with 3-methyladenine notably enhances the therapeutic impact of IM [158].
USP14 inhibitors
Compound b-AP15 is recognized for inducing apoptosis by targeting USP14 and UCHL5 [267]. It is particularly effective in inducing apoptosis in cells overexpressing BCL-2 or lacking functional p53, positioning it as a viable therapeutic approach for BTZ-resistant WM patients [259]. In 2015, the development of VX1570 improved the physicochemical properties of b-AP15 [268]. VX1570 prompts rapid, tumor-specific apoptosis in WM cells resistant to BTZ or ibrutinib, diminishing tumor load and extending survival in WM xenograft models [173]. A subsequent screening of 63,052 compounds identified a novel USP14 inhibitor, IU1, which specifically binds to the active form of USP14, inhibiting its association with the proteasome while sparing other DUBs [269]. When paired with anti-PD-1 therapy, IU1 markedly reduced tumor mass and extended survival in mouse models [228].
USP8 inhibitors
DUBs-IN-2 is an effective USP8 inhibitor with potential in countering various types of immunotherapy resistance. Its application leads to PD-L1 upregulation, which stimulates immune responses and antigen presentation, thus transforming the TME into a more inflamed state. This alteration in TME bolsters the effectiveness of anti-PD-1/PD-L1 immunotherapy across several mouse tumor models [207]. In pancreatic cancer, the combination of DUBs-IN-2 and anti-PD-L1 therapy activates cytotoxic T cells, significantly inhibiting tumor growth [206]. Moreover, a synthesized USP8 inhibitor, 9-Ethyloxyimino-9H-indeno [1,2-b] pyrazine2,3-dicarbonitrile, has been shown to suppress multiple RTKs in gefitinib-resistant NSCLC cells. This inhibitor promotes the colocalization of ubiquitin and target RTKs, effectively overcoming gefitinib resistance in lung cancer [196]. Additionally, in HCC cells and mouse models, 9-Ethyloxyimino-9H-indeno [1,2-b] pyrazine2,3-dicarbonitrile significantly boosts the effectiveness of Dox or sorafenib by reducing RTK expression by approximately 90% [90].
Conclusions and perspectives
In this review, we have thoroughly discussed the intricate mechanisms of USP-mediated drug resistance proceeding from the perspectives of various treatment strategies and specific drugs, and suggested that targeting USPs may offer novel insights into overcoming drug resistance in cancer therapy. Undoubtedly, USP inhibitors have the potential to counteract drug resistance and enhance the responsiveness of cancer cells to anti-cancer treatments, including chemotherapy, molecular targeted therapy, immunotherapy, and radiotherapy. Although the primary focus of our review is to provide insights and perspectives for clinical treatment by exploring USP-mediated cancer therapy resistance within the context of different clinical approaches, it is important to note the inherent interconnectedness between different USPs and drug resistance mechanisms. For instance, USP7 has been implicated in promoting DDR, thus mediating resistance to DNA-damaging chemotherapeutic agents and also radiation therapy. Furthermore, as one of the most extensively studied USPs, USP7 is not only involved in DDR but also participate in EMT, CSC generation, anti-apoptosis, hypoxia, angiogenesis, and modulation of immune cell infiltration within the TME. These biological functions collectively contribute to the development of resistance mechanisms in cancer therapy. Therefore, USP7 mediates resistance to a wide range of chemotherapeutic agents, radiation therapy, and immunotherapy. Another notable example is USP22, which significantly impacts the efficacy of chemotherapy drugs and immunotherapy due to its involvement in EMT, CSC formation, and modulation of TME. The overarching framework of this review focuses on the interaction between USPs and drugs, with a specific emphasis on USP vs. cellular pathway/functional signaling within each particular drug category. Different drug action mechanisms determine the specific resistance signaling mechanisms mediated by USPs, while the USP-mediated signaling pathways, in turn, contribute to varied drug resistance profiles. These relationships exhibit overlapping and reciprocal influences.
Therefore, expanding on these aspects not only deepens the understanding of the complex dynamics underlying USP-mediated resistance but also sheds light on the challenges faced by researchers aiming to unravel these intricate networks and optimize therapeutic outcomes. Given the complexity of USP regulatory network, the exact mechanisms by which USP inhibition can be leveraged to surmount resistance to anti-cancer drugs remain incompletely elucidated. While USPs have demonstrated potential in mediating cancer drug resistance, several challenges and considerations must be addressed.
Firstly, we catalogued the USPs implicated in cancer drug resistance across various cancer types, as illustrated in Fig. 6A. The expression patterns of USPs across different cancer types reflect specific molecular alterations and signaling pathways of each cancer. The presence of multiple USPs within a particular cancer type or the expression and variation of same USP (e.g., USP7, USP14, and USP22) across different cancers suggest functional redundancy. This implies that different USPs may substitute for one another's functions and substrates, adding to their role complexity in cancer and complicating the targeting of a singular USP for treatment. Tumors consist of a heterogeneous mix of cancer cells, each with unique genetic and phenotypic characteristics. Within a tumor, cancer cells can have diverse molecular signatures, including USP expression variations. The impact of USPs on drug resistance is context-dependent, shaped by the specific cellular environment, TME, and genetic landscape, which can also shift in response to external stimuli, such as environmental changes or treatment. This variability introduces further complexity in pinpointing the precise USPs responsible for cancer drug resistance. Overcoming these challenges necessitates extensive profiling of USP expressions and activities across a range of cancer types and stages to track USP dynamics and comprehend their roles. Traditional methods for USP activity assessment, like biochemical assays, may not suit clinical samples or lack necessary sensitivity and specificity. Thus, developing precise and reliable assays for measuring USP activity in patient-derived samples is crucial for identifying USPs pivotal in drug resistance. This endeavor often requires merging multi-omics data, including genomics, transcriptomics, proteomics, and epigenomics to pinpoint USPs linked to drug resistance in particular cancer scenarios. Fostering interdisciplinary collaboration, employing advanced technologies, and analyzing extensive patient cohorts could lead to personalized treatment strategies targeting specific USPs involved in drug resistance for each cancer type.

USPs exhibit overlapping expressions and functional mechanisms in mediating drug resistance during cancer treatment. A The expression and variability of USPs contribute to drug resistance across various cancer types. B Different USPs orchestrate drug resistance through intricate functional mechanisms
Secondly, the USP family consists of numerous members with overlapping function in drug resistance (Fig. 6B). Abovementioned USP7 and USP22 has been implicated in mediating drug resistance through a variety of molecular mechanisms, thus USP7 and USP22 can affect the efficacy of multiple drugs, not just one specific drug (Fig. 7A). In addition, USP2a and USP29 have been shown to influence the therapeutic resistance to paclitaxel, platinum, and Dox. The overlapping functions of different USPs in cancer drug resistance pose challenges for achieving functional specificity and avoiding cross-influence. While some USPs may confer “drug-resistance” roles by stabilizing crucial signaling proteins, others may function as suppressors through deubiquitinating and activating proteins in various molecular pathways. Although multiple USPs may participate in the same cellular processes or drug strategies, they often have unique substrates or regulatory networks that bestow specific resistant functions. Identifying the precise molecular mechanisms that underpin the drug resistance-associated functions of individual USPs is crucial, necessitating a blend of experimental and computational methods. Functional studies, such as RNA interference and CRISPR/Cas9-mediated gene knockout, can modulate the expression of specific USPs in cancer cells or animal models during drug treatment. High-throughput screening can identify downstream substrates or binding partners of USPs relevant to drug resistance. Biochemical assays, like in vitro deubiquitination assays using recombinant USPs and targets, can elucidate specific protein targets and deubiquitination events. These assays, combined with drug treatments, assess the impact of USPs on drug responses. Importantly, computational modeling, including molecular dynamics simulations, docking studies, or network analysis, can predict and elucidate interactions between USPs, their substrates, and drug resistance molecules. Network-based approaches can identify crucial nodes or modules within signaling networks affected by USPs in the context of drug resistance.

Targeting USPs is challenging within the complex UPS to overcome drug resistance. A The overlapping expressions and regulatory functions of USPs are illustrated in mediating drug resistance among different DNA-damage inducing agents. B The essential strategies and specific biochemical approaches are crucial in addressing the challenges of targeting USPs. C The interaction between USPs and E3 ligases maintains the equilibrium between ubiquitination and deubiquitination process in drug resistance
Thirdly, although the identification and creation of strong and specific USP inhibitors is an exciting field with promise for improving cancer outcomes, the challenge of targeting USPs persists. USPs possess conserved catalytic domains, with the active site of USPs featuring a catalytic triad in the Cys and His domains, comprising a cysteine residue, a histidine residue, and an aspartate residue. These residues are essential for the deubiquitinating activity of USPs. The task of designing inhibitors that can specifically target the active site and hinder the catalytic activity of USPs is daunting due to the high conservation of the catalytic triad across various USPs. Additionally, the catalytic domain of USPs can demonstrate structural flexibility, enabling the accommodation of a wide range of substrates. This adaptability presents obstacles in developing small molecule inhibitors that can selectively target individual USPs without impacting other USPs with similar structures. Consequently, many existing USP inhibitors may exhibit off-target effects, affecting cellular processes unrelated to drug resistance, and limiting the therapeutic potential of USP inhibitors. We posit that researchers are pursuing various strategies to address the challenges of targeting USPs (Fig. 7B). 1) Covalent inhibitors: The development of covalent inhibitors that form irreversible bonds with the USP active site can enhance both selectivity and potency. These inhibitors target unique reactive residues within the USP active site for more effective and specific inhibition. 2) Allosteric inhibitors: Instead of the catalytic site, allosteric inhibitors attach to different sites on the USP protein, altering its activity. This method aims to achieve selectivity by focusing on distinctive conformational states or regulatory regions of the USP. 3) Peptide-based or protein-protein interaction inhibitors: Inhibitors derived from USP substrates or interacting proteins can disrupt USP's interactions with its substrates or regulatory proteins, thereby inhibiting its activity. 4) PROTACs and molecular glues: These innovative strategies employ bifunctional molecules to direct USPs towards an E3 ubiquitin ligase or a target protein for ubiquitination and subsequent degradation. This leverages the proteasomal degradation pathway to indirectly diminish USP levels and their activity. 5) Combination therapies: To address potential resistance to USP inhibitors, similar to other targeted therapies, their combination with other treatments could enhance therapeutic efficacy and potentially forestall or delay resistance development. Furthermore, with advancements in science and technology, increasingly sophisticated drug design strategies are being applied (Fig. 7B). It's critical to acknowledge that these strategies are in active research phases, and their success may vary by the specific USP and disease context. Ongoing research in these fields is promising for surmounting USP targeting challenges and improving therapeutic outcomes. We maintain that through the integration of computational modeling and synthesis, USP inhibitors can be identified and optimized to achieve enhanced potency, selectivity, drug-like characteristics, and minimized off-target effects.
