
Abstract
Background
The promise of precision cancer medicine presently centers around the genomic sequence of a patient’s tumor being translated into timely, actionable information to inform clinical care. The analysis of cell-free DNA from liquid biopsy, which contains circulating tumor DNA (ctDNA) in patients with cancer, has proven to be amenable to various settings in oncology. However, open questions surrounding the clinical validity and utility of plasma-based analyses have hindered widespread clinical adoption.
Main body
Owing to the rapid evolution of the field, studies supporting the use of ctDNA as a biomarker throughout a patient’s journey with cancer have accumulated in the last few years, warranting a review of the latest status for clinicians who may employ ctDNA in their precision oncology programs. In this work, we take a step back from the intricate coverage of detection approaches described extensively elsewhere and cover basic concepts around the practical implementation of next generation sequencing (NGS)-guided liquid biopsy. We compare relevant targeted and untargeted approaches to plasma DNA analysis, describe the latest evidence for clinical validity and utility, and highlight the value of genome-wide ctDNA analysis, particularly as it relates to early detection strategies and discovery applications harnessing the non-coding genome.
Conclusions
The maturation of liquid biopsy for clinical application will require interdisciplinary efforts to address current challenges. However, patients and clinicians alike may greatly benefit in the future from its incorporation into routine oncology care.
Similar content being viewed by others
Background
Genomics currently serves as the backbone of the precision medicine construct. In cancer, systematic analyses of tumor genomes have allowed us to describe malignancies at the molecular level, in particular enabling the identification of driver events that propel disease. Novel therapies have been developed to treat these genomic driver events, which has led to improved patient outcomes across a spectrum of tumor types [1,2,3,4]. Given that tumors may evolve under the selective pressure of therapy, rich reservoirs of critical and real-time genomic information can be accessed through non-invasive means, i.e., liquid biopsies. Liquid biopsies rely upon detection of circulating tumor cells, cell-free DNA (cfDNA), which in patients with cancer includes circulating tumor DNA (ctDNA), RNA, proteins, lipids, and metabolites present in biofluids of patients. In principle, bodily fluids other than plasma, such as cerebrospinal fluid, urine, saliva, stool, pleural fluid, and ascites, can be analyzed, but for reasons of brevity, we focus here on blood and only on cfDNA. However, widespread clinical adoption has been slow, which is in part caused by an increasing complexity in selecting the most suitable analyses for the clinical question at hand, interpretation of the results, and lagging clinical trial evidence of utility. In this review, our intention is to especially target clinicians and we therefore cover concepts around practical implementation of next generation sequencing (NGS)-guided liquid biopsy while simultaneously highlighting the emerging new developments in the field. In this work, we break down ready-to-use ctDNA applications, including an overview of present clinical validity and clinical utility, describe early detection strategies, cover industry trends, and highlight exciting future directions and open questions that extend beyond DNA sequence.
Main text
A wealth of information circulating in the peripheral blood
Since the original description of abnormally high levels of circulating cell-free DNA (cfDNA) in the blood of cancer patients [5], further research has demonstrated that extracellular DNA in bodily fluids may reflect an array of pathological processes, including malignant, inflammatory or autoimmune disease, as well as trauma, sepsis and myocardial infarction [6,7,8,9,10], conditions which are outside the scope of this review. In the case of cancer, apoptotic or necrotic cancer cells release DNA into the bloodstream that can then be detected through diverse means, albeit always in the background of cfDNA molecules originating from the hematopoietic system, as these cells are the main contributors of DNA to the circulation in both health and disease [11, 12].
In addition to their eased access, liquid biopsies may capture the tumoral spatial heterogeneity not observed from traditional single-site biopsy genotyping [13,14,15,16], as they may enable the detection of DNA shed from both clonal and subclonal sites within multiple metastatic lesions. An array of studies has established the general concordance between aberrations detected in ctDNA and tumor tissue, ranging approximately between 70 and 90% [17,18,19,20,21]. Some discordance between mutations identified in primary tumor tissue and ctDNA is to be expected, which can be attributed to tumor heterogeneity or evolution, sampling bias, time lapses between sample acquisition, differences in sensitivity of the sequencing assays applied, or even different sequencing platforms [22,23,24,25,26]. However, with suitable and validated workflows, the potential applications of ctDNA are far-reaching, including diagnosing cancers earlier than traditional imaging [27,28,29], customizing treatments detected via genotyping [30,31,32,33], associating DNA levels with response to treatment [34, 35], identifying mechanisms of resistance to therapies [36,37,38,39,40,41] and measuring minimal residual disease after treatment [42,43,44,45,46,47,48]. As new evidence of analytical validity, clinical validity as well as utility continues to accumulate for these applications, strategies and requirements for the integration of ctDNA analysis workflows into clinical oncology programs are taking form [49,50,51,52,53].