Lastly, despite the existence of over 1,000 E3 ligases, there are fewer than 100 DUBs, with USPs constituting a significant subgroup. This discrepancy in numbers suggests that USPs perform multiple roles and participate in various cellular processes critical for overcoming drug resistance. The interactions and competition between E3 ligases and USPs represent a pivotal area of research (Fig. 7C). E3 ligases facilitate ubiquitination and protein degradation, whereas USPs reverse this by deubiquitinating the same substrates, allowing for precise regulation of ubiquitylation status, which affects the stability, localization, and activity of substrate proteins in cancer therapy. Beyond targeting the same protein substrates for their antagonistic effects, USPs and E3 ligases can interact directly, acting as substrates for each other's ubiquitination or deubiquitination activities. Notably, many USPs are linked with E3 ligases, such as USP7 and MDM2, USP15 and Keap1, USP47 and β-Trcp, which are prone to self-ubiquitylate or even to enhance the ubiquitination activities of E3 ligases through deubiquitinating them. Hence, the stabilization of E3 ligases through deubiquitination underscores a key aspect of USP function, while E3 ligases may destabilize their corresponding USPs via ubiquitination. This raises the question of whether specific research strategies could modulate the interaction and competitive dynamics between USPs and E3 ligases to improve the targeting of USPs in drug resistance. Investigating potential reciprocal regulatory networks and biomarkers between USPs and E3 ligases related to drug resistance, are critical initial steps. These efforts can inform drug design and targeted therapy. By precisely mapping interaction sites and employing computational methods, novel drugs could be developed to either simultaneously sensitize E3 ligase and inhibit USP or co-inhibit both, enabling dual or multiple substrate protein degradation for augmented anti-cancer effects. However, strategies to regulate USP and E3 ligase interactions are still nascent, necessitating further research to confirm their efficacy and safety. Sensitizing or inhibiting specific E3 ligases might also trigger unintended degradation of unknown substrate proteins, highlighting the need for additional investigation and consideration.
It is crucial to emphasize that, while detailed mechanistic roles have shown promise in preclinical studies, the clinical validation of USP inhibitors for overcoming drug resistance remains nascent, necessitating further investigation into their safety, efficacy, and long-term impacts. Executing well-designed clinical trials with meticulous patient selection and rigorous outcome measures is vital to ascertain the clinical utility of USP inhibitors in combating drug resistance. The identification and validation of predictive biomarkers that can categorize patients based on their likelihood of responding to USP inhibitors will enhance patient selection and facilitate monitoring of treatment responses.
In conclusion, this review advances current understanding of USPs' complex roles, suggesting that targeting USPs could be a strategic approach to tackling tumor resistance. It may also uncover new clinical applications and provide a framework for the future improvement of USP inhibitors. However, comprehensive research is required to elucidate the complex mechanisms by which USPs influence drug resistance. This includes additional studies to decipher the USP family's complexity and redundancy, develop personalized treatment modalities based on tissue-specific USP profiling, enhance the selectivity and specificity of USP inhibitors, investigate combination therapies to circumvent resistance, and implement rigorous clinical trials with strategic patient selection and biomarker validation. By confronting these challenges, the potential of USPs as therapeutic targets for countering drug resistance in cancer can be more fully realized.
Availability of data and materials
No datasets were generated or analysed during the current study.
Abbreviations
- PTM:
-
Post-translational modification
- DUB:
-
Deubiquitinating enzyme
- UPS:
-
Ubiquitin proteasome system
- USP:
-
Ubiquitin-specific protease
- UBL:
-
Ubiquitin-like
- ZnF-UBP:
-
Zinc finger ubiquitin-binding
- DUSP:
-
Domains specific to USP
- UIM:
-
Ubiquitin-interacting motifs
- UBA:
-
Ubiquitin-associated
- CSC:
-
Cancer stem cell
- EMT:
-
Epithelial mesenchymal transformation
- DDR:
-
DNA damage response
- CHK1:
-
Checkpoint kinase 1
- DSBs:
-
Double strand breaks
- PALB2:
-
partner and localizer of BRCA2
- CtIP:
-
C-terminal binding protein-interacting protein
- NSCLC:
-
Non-small cell lung cancer
- UAF1:
-
USP1 associated factor 1
- ZEB1:
-
Zinc finger E-box binding homeobox 1
- OC:
-
Ovarzian cancer
- ESCC:
-
Esophageal squamous cell carcinoma
- PARP:
-
Poly‑ADP ribose polymerase
- IAP:
-
Inhibitor of apoptosis protein
- SIRT1:
-
Sirtuin 1
- TNBC:
-
Triple-negative breast cancer
- ALDH:
-
Aldehyde dehydrogenase
- SOC:
-
Serous ovarian cancer
- BC:
-
Breast cancer
- CAF:
-
Cancer-associated fibroblast
- GC:
-
Gastric cancer
- LSCC:
-
Laryngeal squamous cell carcinoma
- 5-Fu:
-
5-fluorouracil
- lncRNA:
-
Long non-coding RNA
- CRC:
-
Colorectal cancer
- HIF-1α:
-
Hypoxia-inducible factor
- AR:
-
Androgen receptor
- HCC:
-
Hepatocellular carcinoma
- FUCA1:
-
α-L-fucosidase 1
- NER:
-
Nucleotide excision repair
- HDAC3:
-
Histone deacetylase 3
- Dox:
-
Doxorubicin
- TME:
-
Tumor microenvironment
- PDAC:
-
Pancreatic ductal adenocarcinoma
- NB:
-
Neuroblastoma
- β-Trcp:
-
β-transducin repeat-containing protein
- RTK:
-
Receptor tyrosine kinase
- ABC:
-
ATP-binding cassette,
- Pgp:
-
P-glycoprotein
- Ct-HBx:
-
Carboxyl-terminal truncated form of the Hepatitis B virus X protein
- CAM-DR:
-
Cell adhesion-mediated drug resistance
- MM:
-
Multiple myeloma
- ROS:
-
Reactive oxygen species
- CRPC:
-
Castration-resistant prostate cancer
- JNK:
-
cJUN NH2-terminal kinase
- OS:
-
Oxidative stress
- Nrf2:
-
NF-E2-related factor 2
- lipid-ROS:
-
Lipid reactive oxygen species
- hnRNPA1:
-
Heterogeneous nuclear ribonucleoprotein A1
- ALOX15:
-
Arachidonate lipoxygenase 15
- TS:
-
Thymidylate synthase
- PcG:
-
Polycomb group
- HNSCC:
-
Head and neck squamous cell carcinoma
- MRP1:
-
Multidrug resistance-associated protein 1
- GBM:
-
Glioblastoma
- GSC:
-
Glioma stem cell
- USP6NL:
-
USP 6 N-terminal-like protein
- CAV1:
-
Caveolin-1
- SSBs:
-
Single strand breaks
- NHEJ:
-
Nonhomologous end-joining
- HR:
-
Homologous recombination
- RAP80:
-
Receptor-associated protein 80
- TLS:
-
Translesion synthesis
- FBP1:
-
Fructose-1,6-bisphosphatase 1
- DNMT1:
-
DNA (cytosine-5)-methyltransferase 1
- TKI:
-
Tyrosine kinase inhibitor
- IM:
-
Imatinib
- CML:
-
Chronic myeloid leukemia
- GIST:
-
Gastrointestinal stromal tumor
- PBMC:
-
Peripheral blood mononuclear cell
- GLS1:
-
Glutaminase-1
- hucMSC:
-
Human umbilical cord mesenchymal stem cell
- YB-1:
-
Y-box binding protein 1
- ATG5:
-
Autophagy-related protein 5
- METTL3:
-
N6-methyladenosine methyltransferase-like 3
- Rab35:
-
Ras-related protein Rab-35
- Gal-SLP:
-
Galactose-decorated lipopolyplex
- PDX:
-
Patient-derived xenograft
- HK2:
-
Hexokinase 2
- BTK:
-
Bruton's tyrosine kinase
- BCR:
-
B-cell receptor
- CLL:
-
Chronic lymphocytic leukemia
- WM:
-
Waldenstrom macroglobulinemia
- ADT:
-
Androgen deprivation therapy
- HSPC:
-
Hormone-sensitive prostate cancer
- KIF15:
-
Kinesin family member 15
- GRP75:
-
Glucose-regulated protein 75
- HSP:
-
Heat shock protein
- SIX1:
-
Sine oculis homeobox homolog 1
- ER:
-
Estrogen receptor
- OS:
-
Overall survival
- T-DM1:
-
Ado-trastuzumab emtansine
- PI:
-
Proteasome inhibitor
- BTZ:
-
Bortezomib
- HMGB1:
-
high mobility group box-1
- PD-1:
-
Programmed death protein-1
- PD-L1:
-
Ligand programmed death-ligand
- CTLA-4:
-
Cytotoxic T lymphocyte-associated antigen-4
- GSDME:
-
Gasdermin E
- TAMs:
-
Tumor-associated macrophages
- ISGs:
-
IFN-stimulated genes
- TET2:
-
Ten-eleven translocation 2
- IMiDs:
-
Immunomodulatory drugs
- PROTACs:
-
Proteolysis-targeting chimeras
- TRP:
-
tryptophan
- KYN:
-
Kynurenine
- IDO1:
-
Indoleamine 2,3-dioxygenase 1
- IR:
-
Ionizing radiation
- DDB2:
-
DNA damage binding protein 2
- TopBP1:
-
Topoisomerase 2 binding protein 1
- HU:
-
Hydroxyurea
- CPT:
-
Camptothecin
- UV:
-
Ultraviolet
References
International Agency for Research on Cancer. Latest global cancer data: Cancer burden rises to 19.3 million new cases and 10.0 million cancer deaths in 2020. World Health Organization. 2020.