Next-generation sequencing of plasma DNA allows for diverse detection modalities of ctDNA
In order to better understand the novel ctDNA profiling strategies described herein later on, we briefly summarize basic concepts and assays used in NGS-based detection of ctDNA, as other diverse detection methodologies and their features have been reviewed in depth elsewhere [49, 50, 54,55,56].
Sampling, sequencing and detection of alterations from cfDNA
Sample acquisition begins with the collection of the peripheral blood, typically drawn in specialized collection tubes, e.g. PAXgene Blood ccfDNA tubes or those provided by a commercial provider, which contain an additive that stabilizes blood cells and prevents cell lysis. This aspect is critical, as any lysis of healthy blood cells will generate even more background signal that dilutes the probability of capturing the essential tumor-specific signal downstream [57,58,59,60,61,62]. After plasma separation, DNA extraction and quantification (Fig. 1 A), the selection of the approach to library preparation dictates what type of information can be harvested from the analysis. All NGS approaches, regardless of analyte, i.e., DNA from tissue or plasma DNA from whole blood, typically follow the same general workflow [63, 64]. The basic protocol begins when adapters of known sequence are added to the isolated DNA, which is then amplified into a library of DNA fragments (Fig. 1B). These adapters serve a technical rather than a biological purpose, as they enable the binding of amplified library fragments to the glass flow cell on which—in the case of Illumina instruments—sequencing-by-synthesis (SBS) takes place. The sequencer then converts the nucleotide-level biological information into a digital readout, which is stored in a large text file consisting of individual reads of pre-determined length (Fig. 1C). These reads, essentially a string of A’s, T’s, C’s and G’s, are then processed through various computational analysis pipelines to derive genomic variance from a reference genome (Fig. 1D). This variance must be interpreted within the context of the tumor and ctDNA fraction in plasma. For discussion at molecular tumor boards (MTBs), only the most pertinent information is summarized and prioritized in a clinical report such that actionability of alterations is displayed clearly for the expert panel (Fig. 1E) [65,66,67,68].

NGS technology as the backbone of ctDNA analysis. A Starting with whole blood collected in specialized cfDNA collection tubes, the plasma layer containing cfDNA is separated via centrifugation, followed by extraction of cfDNA from plasma. Typically, two vials of blood corresponding to ~17-20ml are submitted for analysis for both research studies or analysis by commercial vendors to ensure that sufficient amounts of plasma are available for extraction and harvesting of the ctDNA signal. B Simplified theoretical (Illumina) library fragment as a result of NGS library preparation. The dark green and dark blue bars represent the Illumina adapters P5 and P7, respectively, which enable hybridization to the sequencing flow cell and subsequent bridge amplification after ligation to the cfDNA fragment (gray bars). Sample-specific indexes, which are used to identify the patient sample, are typically in dual format and are shown here as i5 and i7. Additionally, unique molecular identifiers (UMI) serve as molecule-specific barcodes that enable the bioinformatics filtration of amplification or sequencing errors to ensure high-quality variant calling. C Sequencing-by-synthesis (SBS) on an Illumina instrument allows for one fluorescently-tagged nucleotide to be added to the growing read per cycle. Here, G’s, A’s, C’s and T’s are tagged with pink, blue, green and yellow fluorochromes, respectively. After the instrument has converted the captured images to base calls, the data is converted into a FASTQ file containing reads and quality scores. D Since the reads in the FASTQ file do not describe genomic location of the read, the data must first be aligned to a reference genome. This alignment is referred to as a SAM file, which has a binary counterpart called a BAM file. The BAM file contains all information in the original FASTQ file along with the mapping information of the read, i.e. the genomic coordinates to which it aligned. The BAM (alignment) file serves as the core data for diverse downstream analyses, e.g. calling of SCNAs or variants, estimation of tumor fraction from plasma, calculation of fragment size distributions, or nucleosome mapping. E Example of a clinical report summarizing the interpretation of genomic alterations detected from cfDNA. Such a clinical report describes the detected genomic alterations alongside their variant allele frequencies (VAF) and pathogenicity with potential clinical implications. Such findings should be discussed at a molecular tumor board and aligned to the patient’s clinical status
The impact of ctDNA levels, sequencing depth and breadth on ctDNA detection capabilities
Factors that critically affect the probability of ctDNA detection include the ctDNA levels in a plasma sample, the sequencing depth at mutant positions and the number of alterations tracked, i.e., the breadth of analysis.