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58.
Williams PA, Zaidi SK, Sengupta R. AACR cancer progress report 2023: advancing the frontiers of cancer science and medicine. Clin Cancer Res. 2023;29(19):3850–1.
Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81–94.
Marine JC, Dawson SJ, Dawson MA. Non-genetic mechanisms of therapeutic resistance in cancer. Nat Rev Cancer. 2020;20(12):743–56.
Musyuni P, Bai J, Sheikh A, Vasanthan KS, Jain GK, Abourehab MAS, et al. Precision medicine: ray of hope in overcoming cancer multidrug resistance. Drug Resist Updat. 2022;65:100889.
Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575(7782):299–309.
Nussinov R, Tsai CJ, Jang H. Anticancer drug resistance: an update and perspective. Drug Resist Updat. 2021;59:100796.
Wang N, Ma T, Yu B. Targeting epigenetic regulators to overcome drug resistance in cancers. Signal Transduct Target Ther. 2023;8(1):69.
Huang X, Dixit VM. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 2016;26(4):484–98.
Dai C, Gu W. P53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16(11):528–36.
Mansour MA. Ubiquitination: friend and foe in cancer. Int J Biochem Cell Biol. 2018;101:80–93.
Park J, Cho J, Song EJ. Ubiquitin–proteasome system (UPS) as a target for anticancer treatment. Arch Pharm Res. 2020;43(11):1144–61.
Tang R, Langdon WY, Zhang J. Regulation of immune responses by E3 ubiquitin ligase Cbl-b. Cell Immunol. 2019;340:103878.
Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, Martin A. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482(7384):186–91.
Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019;366(6467):818–22.
Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10(8):550–63.
Snyder NA, Silva GM. Deubiquitinating Enzymes (DUBs): Regulation, Homeostasis, and Oxidative Stress Response. J Biol Chem. 2021;297(3):101077.
Mennerich D, Kubaichuk K, Kietzmann T. DUBs, hypoxia, and cancer. Trends Cancer. 2019;5(10):632–53.
Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discov. 2018;17(1):57–78.
Tanguturi P, Kim KS, Ramakrishna S. The role of deubiquitinating enzymes in cancer drug resistance. Cancer Chemother Pharmacol. 2020;85(4):627–39.
Qiu GZ, Sun W, Jin MZ, Lin J, Lu PG, Jin WL. The bad seed gardener: deubiquitinases in the cancer stem-cell signaling network and therapeutic resistance. Pharmacol Ther. 2017;172:127–38.
Ge F, Li Y, Yuan T, Wu Y, He Q, Yang BD, et al. Deubiquitinating enzymes: promising targets for drug resistance. Drug Discov Today. 2022;27(9):2603–13.
Rottenberg S, Disler C, Perego P. The rediscovery of platinum-based cancer therapy. Nat Rev Cancer. 2021;21(1):37–50.
Wangpaichitr M, Theodoropoulos G, Nguyen DJM, Wu C, Spector SA, Feun LG, et al. Cisplatin resistance and redox-metabolic vulnerability: a second alteration. Int J Mol Sci. 2021;22(14):7379.
Scully A, Panday R, Elango NA. Willis, DNA double-strand break repair pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 2019;20:698–714.
Adam-Zahir S, Plowman PN, Bourton EC, Sharif F, Parris CN. Increased gamma-H2AX and Rad51 DNA repair biomarker expression in human cell lines resistant to the chemotherapeutic agents nitrogen mustard and cisplatin. Chemotherapy. 2014;60(5–6):310–20.
Zhou F, Lu J, Jin W, Li Z, Xu D, Gu L. The role of USP51 in attenuating chemosensitivity of lung cancer cells to cisplatin by regulating DNA damage response. Biotechnol Appl Biochem. 2023;70(2):634–44.
Wang A, Ning Z, Lu C, Gao W, Liang J, Yan Q, et al. USP22 induces cisplatin resistance in lung adenocarcinoma by regulating γH2AX-Mediated DNA damage repair and Ku70/Bax-mediated apoptosis. Front Pharmacol. 2017;17(8):274.
Nardi IK, Stark JM, Larsen A, Salgia R, Raz DJ. USP22 interacts with PALB2 and promotes chemotherapy resistance via homologous recombination of DNA double-strand breaks. Mol Cancer Res. 2020;18(3):424–35.
Su D, Ma S, Shan L, Wang Y, Wang Y, Cao C, et al. Ubiquitin-specific protease 7 sustains DNA damage response and promotes cervical carcinogenesis. J Clin Invest. 2018;128(10):4280–96.
Alonso-de Vega I, Martín Y, Smits VA. USP7 controls Chk1 protein stability by direct deubiquitination. Cell Cycle. 2014;13(24):3921–6.
Zlatanou A, Sabbioneda S, Miller ES, Greenwalt A, Aggathanggelou A, Maurice MM, et al. USP7 is essential for maintaining Rad18 stability and DNA damage tolerance. Oncogene. 2016;35(8):965–76.
Zhu Q, Sharma N, He J, Wani G, Wani AA. USP7 deubiquitinase promotes ubiquitin-dependent DNA damage signaling by stabilizing RNF168. Cell Cycle. 2015;14(9):1413–25.
Das S, Chandrasekaran AP, Jo KS, Ko NR, Oh SJ, Kim KS, et al. HAUSP stabilizes Cdc25A and protects cervical cancer cells from DNA damage response. Biochim Biophys Acta Mol Cell Res. 2020;1867(12):118835.
Liu J, Zhou T, Dong X, Guo Q, Zheng L, Wang X, et al. De-ubiquitination of SAMHD1 by USP7 promotes DNA damage repair to overcome oncogenic stress and affect chemotherapy sensitivity. Oncogene. 2023;42(22):1843–56.
Sourisseau T, Helissey C, Lefebvre C, Ponsonnailles F, Malka-Mahieu H, Olaussen KA, et al. Translational regulation of the mRNA encoding the ubiquitin peptidase USP1 involved in the DNA damage response as a determinant of Cisplatin resistance. Cell Cycle. 2016;15(2):295–302.
Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, et al. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol. 2006;8(4):339–47.
Chen J, Dexheimer TS, Ai Y, Liang Q, Villamil MA, Inglese J, et al. Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem Biol. 2011;18(11):1390–400.
Pan C, Chun J, Li D, Boese AC, Li J, Kang J, et al. Hsp90B enhances MAST1-mediated cisplatin resistance by protecting MAST1 from proteosomal degradation. J Clin Invest. 2019;129(10):4110–23.
Pan C, Kang J, Hwang JS, Li J, Boese AC, Wang X, et al. Cisplatin-mediated activation of glucocorticoid receptor induces platinum resistance via MAST1. Nat Commun. 2021;12(1):4960.
Tyagi A, Kaushal K, Chandrasekaran AP, Sarodaya N, Das S, Park CH, et al. CRISPR/Cas9-based genome-wide screening for deubiquitinase subfamily identifies USP1 regulating MAST1-driven cisplatin-resistance in cancer cells. Theranostics. 2022;12(13):5949–70.
Srivastava M, Raghavan SC. DNA double-strand break repair inhibitors as cancer therapeutics. Chem Biol. 2015;22(1):17–29.
Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J, et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. 2014;16(9):864–75.
Zhou F, Du C, Xu D, Lu J, Zhou L, Wu C, et al. Knockdown of ubiquitin-specific protease 51 attenuates cisplatin resistance in lung cancer through ubiquitination of zinc-finger E-box binding homeobox 1. Mol Med Rep. 2020;22(2):1382–90.
Wang M, He SF, Liu LL, Sun XX, Yang F, Ge Q, et al. Potential role of ZEB1 as a DNA repair regulator in colorectal cancer cells revealed by cancer-associated promoter profiling. Oncol Rep. 2017;38(4):1941–8.
Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869–83.
Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–78.
Corno C, D’Arcy P, Bagnoli M, Paolini B, Costantino M, Carenini N, et al. The deubiquitinase USP8 regulates ovarian cancer cell response to cisplatin by suppressing apoptosis. Front Cell Dev Biol. 2022;10:1055067.
Shen J, Hong L, Chen L. Ubiquitin-specific protease 14 regulates ovarian cancer cisplatin resistance by stabilizing BCL6 oncoprotein. Biochem Biophys Res Commun. 2020;524(3):683–8.
Xu L, Zhang B, Li W. Downregulated expression levels of USP46 promote the resistance of ovarian cancer to cisplatin and are regulated by PUM2. Mol Med Rep. 2021;23(4):263.