The variable fractions of ctDNA among total cfDNA populations naturally have an effect on variant allele frequencies (VAFs). VAFs, reported as percentages, define the variant mutant reads over the total number of reads in a sample and are dependent not only on a patient’s tumor burden, but also on tumor fraction in plasma and tumor heterogeneity. However, a general problem is the low abundance of ctDNA fragments in many samples, particularly in early-stage disease. For this reason, detecting lower allelic frequencies—i.e. VAFs < 1% or even lower—is a basic requirement for cfDNA assays. Hence, highly sensitive approaches are needed to attain low detection limits for analyzing minute amounts within cfDNA. Importantly, the presence of ctDNA even at low levels, such as 0.1%, means that millions of actively dividing cancer cells are present within the body [69]. In contrast, in advanced disease, higher VAFs facilitate robust detection in most samples [24, 70].
Sequencing coverage, sometimes referred to as “depth”, describes the number of unique reads that align to, i.e., cover, nucleotide bases in a reference genome. After alignment of reads to the reference genome, we can observe how many of these reads support a particular locus. At higher levels of coverage, each base is covered by a greater number of aligned sequencing reads, therefore increasing the degree of confidence that base calls can be made at a given position or region. Importantly, deeper coverage improves the sensitivity of calling alterations and especially enhances the detection of rare variants that may be attributed to both tumor heterogeneity and/or low tumor fraction in a plasma sample.
The breadth refers to increasing the number of detectable sites, which can be achieved by increasing the number of regions in targeted panels or conducting whole-exome (WES) or whole-genome sequencing (WGS). The reasoning is that the detection of a single somatic mutation depends on the probability that the mutated fragment is actually sampled within the limited number of genome equivalents (GEs) present in a typical plasma sample [50]. In contrast, the probability to detect at least one somatic tumor-associated variant increases with the number of mutations analyzed so that the breadth of sequencing can compensate the limitation of low numbers of ctDNA fragments in plasma samples.
Selecting the most appropriate assay
In general, ctDNA analyses have two key objectives. One is to detect evidence for the presence of ctDNA in the circulation with the highest possible specificity and sensitivity. The second is, in addition, a detailed characterization of the tumor genome, for example to identify druggable targets, resistance markers, or to follow the evolution of the tumor genome during a disease course. Key concepts in assay design (Fig. 2A) include first, whether an assay is targeted, i.e., focusing on particular regions in the genome according to specific criteria, or whether it is untargeted, which may be the case when WES or WGS is conducted. Second, assays can be personalized, i.e., tailored for an individual patient based on sequencing data obtained from the primary tumor or a baseline plasma sample (Fig. 2A,B), or they can be non-personalized, i.e., tumor-agnostic, which means that they are conducted without a priori knowledge of alterations. The selection of the assay depends on the objective and the associated costs plus turnaround time to obtain the results.