Zhu X, Ma J, Lu M, Liu Z, Sun Y, Chen H. The Deubiquitinase USP39 Promotes Esophageal Squamous Cell Carcinoma Malignancy as a Splicing Factor. Genes (Basel). 2022;13(5):819.
Yuan J, Li X, Zhang Y, Zhang G, Cheng W, Wang W, et al. USP39 attenuates the antitumor activity of cisplatin on colon cancer cells dependent on p53. Cell Biol Toxicol. 2023;39(5):1995–2010.
Liu C, Chen Z, Ding X, Qiao Y, Li B. Ubiquitin-specific protease 35 (USP35) mediates cisplatin-induced apoptosis by stabilizing BIRC3 in non-small cell lung cancer. Lab Invest. 2022;102(5):524–33.
Hou Y, Fan Y, Xia X, Pan J, Hou J, Liu X, et al. USP31 acetylation at Lys1264 is essential for its activity and cervical cancer cell growth. Acta Biochim Biophys Sin (Shanghai). 2021;53(8):1037–43.
Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20(2):69–84.
Brabletz S, Schuhwerk H, Brabletz T, Stemmler MP. Dynamic EMT: a multi-tool for tumor progression. EMBO J. 2021;40(18):e108647.
Brozovic A. The relationship between platinum drug resistance and epithelial-mesenchymal transition. Arch Toxicol. 2017;91(2):605–19.
Li J, Gao R, Zhang J. USP22 contributes to chemoresistance, stemness, and EMT phenotype of triple-negative breast cancer cells by egulating the Warburg effect via c-Myc deubiquitination. Clin Breast Cancer. 2023;23(2):162–75.
Yun X, Zhang K, Wang J, Pangeni RP, Yang L, Bonner M, et al. Targeting USP22 suppresses tumorigenicity and enhances cisplatin sensitivity through ALDH1A3 downregulation in cancer-initiating cells from lung adenocarcinoma. Mol Cancer Res. 2018;16(7):1161–71.
Baulida J, Garcia de Herreros A. Snail1-driven plasticity of epithelial and mesenchymal cells sustains cancer malignancy. Biochim Biophys Acta. 2015;1856(1):55–61.
Guan T, Li M, Song Y, Chen J, Tang J, Zhang C, et al. Phosphorylation of USP29 by CDK1 Governs TWIST1 Stability and Oncogenic Functions. Adv Sci (Weinh). 2023;10(11):e2205873.
Sonego M, Pellarin I, Costa A, Vinciguerra GLR, Coan M, Kraut A, et al. USP1 links platinum resistance to cancer cell dissemination by regulating Snail stability. Sci Adv. 2019;5(5):eaav3235.
Liu L, Chen C, Liu P, Li J, Pang Z, Zhu J, et al. MYH10 combines with MYH9 to recruit USP45 by deubiquitinating snail and promotes serous ovarian cancer carcinogenesis, progression, and cisplatin resistance. Adv Sci (Weinh). 2023;10(14):e2203423.
Lambies G, Miceli M, Martínez-Guillamon C, Olivera-Salguero R, Peña R, Frías CP, et al. TGFβ-activated USP27X deubiquitinase regulates cell migration and chemoresistance via stabilization of Snail1. Cancer Res. 2019;79(1):33–46.
Zhu H, Gu X, Xia L, Zhou Y, Bouamar H, Yang J, et al. A novel TGFbeta trap blocks chemotherapeutics-induced TGFbeta1 signaling and enhances their anticancer activity in gynecologic cancers. Clin Cancer Res. 2018;24(12):2780–93.
Dou N, Hu Q, Li L, Wu Q, Li Y, Gao Y. USP32 promotes tumorigenesis and chemoresistance in gastric carcinoma via upregulation of SMAD2. Int J Biol Sci. 2020;16(9):1648–57.
Dai WL, Yuan SX, Cao JP. The deubiquitinase USP34 stabilizes SOX2 and induces cell survival and drug resistance in laryngeal squamous cell carcinoma. Kaohsiung J Med Sci. 2020;36(12):983–9.
Hashemi M, Esbati N, Rashidi M, Gholami S, Raesi R, Bidoki SS, et al. Biological landscape and nanostructural view in development and reversal of oxaliplatin resistance in colorectal cancer. Transl Oncol. 2023;40:101846.
Tavakoli Pirzaman A, Ebrahimzadeh Pirshahid M, Babajani B, Rahmati A, Niknezhad S. The role of microRNAs in regulating cancer cell response to oxaliplatin-containing regimens. Technol Cancer Res Treat. 2023;22:15330338231206004.
Li Q, Sun H, Luo D, Gan L, Mo S, Dai W, et al. Lnc-RP11-536 K7.3/SOX2/HIF-1α signaling axis regulates oxaliplatin resistance in patientderived colorectal cancer organoids. J Exp Clin Cancer Res. 2021;40(1):348.
Zheng Z, Wu M, Li H, Xu W, Yang M, Pan K, et al. Downregulation of AC092894.1 promotes oxaliplatin resistance in colorectal cancer via the USP3/AR/RASGRP3 axis. BMC Med. 2023;21(1):132.
Xiong H, Ni Z, He J, Jiang S, Li X, He J, et al. LncRNA HULC triggers autophagy via stabilizing Sirt1 and attenuates the chemosensitivity of HCC cells. Oncogene. 2017;36(25):3528–40.
Xiao Y, Jiang X, Yin K, Miao T, Lu H, Wang W, et al. USP35 promotes cell proliferation and chemotherapeutic resistance through stabilizing FUCA1 in colorectal cancer. Oncogenesis. 2023;12(1):12.
Zhan W, Liao X, Liu J, Tian T, Yu L, Li R. USP38 regulates the stemness and chemoresistance of human colorectal cancer via regulation of HDAC3. Oncogenesis. 2020;9(5):48.
Zhang C, Xu C, Gao X, Yao Q. Platinum-based drugs for cancer therapy and anti-tumor strategies. Theranostics. 2022;12(5):2115–32.
Wang L, Chen T, Li X, Yan W, Lou Y, Liu Z, et al. USP39 promotes ovarian cancer malignant phenotypes and carboplatin chemoresistance. Int J Oncol. 2019;55(1):277–88.
Lei X, Li X, Chen H, Liu Z. USP48 sustains chemoresistance and metastasis in ovarian cancer. Curr Cancer Drug Targets. 2020;20(9):689–99.
Bagdasaryan AA, Chubarev VN, Smolyarchuk EA, Drozdov VN, Krasnyuk II, Liu J, et al. Pharmacogenetics of drug metabolism: the role of gene polymorphism in the regulation of doxorubicin safety and efficacy. Cancers (Basel). 2022;14(21):5436.
Mattioli R, Ilari A, Colotti B, Mosca L, Fazi F, Colotti G. Doxorubicin and other anthracyclines in cancers: activity, chemoresistance and its overcoming. Mol Aspects Med. 2023;93:101205.
Zhang W, Zhang J, Xu C, Zhang S, Bian S, Jiang F, et al. Ubiquitin-specific protease 7 is a drug-able target that promotes hepatocellular carcinoma and chemoresistance. Cancer Cell Int. 2020;28(20):28.
Chen H, Zhu X, Sun R, Ma P, Zhang E, Wang Z, et al. Ubiquitin-specific protease 7 is a druggable target that is essential for pancreatic cancer growth and chemoresistance. Invest New Drugs. 2020;38(6):1707–16.
Fan YH, Cheng J, Vasudevan SA, Dou J, Zhang H, Patel RH, et al. USP7 inhibitor P22077 inhibits neuroblastoma growth via inducing p53-mediated apoptosis. Cell Death Dis. 2013;4(10):e867.
Yu Y, Zhao Y, Fan Y, Chen Z, Li H, Lu J, et al. Inhibition of Ubiquitin-specific protease 14 suppresses cell proliferation and synergizes with chemotherapeutic agents in neuroblastoma. Mol Cancer Ther. 2019;18(6):1045–56.
Qin T, Cui XY, Xiu H, Huang C, Sun ZN, Xu XM, et al. USP37 downregulation elevates the chemical sensitivity of human breast cancer cells to Adriamycin. Int J Med Sci. 2021;18(2):325–34.
Srivastava R, Fernández-Ginés R, Encinar JA, Cuadrado A, Wells G. The current status and future prospects for therapeutic targeting of KEAP1-NRF2 and β-TrCP-NRF2 interactions in cancer chemoresistance. Free Radic Biol Med. 2022;192:246–60.
Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, et al. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature. 2003;426(6962):87–91.
Peschiaroli A, Skaar JR, Pagano M, Melino G. The ubiquitin-specific protease USP47 is a novel β-TRCP interactor regulating cell survival. Oncogene. 2010;29(9):1384–93.
Zhu Y, Xu J, Hu W, Wang F, Zhou Y, Gong W, et al. Inhibiting USP8 overcomes hepatocellular carcinoma resistance via suppressing receptor tyrosine kinases. Aging (Albany NY). 2021;13(11):14999–5012.
Krech T, Scheuerer E, Geffers R, Kreipe H, Lehmann U, Christgen M. ABCB1/MDR1 contributes to the anticancer drug-resistant phenotype of IPH-926 human lobular breast cancer cells. Cancer Lett. 2012;315(2):153–60.
Zelnak A. Overcoming taxane and anthracycline resistance. Breast J. 2010;16(3):309–12.
Lin YT, Lin J, Liu YE, Chen YC, Liu ST, Hsu KW, et al. USP7 induces chemoresistance in triple-negative breast cancer via deubiquitination and stabilization of ABCB1. Cells. 2022;11(20):3294.
Wu Y, Zhang Y, Wang D, Zhang Y, Zhang J, Zhang Y, et al. USP29 enhances chemotherapy-induced stemness in non-small cell lung cancer via stabilizing Snail1 in response to oxidative stress. Cell Death Dis. 2020;11(9):796.
Tu X, Li C, Sun W, Tian X, Li Q, Wang S, et al. Suppression of cancer cell stemness and drug resistance via MYC destabilization by deubiquitinase USP45 inhibition with a natural small molecule. Cancers (Basel). 2023;15(3):930.
Qian Y, Wang B, Ma A, Zhang L, Xu G, Ding Q, et al. USP16 downregulation by carboxyl-terminal truncated HBx promotes the growth of hepatocellular carcinoma cells. Sci Rep. 2016;6:33039.
Suzuki K, Nishiwaki K, Yano S. Treatment strategies considering micro-environment and clonal evolution in multiple myeloma. Cancers (Basel). 2021;13(2):215.
Xu X, Liu J, Shen C, Ding L, Zhong F, Ouyang Y, et al. The role of ubiquitin-specific protease 14 (USP14) in cell adhesion-mediated drug resistance (CAM-DR) of multiple myeloma cells. Eur J Haematol. 2017;98(1):4–12.
Mosca L, Ilari A, Fazi F, Assaraf YG, Colotti G. Taxanes in cancer treatment: activity, chemoresistance and its overcoming. Drug Resist Updat. 2021;54:100742.
Nawara HM, Afify SM, Hassan G, Zahra MH, Seno A, Seno M, et al. Paclitaxel-based chemotherapy targeting cancer stem cells from mono- to combination therapy. Biomedicines. 2021;9(5):500.
He ZL, Zheng H, Lin H, Miao XY, Zhong DW. Overexpression of polo-like kinase1 predicts a poor prognosis in hepatocellular carcinoma patients. World J Gastroenterol. 2009;15(33):4177–82.
Feng Y, Lin J, Liu Y, Tang Y, Zhou Y, Zhong M. Investigation of expressions of PDK1, PLK1 and c-Myc in diffuse large B-cell lymphoma. Int J Exp Pathol. 2019;100(1):32–40.
Qin N, Han F, Li L, Ge Y, Lin W, Wang J, et al. Deubiquitinating enzyme 4 facilitates chemoresistance in glioblastoma by inhibiting P53 activity. Oncol Lett. 2019;17(1):958–64.
Shin SB, Kim CH, Jang HR, Yim H. Combination of inhibitors of USP7 and PLK1 has a strong synergism against paclitaxel resistance. Int J Mol Sci. 2020;21(22):8629.
Guo F, Zhang C, Wang F, Zhang W, Shi X, Zhu Y, et al. Deubiquitinating enzyme USP33 restrains docetaxel-induced apoptosis via stabilising the phosphatase DUSP1 in prostate cancer. Cell Death Differ. 2020;27(6):1938–51.
Horn-Ghetko D, Krist DT, Prabu JR, Baek K, Mulder MPC, Klügel M, et al. Ubiquitin ligation to F-box protein targets by SCF-RBR E3–E3 super-assembly. Nature. 2021;590(7847):671–6.
Xu M, Takanashi M, Oikawa K, Tanaka M, Nishi H, Isaka K, et al. USP15 plays an essential role for caspase-3 activation during Paclitaxel-induced apoptosis. Biochem Biophys Res Commun. 2009;388(2):366–71.
Benassi B, Marani M, Loda M, Blandino G. USP2a alters chemotherapeutic response by modulating redox. Cell Death Dis. 2013;4(9):e812.
Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–26.
Yu C, Xiao JH. The Keap1-Nrf2 System: a mediator between oxidative stress and aging. Oxid Med Cell Longev. 2021;2021:6635460.
Wu T, Harder BG, Wong PK, Lang JE, Zhang DD. Oxidative stress, mammospheres and Nrf2-new implication for breast cancer therapy? Mol Carcinog. 2015;54(11):1494–502.
Villeneuve NF, Tian W, Wu T, Sun Z, Lau A, Chapman E, et al. USP15 negatively regulates Nrf2 through deubiquitination of Keap1. Mol Cell. 2013;51(1):68–79.
Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemoresistance in gastric cancer. Mol Cancer. 2020;19(1):43.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death Nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–85.
Ligorio M, Sil S, Malagon-Lopez J, Nieman LT, Misale S, Di Pilato M, et al. Stromal microenvironment shapes the Intratumoral architecture of pancreatic Cancer. Cell. 2019;178(1):160–175. e127.
Sethy C, Kundu CN. 5-Fluorouracil (5-FU) resistance and the new strategy to enhance the sensitivity against cancer: implication of DNA repair inhibition. Biomed Pharmacother. 2021;137:111285.
Jiang S, Song C, Gu X, Wang M, Miao D, Lv J, et al. Ubiquitin-specific peptidase 22 contributes to colorectal cancer stemness and chemoresistance via Wnt/β-catenin pathway. Cell Physiol Biochem. 2018;46(4):1412–22.
Jiang S, Miao D, Wang M, Lv J, Wang Y. Tong JMiR-30-5p suppresses cell chemoresistance and stemness in colorectal cancer through USP22/Wnt/β-catenin signaling axis. J Cell Mol Med. 2019;23(1):630–40.
Siddique HR, Saleem M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells. 2012;30(3):372–8.
Jia L, Zhang W, Wang CY. BMI1 inhibition eliminates residual cancer stem cells after PD1 blockade and activates antitumor immunity to prevent metastasis and relapse. Cell Stem Cell. 2020;27(2):238–253.e6.
Zhai R, Tang F, Gong J, Zhang J, Lei B, Li B, et al. The relationship between the expression of USP22, BMI1, and EZH2 in hepatocellular carcinoma and their impacts on prognosis. Onco Targets Ther. 2016;9:6987–98.
Armour SM, Bennett EJ, Braun CR, Zhang XY, McMahon SB, Gygi SP, et al. A high-confidence interaction map identifies SIRT1 as a mediator of acetylation of USP22 and the SAGA coactivator complex. Mol Cell Biol. 2013;33(8):1487–502.
Ma S, Sun L, Wu W, Wu J, Sun Z, Ren J. USP22 Protects against myocardial ischemia-reperfusion injury via the SIRT1-P53/SLC7A11-dependent inhibition of ferroptosis-induced cardiomyocyte death. Front Physiol. 2020;11:551318.
Ling S, Li J, Shan Q, Dai H, Lu D, Wen X, et al. USP22 mediates the multidrug resistance of hepatocellular carcinoma via the SIRT1/AKT/MRP1 signaling pathway. Mol Oncol. 2017;11(6):682–95.
Wen X, Ling S, Wu W, Shan Q, Liu P, Wang C, et al. Ubiquitin-specific protease 22/silent information regulator 1 axis plays a pivotal role in the prognosis and 5-fluorouracil resistance in hepatocellular carcinoma. Dig Dis Sci. 2020;65(4):1064–73.
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–66.
Messaoudi K, Clavreul A, Lagarce F. Toward an effective strategy in glioblastoma treatment. Part I: Resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov Today. 2015;20(7):899–905.
Tirosh I, Suva ML. Tackling the many facets of glioblastoma heterogeneity. Cell Stem Cell. 2020;26(3):303–4.
Avanzato D, Pupo E, Ducano N, Isella C, Bertalot G, Luise C, et al. High USP6NL levels in breast cancer sustain chronic AKT phosphorylation and GLUT1 stability fueling aerobic glycolysis. Cancer Res. 2018;78(13):3432–44.
Su IC, Su YK, Chuang HY, Yadav VK, Setiawan SA, Fong IH, et al. Ubiquitin-Specific Protease 6 n-Terminal-like Protein (USP6NL) and the Epidermal Growth Factor Receptor (EGFR) signaling axis regulates ubiquitin-mediated DNA repair and temozolomide-resistance in glioblastoma. Biomedicines. 2022;10(7):1531.
Chang G, Xie GS, Ma L, Li P, Li L, Richard HT. USP36 promotes tumorigenesis and drug sensitivity of glioblastoma by deubiquitinating and stabilizing ALKBH5. Neuro Oncol. 2023;25(5):841–53.
Li Y, Wang T, Wan Q, Wang Q, Chen Z, Gao Y, et al. TRAF4 maintains deubiquitination of caveolin-1 to drive glioblastoma stemness and temozolomide resistance. Cancer Res. 2022;82(19):3573–87.
Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58(5):235–63.
Muvarak NE, Chowdhury K, Xia L, Robert C, Choi EY, Cai Y, et al. Enhancing the cytotoxic effects of PARP inhibitors with DNA demethylating agents - a potential therapy for cancer. Cancer Cell. 2016;30(4):637–50.
Silver DP, Livingston DM. Mechanisms of BRCA1 tumour suppression. Cancer Discov. 2012;2(8):679–84.
Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316(5828):1202–5.
Li Y, Luo K, Yin Y, Wu C, Deng M, Li L, et al. USP13 regulates the RAP80-BRCA1 complex dependent DNA damage response. Nat Commun. 2017;8:15752.
Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136(3):435–46.
Fabbro M, Rodriguez JA, Baer R, Henderson BR. BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J Biol Chem. 2002;277(24):21315–24.
Peng Y, Liao Q, Tan W, Peng C, Hu Z, Chen Y, et al. The deubiquitylating enzyme USP15 regulates homologous recombination repair and cancer cell response to PARP inhibitors. Nat Commun. 2019;10(1):1224.
Andres SN, Williams RS. CtIP/Ctp1/Sae2, molecular form fit for function. DNA Repair (Amst). 2017;56:109–17.
Gao M, Guo G, Huang J, Kloeber JA, Zhao F, Deng M, et al. USP52 regulates DNA end resection and chemosensitivity through removing inhibitory ubiquitination from CtIP. Nat Commun. 2020;11(1):5362.
Lim KS, Li H, Roberts EA, Gaudiano EF, Clairmont C, Sambel LA, et al. USP1 is required for replication fork protection in BRCA1Deficient tumors. Mol Cell. 2018;72(6):925–941.e4.
Cheng X, Zhang B, Guo F, Wu H, Jin X. Deubiquitination of FBP1 by USP7 blocks FBP1–DNMT1 interaction and decreases the sensitivity of pancreatic cancer cells to PARP inhibitors. Mol Oncol. 2022;16(7):1591–607.
Morra F, Merolla F, Napolitano V, Ilardi G, Miro C, Paladino S, et al. The combined effect of USP7 inhibitors and PARP inhibitors in hormone-sensitive and castration-resistant prostate cancer cells. Oncotarget. 2017;8(19):31815–29.
Morra F, Luise C, Merolla F, Poser I, Visconti R, Ilardi G, et al. FBXW7 and USP7 regulate CCDC6 turnover during the cell cycle and affect cancer drugs susceptibility in NSCLC. Oncotarget. 2015;6(14):12697–709.
Malapelle U, Morra F, Ilardi G, Visconti R, Merolla F, Cerrato A, et al. USP7 inhibitors, downregulating CCDC6, sensitize lung neuroendocrine cancer cells to PARP-inhibitor drugs. Lung Cancer. 2017;107:41–9.
Morra F, Merolla F, Damia G, Ricci F, Varricchio S, Ilardi G, et al. The disruption of the CCDC6 – PP4 axis induces a BRCAness like phenotype and sensitivity to PARP inhibitors in high-grade serous ovarian carcinoma. J Exp Clin Cancer Res. 2022;41(1):245.
Castagnetti F, Gugliotta G, Breccia M, Stagno F, Iurlo A, Albano F, et al. Long-term outcome of chronic myeloid leukemia patients treated frontline with imatinib. Leukemia. 2015;29(9):1823–31.
Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long-term outcomes of Imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376(10):917–27.
Nie ZY, Yao M, Yang Z, Yang L, Liu XJ, Yu J, et al. De-regulated STAT5A/miR-202-5p/USP15/ Caspase-6 regulatory axis suppresses CML cell apoptosis and contributes to Imatinib resistance. J Exp Clin Cancer Res. 2020;39(1):17.
Chen X, Chen Y, Zhang M, Cheng H, Mai H, Yi M, et al. HucMSC exosomes promoted imatinib-induced apoptosis in K562-R cells via a miR-145a-5p/USP6/GLS1 axis. Cell Death Dis. 2022;13(1):92.
Ren L, Ruiz-Rodado V, Dowdy T, Huang S, Issaq SH, Beck J, et al. Glutaminase-1 (GLS1) inhibition limits metastatic progression in osteosarcoma. Cancer Metab. 2020;8:4.
Lei H, Xu HZ, Shan HZ, Liu M, Lu Y, Fang ZX, et al. Targeting USP47 overcomes tyrosine kinase inhibitor resistance and eradicates leukemia stem/ progenitor cells in chronic myelogenous leukemia. Nat Commun. 2021;12(1):51.
Farag S, van Coevorden F, Sneekes E, Grunhagen DJ, Reyners AKL, Boonstra PA, et al. Elderly patients with gastrointestinal stromal tumour (GIST) receive less treatment irrespective of performance score or comorbidity - A retrospective multicentre study in a large cohort of GIST patients. Eur J Cancer. 2017;86:318–25.
Soreide K, Sandvik OM, Soreide JA, Giljaca V, Jureckova A, Bulusu VR. Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population-based cohort studies. Cancer Epidemiol. 2016;40:39–46.
Ni B, Li Q, Zhuang C, Huang P, Xia X, Yang L, et al. The nerve-tumour regulatory axis GDNF-GFRA1 promotes tumour dormancy, imatinib resistance and local recurrence of gastrointestinal stromal tumours by achieving autophagic flux. Cancer Lett. 2022;535:215639.
Gao Z, Li C, Sun H, Bian Y, Cui Z, Wang N, et al. N6-methyladenosine-modified USP13 induces pro-survival autophagy and imatinib resistance via regulating the stabilization of autophagy-related protein 5 in gastrointestinal stromal tumors. Cell Death Differ. 2023;30(2):544–59.
Pegtel DM, Gould SJ. Exosomes. Annu Rev Biochem. 2019;88:487–514.
Yousefi H, Maheronnaghsh M, Molaei F, Mashouri L, Reza Aref A, Momeny M, et al. Long noncoding RNAs and exosomal lncRNAs: classification, and mechanisms in breast cancer metastasis and drug resistance. Oncogene. 2020;39(5):953–74.
Li C, Gao Z, Cui Z, Liu Z, Bian Y, Sun H, et al. Deubiquitylation of Rab35 by USP32 promotes the transmission of imatinib resistance by enhancing exosome secretion in gastrointestinal stromal tumours. Oncogene. 2023;42(12):894–910.
Tang W, Chen Z, Zhang W, Cheng Y, Zhang B, Wu F, et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther. 2020;5(1):87.
Mendez-Blanco C, Fondevila F, Garcia-Palomo A, Gonzalez-Gallego J, Mauriz JL. Sorafenib resistance in hepatocarcinoma: role of hypoxia-inducible factors. Exp Mol Med. 2018;50(10):1–9.
Zhang HL, Wang MD, Zhou X, Qin CJ, Fu GB, Tang L, et al. Blocking preferential glucose uptake sensitizes liver tumor-initiating cells to glucose restriction and sorafenib treatment. Cancer Lett. 2017;388:1–11.
Ling S, Shan Q, Zhan Q, Ye Q, Liu P, Xu S, et al. USP22 promotes hypoxia- induced hepatocellular carcinoma stemness by a HIF1α/USP22 positive feedback loop upon TP53 inactivation. Gut. 2020;69(7):1322–34.
Xu S, Ling S, Shan Q, Ye Q, Zhan Q, Jiang G, et al. Self-activated cascade-responsive sorafenib and USP22 shRNA co-delivery system for synergetic hepatocellular carcinoma therapy. Adv Sci (Weinh). 2021;8(5):2003042.
Chang YS, Su CW, Chen SC, Chen YY, Liang YJ, Wu JC. Upregulation of USP22 and ABCC1 during sorafenib treatment of hepatocellular carcinoma contribute to development of resistance. Cells. 2022;11(4):634.
Gao R, Buechel D, Kalathur RKR, Morini MF, Coto-Llerena M, Ercan C, et al. USP29-mediated HIF1α stabilization is associated with Sorafenib resistance of hepatocellular carcinoma cells by upregulating glycolysis. Oncogenesis. 2021;10(7):52.
Sutton KA, Jungnickel MK, Wang Y, Cullen K, Lambert S, Florman HM. Enkurin is a novel calmodulin and TRPC channel binding protein in sperm. Dev Biol. 2004;274(2):426–35.
Hou R, Li Y, Luo X, Zhang W, Yang H, Zhang Y, et al. ENKUR expression induced by chemically synthesized cinobufotalin suppresses malignant activities of hepatocellular carcinoma by modulating β-catenin/ c-Jun/MYH9/USP7/c-Myc axis. Int J Biol Sci. 2022;18(6):2553–67.
Wang E, Mi X, Thompson MC, Montoya S, Notti RQ, Afaghani J, et al. Mechanisms of resistance to noncovalent Bruton’s tyrosine kinase inhibitors. N Engl J Med. 2022;386(8):735–43.
Agathanggelou A, Smith E, Davies NJ, Kwok M, Zlatanou A, Oldreive CE, et al. USP7 inhibition alters homologous recombination repair and targets CLL cells independent of ATM/p53 functional status. Blood. 2017;130(2):156–66.
Paulus A, Akhtar S, Caulfield TR, Samuel K, Yousaf H, Bashir Y, et al. Coinhibition of the deubiquitinating enzymes, USP14 and UCHL5, with VLX1570 is lethal to ibrutinib- or bortezomib-resistant Waldenstrom macroglobulinemia tumor cells. Blood Cancer J. 2016;6(11):e492.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl Androl Urol. 2015;4(3):365–80.
Okegawa T, Ninomiya N, Masuda K, Nakamura Y, Tambo M, Nutahara K. AR-V7 in circulating tumor cells cluster as a predictive biomarker of abiraterone acetate and enzalutamide treatment in castration-resistant prostate cancer patients. Prostate. 2018;78(8):576–82.
Schrecengost RS, Dean JL, Goodwin JF, Schiewer MJ, Urban MW, Stanek TJ, et al. USP22 regulates oncogenic signaling pathways to drive lethal cancer progression. Cancer Res. 2014;74(1):272–86.
Zhang B, Zhang M, Shen C, Liu G, Zhang F, Hou J, et al. LncRNA PCBP1-AS1-mediated AR/AR-V7 deubiquitination enhances prostate cancer enzalutamide resistance. Cell Death Dis. 2021;12(10):856.
Liao Y, Xia X, Liu N, Cai J, Guo Z, Li Y, et al. Growth arrest and apoptosis induction in androgen receptor-positive human breast cancer cells by inhibition of USP14-mediated androgen receptor deubiquitination. Oncogene. 2018;37(14):1896–910.
Gao L, Zhang W, Zhang J, Liu J, Sun F, Liu H, et al. KIF15-mediated stabilization of AR and AR-V7 contributes to enzalutamide resistance in prostate cancer. Cancer Res. 2021;81(4):1026–39.
Liu Y, Yu C, Shao Z, Xia X, Hu T, Kong W, et al. Selective degradation of AR-V7 to overcome castration resistance of prostate cancer. Cell Death Dis. 2021;12(10):857.
Liao Y, Liu Y, Shao Z, Xia X, Deng Y, Cai J, et al. A new role of GRP75-USP1-SIX1 protein complex in driving prostate cancer progression and castration resistance. Oncogene. 2021;40(25):4291–306.
Tang Y, Wang Y, Kiani MF, Wang B. Classification, treatment strategy, and associated drug resistance in breast cancer. Clin Breast Cancer. 2016;16(5):335–43.
Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. 2018;34(3):427–438.e6.
Wang S, Zhong X, Wang C, Luo H, Lin L, Sun H, et al. USP22 positively modulates ERα action via its deubiquitinase activity in breast cancer. Cell Death Differ. 2020;27(11):3131–45.
Xia X, Huang C, Liao Y, Liu Y, He J, Shao Z, et al. The deubiquitinating enzyme USP15 stabilizes ERα and promotes breast cancer progression. Cell Death Dis. 2021;12(4):329.
Cao J, Wu D, Wu G, Wang Y, Ren T, Wang Y, et al. USP35, regulated by estrogen and AKT, promotes breast tumorigenesis by stabilizing and enhancing transcriptional activity of estrogen receptor α. Cell Death Dis. 2021;12(6):619.
Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Jänne PA, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28(2):357–60.
Byun S, Lee SY, Lee J, Jeong CH, Farrand L, Lim S, et al. USP8 is a novel target for overcoming gefitinib resistance in lung cancer. Clin Cancer Res. 2013;19(14):3894–904.
Giron P, Eggermont C, Noeparast A, Vandenplas H, Teugels E, Forsyth R, et al. Targeting USP13-mediated drug tolerance increases the efficacy of EGFR inhibition of mutant EGFR in non-small cell lung cancer. Int J Cancer. 2021;148(10):2579–93.
Zhang H, Han B, Lu H, Zhao Y, Chen X, Meng Q, et al. USP22 promotes resistance to EGFR-TKIs by preventing ubiquitination mediated EGFR degradation in EGFR-mutant lung adenocarcinoma. Cancer Lett. 2018;433:186–98.
Yu F, Liu JB, Wu ZJ, Xie WT, Zhong XJ, Hou LK, et al. Tumor suppressive microRNA-124a inhibits stemness and enhances gefitinib sensitivity of non-small cell lung cancer cells by targeting ubiquitin-specific protease 14. Cancer Lett. 2018;427:74–84.
Swain SM, Shastry M, Hamilton E. Targeting HER2-positive breast cancer: advances and future directions. Nat Rev Drug Discov. 2023;22(2):101–26.
Alam S, Zunic A, Venkat S, Feigin ME, Atanassov BS. Regulation of Cyclin D1 degradation by ubiquitin specific protease 27X is critical for cancer cell proliferation and tumor growth. Mol Cancer Res. 2022;20(12):1751–62.
Zhang J, Liu S, Li Q, Shi Y, Wu Y, Liu F, et al. The deubiquitylase USP2 maintains ErbB2 abundance via counteracting endocytic degradation and represents a therapeutic target in ErbB2-positive breast cancer. Cell Death Differ. 2020;27(9):2710–25.
Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61(7):3071–6.
Franqui-Machin R, Hao M, Bai H, Gu Z, Zhan X, Habelhah H, et al. Destabilizing NEK2 overcomes resistance to proteasome inhibition in multiple myeloma. J Clin Invest. 2018;128(7):2877–93.
Yao Y, Zhang Y, Shi M, Sun Y, Chen C, Niu M, et al. Blockade of deubiquitinase USP7 overcomes bortezomib resistance by suppressing NF-κB signaling pathway in multiple myeloma. J Leukoc Biol. 2018;104(6):1105–15.
Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22(3):345–58.
Zhang H, Chen J, Zhang M, Zhao M, Zhang L, Liu B, et al. Tetrahydrobiopterin induces proteasome inhibitor resistance and tumor progression in multiple myeloma. Med Oncol. 2022;39(5):55.
Lu Y, Wang Y, Xu H, Shi C, Jin FY, Li W. Profilin 1 induces drug resistance through Beclin1 complex-mediated autophagy in multiple myeloma. Cancer Sci. 2018;109(9):2706–16.
Li H, Roy M, Liang L, Cao W, Hu B, Li Y, et al. Deubiquitylase USP12 induces pro-survival autophagy and bortezomib resistance in multiple myeloma by stabilizing HMGB1. Oncogene. 2022;41(9):1298–308.
Waldman AD, Fritz JM, Lenardo MJ. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat Rev Immunol. 2020;20(11):651–68.
Borcoman E, Kanjanapan Y, Champiat S, Kato S, Servois V, Kurzrock R, et al. Novel patterns of response under immunotherapy. Ann Oncol. 2019;30(3):385–96.
Gao H, Yin J, Ji C, Yu X, Xue J, Guan X, et al. Targeting ubiquitin specific proteases (USPs) in cancer immunotherapy: from basic research to preclinical application. J Exp Clin Cancer Res. 2023;42(1):225.
Yang H, Zhang X, Lao M, Sun K, He L, Xu J, et al. Targeting ubiquitin-specific protease 8 sensitizes antiprogrammed death-ligand 1 immunotherapy of pancreatic cancer. Cell Death Differ. 2023;30(2):560–75.
Xiong W, Gao X, Zhang T, Jiang B, Hu MM, Bu X, et al. USP8 inhibition reshapes an inflamed tumor microenvironment that potentiates the immunotherapy. Nat Commun. 2022;13(1):1700.
Wang Z, Kang W, Li O, Qi F, Wan J, You Y, et al. Abrogation of USP7 is an alternative strategy to downregulate PD-L1 and sensitize gastric cancer cells to T cells killing. Acta Pharm Sin B. 2021;11(3):694–707.
Dai X, Lu L, Deng S, Meng J, Wan C, Huang J, et al. USP7 targeting modulates anti-tumor immune response by reprogramming Tumor-associated Macrophages in Lung Cancer. Theranostics. 2020;10(20):9332–47.
Huang X, Zhang Q, Lou Y, Wang J, Zhao X, Wang L, et al. USP22 deubiquitinates CD274 to suppress anti-cancer immunity. Cancer Immunol Res. 2019;7(10):1580–90.
Li J, Yuan S, Norgard RJ, Yan F, Yamazoe T, Blanco A, et al. Tumor cell–intrinsic USP22 suppresses antitumor immunity in pancreatic cancer. Cancer Immunol Res. 2020;8(3):282–91.
Li M, Xu Y, Liang J, Lin H, Qi X, Li F, et al. USP22 deficiency in melanoma mediates resistance to T cells through IFNg-JAK1-STAT1 signal axis. Mol Ther. 2021;29(6):2108–20.
Wang Q, Li G, Ma X, Liu L, Liu J, Yin Y, et al. LncRNA TINCR impairs the efficacy of immunotherapy against breast cancer by recruiting DNMT1 and downregulating MiR-199a-5p via the STAT1–TINCR-USP20-PD-L1 axis. Cell Death Dis. 2023;14(2):76.
Xiao X, Shi J, He C, Bu X, Sun Y, Gao M, et al. ERK and USP5 govern PD-1 homeostasis via deubiquitination to modulate tumor immunotherapy. Nat Commun. 2023;14(1):2859.
Hsu SK, Li CY, Lin IL, Syue WJ, Chen YF, Cheng KC, et al. Inflammation-related pyroptosis, a novel programmed cell death pathway, and its crosstalk with immune therapy in cancer treatment. Theranostics. 2021;11(18):8813–35.
Arimoto KI, Miyauchi S, Troutman TD, Zhang Y, Liu M, Stoner SA, et al. Expansion of interferon inducible gene pool via USP18 inhibition promotes cancer cell pyroptosis. Nat Commun. 2023;14(1):251.
Ren Y, Feng M, Hao X, Liu X, Li J, Li P, et al. USP48 stabilizes gasdermin E to promote pyroptosis in cancer. Cancer Res. 2023;83(7):1074–93.
Arimoto KI, Löchte S, Stoner SA, Burkart C, Zhang Y, Miyauchi S, et al. STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nat Struct Mol Biol. 2017;24(3):279–89.
Wang W, Zhang L, Sun Z. Eliciting pyroptosis to fuel cancer immunotherapy: mechanisms and strategies. Cancer Biol Med. 2022;19(7):948–64.
Xu YP, Lv L, Liu Y, Smith MD, Li WC, Tan XM, et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J Clin Invest. 2019;129(10):4316–31.
Chen LL, Smith MD, Lv L, Nakagawa T, Li Z, Sun SC, et al. USP15 suppresses tumor immunity via deubiquitylation and inactivation of TET2. Sci Adv. 2020;6(38):eabc9730.
Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343(6168):305–9.
Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, et al. Immunomodulatory agents lenalidomide and pomalidomide costimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br J Haematol. 2014;164(6):811–21.
Nguyen TV. USP15 antagonizes CRL4CRBN-mediated ubiquitylation of glutamine synthetase and neosubstrates. Proc Natl Acad Sci USA. 2021;118(40):e2111391118.
Reck M, Carbone DP, Garassino M, Barlesi F. Targeting KRAS in non-small-cell lung cancer: recent progress and new approaches. Ann Oncol. 2021;32(9):1101–10.
Yang Z, Xu G, Wang B, Liu Y, Zhang L, Jing T, et al. USP12 downregulation orchestrates a protumourigenic microenvironment and enhances lung tumour resistance to PD-1 blockade. Nat Commun. 2021;12(1):4852.
Cheong JE, Sun L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy - Challenges and Opportunities. Trends Pharmacol Sci. 2018;39(3):307–25.
Shi D, Wu X, Jian Y, Wang J, Huang C, Mo S, et al. USP14 promotes tryptophan metabolism and immune suppression by stabilizing IDO1 in colorectal cancer. Nat Commun. 2022;13(1):5644.
Bareche Y, Kelly D Abbas-Aghababazadeh F, Nakano M, Esfahani PN, Tkachuk D, et al. Leveraging big data of immune checkpoint blockade response identifies novel potential targets. Ann Oncol 2022;33(12):1304-1317.
Zhang Q, Liu YJ, Li JP, Zeng SH, Shen H, Han M, et al. USP35 is a potential immunosuppressive factor in skin cutaneous melanoma. J Inflamm Res. 2022;15:3065–82.
Liu YJ, Zeng SH, Zhang W, Li JP, Yin Y, Zhuang YW, et al. USP51/ZEB1/ACTA2 axis promotes mesenchymal phenotype in gastric cancer and is associated with low cohesion characteristics. Pharmacol Res. 2023;188:106644.
Huang RX, Zhou PK. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther. 2020;5(1):60.
Sharma A, Almasan A. USP14 regulates DNA damage response and is a target for radiosensitization in non-small cell lung cancer. Int J Mol Sci. 2020;21(17):6383.
Hang Q, Zeng L, Wang L, Nie L, Yao F, Teng H, et al. Non-canonical function of DGCR8 in DNA doublestrand break repair signaling and tumor radioresistance. Nat Commun. 2021;12(1):4033.
Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol. 2016;8(4):a019521.
Wang Q, Ma S, Song N, Li X, Liu L, Yang S, et al. Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J Clin Invest. 2016;126(6):2205–20.
Yang Y, Yang C, Li T, Yu S, Gan T, Hu J, et al. The deubiquitinase USP38 promotes NHEJ repair through regulation of HDAC1 activity and regulates cancer cell response to genotoxic insults. Cancer Res. 2020;80(4):719–31.
Wang B, Zheng J, Li R, Tian Y, Lin J, Liang Y, et al. Long noncoding RNA LINC02582 acts downstream of miR-200c to promote radioresistance through CHK1 in breast cancer cells. Cell Death Dis. 2019;10(10):764.
Wu J, Chen Y, Geng G, Li L, Yin P, Nowsheen S, et al. USP39 regulates DNA damage response and chemo-radiation resistance by deubiquitinating and stabilizing CHK2. Cancer Lett. 2019;449:114–24.
Weller M, van den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. 2021;18(3):170–86.
Lee JK, Chang N, Yoon Y, Yang H, Cho H, Kim E, et al. USP1 targeting impedes GBM growth by inhibiting stem cell maintenance and radioresistance. Neuro Oncol. 2016;18(1):37–47.
Zheng J, Wang B, Zheng R, Zhang J, Huang C, Zheng R, et al. Linc-RA1 inhibits autophagy and promotes radioresistance by preventing H2Bub1/USP44 combination in glioma cells. Cell Death Dis. 2020;11(9):758.
Tu Y, Chen Z, Zhao P, Sun G, Bao Z, Chao H, et al. Smoothened promotes glioblastoma radiation resistance via activating USP3-mediated claspin deubiquitination. Clin Cancer Res. 2020;26(7):1749–62.
Wolfsperger F, Hogh-Binder SA, Schittenhelm J, Psaras T, Ritter V, Bornes L, et al. Deubiquitylating enzymeUSP9x regulates radiosensitivity in glioblastoma cells by Mcl-1-dependent and -independent mechanisms. Cell Death Dis. 2016;7(1):e2039.
Kushwaha D, O’Leary C, Cron KR, Deraska P, Zhu K, D’Andrea AD, et al. USP9X inhibition promotes radiation-induced apoptosis in non-small cell lung cancer cells expressing mid-tohigh MCL1. Cancer Biol Ther. 2015;16(3):392–401.
Jie X, Fong WP, Zhou R, Zhao Y, Zhao Y, Meng R, et al. USP9X-mediated KDM4C deubiquitination promotes lung cancer radioresistance by epigenetically inducing TGF-β2 transcription. Cell Death Differ. 2021;28(7):2095–111.
Niu H, Zhu Y, Wang J, Wang T, Wang X, Yan L. Effects of USP7 on radiation sensitivity through p53 pathway in laryngeal squamous cell carcinoma. Transl Oncol. 2022;22:101466.
Zhang L, Nemzow L, Chen H, Lubin A, Rong X, Sun Z, et al. The Deubiquitinating Enzyme USP24 Is a Regulator of the UV Damage Response. Cell Rep. 2015;10(2):140–7.
Li X, Xie L, Zhou L, Gan Y, Han S, Zhou Y, et al. Bergenin inhibits tumor growth and overcomes radioresistance by targeting aerobic glycolysis. Am J Chin Med. 2023;51(7):1905–25.
Zhou Q, Yao X, Wu C, Chen S, Fan D. Knockdown of ubiquitin-specific protease 53 enhances the radiosensitivity of human cervical squamous cell carcinoma by regulating DNA damage-binding protein 2. Technol Cancer Res Treat. 2020;19:1533033820929792.
Weili Z, Zhikun L, Jianmin W, Qingbao T. Knockdown of USP28 enhances the radiosensitivity of esophageal cancer cells via the c-Myc/hypoxia-inducible factor-1 alpha pathway. J Cell Biochem. 2019;120(1):201–12.
Chen S, Liu Y, Zhou H. Advances in the development Ubiquitin-Specific Peptidase (USP) inhibitors. Int J Mol Sci. 2021;22(9):4546.
Lange SM, Armstrong LA, Kulathu Y. Deubiquitinases: from mechanisms to their inhibition by small molecules. Mol Cell. 2022;82(1):15–29.
Lee G, Oh TI, Um KB, Yoon H, Son J, Kim BM, et al. Inhibition of ubiquitin-specific protease 7 sensitizes acute myeloid leukemia to chemotherapy. Biochem Biophys Res Commun. 2016;470(1):181–6.
Das DS, Ray A, Das A, Song Y, Tian Z, Oronsky B, et al. A novel hypoxia-selective epigenetic agent RRx-001 triggers apoptosis and overcomes drug resistance in multiple myeloma cells. Leukemia. 2016;30(11):2187–97.
Grunblatt E, Wu N, Zhang H, Liu X, Norton JP, Ohol Y, et al. MYCN drives chemoresistance in small cell lung cancer while USP7 inhibition can restore chemosensitivity. Genes Dev. 2020;34(17–18):1210–26.
Li XY, Wu JC, Liu P, Li ZJ, Wang Y, Chen BY, et al. Inhibition of USP1 reverses the chemotherapy resistance through destabilization of MAX in the relapsed/refractory B-cell lymphoma. Leukemia. 2023;37(1):164–77.
Das DS, Das A, Ray A, Song Y, Samur MK, Munshi NC, et al. Blockade of deubiquitylating enzyme USP1 inhibits DNA repair and triggers apoptosis in multiple myeloma cells. Clin Cancer Res. 2017;23(15):4280–9.
Chitta K, Paulus A, Akhtar S, Blake MK, Caulfield TR, Novak AJ, et al. Targeted inhibition of the deubiquitinating enzymes, USP14 and UCHL5, induces proteotoxic stress and apoptosis in Waldenström macroglobulinaemia tumor cells. Br J Haematol. 2015;169(3):377–90.
Small-molecule inhibitors of USP7 induce apoptosis through oxidative and endoplasmic reticulum stress in cancer cells. Biochem Biophys Res Commun. 2016,470, 181–186.
Kategaya L, Di Lello P, Rougé L, Pastor R, Clark KR, Drummond J, et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature. 2017;550(7677):534–8.
Leger PR, Hu DX, Biannic B, Bui M, Han X, Karbarz E, et al. Discovery of potent, selective, and orally bioavailable inhibitors of USP7 with in vivo antitumor activity. J Med Chem. 2020;63(10):5398–420.
Mistry H, Hsieh G, Buhrlage SJ, Huang M, Park E, Cuny GD, et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol Cancer Ther. 2013;12(12):2651–62.
Liang Q, Dexheimer TS, Zhang P, Rosenthal AS, Villamil MA, You CA, et al. Selective usp1–uaf1 inhibitor links deubiquitination to DNA damage responses. Nat Chem Biol. 2014;10(4):298–304.
Kim W, Zhao F, Gao H, Qin S, Hou J, Deng M, et al. USP13 regulates the replication stress response by deubiquitinating TopBP1. DNA Repair (Amst). 2021;100:103063.
Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147(1):223–34.
Tian Z, D’Arcy P, Wang X, Ray A, Tai YT, Hu Y, et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood. 2014;123(5):706–16.
Wang X, D’Arcy P, Caulfield TR, Paulus A, Chitta K, Mohanty C, et al. Synthesis and evaluation of derivatives of the proteasome deubiquitinase inhibitor b-AP15. Chem Biol Drug Des. 2015;86(5):1036–48.
Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467(7312):179–84.
Acknowledgments
We express our sincere gratitude to these researchers for their valuable contributions to the understanding of USP and drug resistance mechanisms, as well as the development of USP inhibitors.
Funding
This work was supported by grants from the 345 Talent Project of Shengjing Hospital of China Medical University, Science and Technology Plan Joint Plan Project of Liaoning Province (2023JH2/101700153), and the National Natural Science Foundation of China (No. 81701570).
Author information
Authors and Affiliations
Contributions
GHL, XZ, and DJW drafted the manuscript, generated the graphs, completed all modifications throughout the revision process together, and contributed equally to this manuscript; XF, CZG and ZL designed and conducted this study; XJQ and GX provided comments to the figures. XF and CZG revised the manuscript text and obtained funding. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent of publication
All listed authors have read and approved this manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gao, H., Xi, Z., Dai, J. et al. Drug resistance mechanisms and treatment strategies mediated by Ubiquitin-Specific Proteases (USPs) in cancers: new directions and therapeutic options. Mol Cancer 23, 88 (2024). https://doi.org/10.1186/s12943-024-02005-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-024-02005-y