Choosing the right ctDNA assay based on sensitivity and breadth of genome coverage. A The number of detectable alterations critically depends on the selected cfDNA assay. The first row illustrates a DNA segment with various alterations (explained in the bottom legend). The second row (untargeted profiling) indicates the use of an “off the shelf” panel, which is capable of identifying a number of alterations, but as it represents a rather general assay, it may miss a considerable number of alterations (indicated by empty symbols). The third row (targeted profiling) indicates the use of a panel tailored for a specific tumor entity. For example, after screening of databases such as COSMIC and TCGA, panels can be designed that will identify specific alterations for this particular tumor entity with high likelihood. However, mutations “private”, i.e. unique, to the patient’s tumor will be missed. The fourth row (targeted, personalized profiling) indicates the use of a patient-specific multiplex assay, which was individually designed based on sequencing information from the primary tumor. In theory, all mutations from the primary tumor are detectable; however, new alterations that may have occurred at a later timepoint will be missed. The fifth row shows the use of whole-exome sequencing (WES) or whole-genome sequencing (WGS), which enables comprehensive coverage of all coding regions of the genome and, in case of WGS, also of all non-coding regions. The actual capability of detecting variants does not solely depend on the selected assay but also on other factors such as the ctDNA levels. B For a personalized approach, the tumor or a baseline plasma sample needs to be sequenced first. The observed mutations can then be leveraged for subsequent cfDNA analyses. The triangle in the center indicates the various breadth of such analyses. Advantages of analyzing only a single locus include low costs and easy interpretation without the necessity of sophisticated bioinformatics. However, sensitivity is limited, as sampling issues represent a significant confounding factor. In contrast, analyses of hundreds or thousands of targets requires some error-suppression means, i.e., bioinformatics tools. At the same time, the likelihood for the detection of evidence for the presence of ctDNA increases tremendously, making such approaches the most sensitive for MRD detection. In fact, while sequencing depth remains a critical factor for ctDNA detection, sequencing breadth may supplant the importance of high coverage analyses. C Some clinics may have access to their own academic or partner laboratory, which may develop and apply its own tests and address liquid biopsy related research questions. Alternatively, samples can be sent to a commercial end-to-end provider. Regardless which laboratory conducts the analyses, the aim is to provide the MTB with all relevant information at hand so that the best decisions can be made for patients
Targeted, non-personalized approaches: from single locus to panel applications
Single-locus PCR assays for known mutations have the potential to detect those with high sensitivity, in particular if analyzed with a several thousand-fold coverage (i.e., high depth, low breadth). For example, hotspot mutations in KRAS can easily be tracked in cases where KRAS is involved with high prevalence, such as in pancreatic or colon cancer. However, sampling issues may affect single-locus approaches, e.g., the region may be missed in samples that have low fractional concentrations of ctDNA, such that usually only a reliable detection limit of ~ 0.1% can be achieved. Hence, targeting a larger number of variants, i.e., increasing the breadth, has the potential to increase the sensitivity of ctDNA assays. For this reason, a number of panel sequencing assays capable of deeply sequencing a variable number of actionable mutations and genes have been developed.
Targeted sequencing approaches offer a cost-effective solution to interrogate only those loci of interest, such as clinically relevant hotspot mutations, that will guide treatment decisions, as the majority of the genome still remains undruggable [71]. Essentially, these protocols select for particular subsets of the genome to be subsequently sequenced, a process referred to as target enrichment [72]. For example, for the design of targeted but not personalized assays, recurrent mutations in driver genes from the Catalog of Somatic Mutations in Cancer (COSMIC) may be selected [73].
In terms of assay technology, targeted sequencing can either be amplicon-based or hybrid capture-based. Examples for amplicon-based assays include tagged-amplicon deep sequencing (TAm-Seq) [74], which is now commercially offered by Inivata as Enhanced TAm-Seq [75], Safe-Sequencing System (Safe-SeqS) [76] and Simple, Multiplexed, PCR-based barcoding of DNA for Sensitive mutation detection using Sequencing (SiMSen-seq) [77, 78]. With hybrid capture-based targeted sequencing, select regions within the library are captured using long, biotinylated oligonucleotide baits, or probes. These biotinylated baits have been designed to hybridize to regions of interest (e.g., cancer-related genes, exonic regions) within the fragmented cfDNA and streptavidin is subsequently used to separate the baits bound to target DNA from other fragments which were not bound. An example of this is the pan-cancer AVENIO ctDNA Expanded liquid biopsy panel from Roche—the commercial adaptation of CAncer Personalized Profiling by deep Sequencing (CAPP-Seq) [79]—which contains 17 biomarkers in the U.S. National Comprehensive Cancer Network (NCCN) and other guidelines in addition to 60 biomarkers currently being investigated in clinical trials. Several of these targeted sequencing assays represent what is known as comprehensive genomic profiling (CGP) (Fig. 2A), which refers to a single assay that can detect all major classes of genomic alterations known to drive cancer growth: base substitutions, insertions and deletions, somatic copy number alterations (SCNA) and structural rearrangements.
The largest “panel” that can be applied to cfDNA is WES, i.e., targeting all protein coding genes of the human genome. Indeed, the first landmark studies applying WES to plasma DNA demonstrated that acquired resistance to cancer therapy and disease monitoring is possible in patients with breast cancer [80, 81].
As the development and validation of in-house CGP cfDNA assays require a considerable amount of effort and resources, it simply is neither realistic nor feasible for many labs to design their own comprehensive multi-gene approaches [52] (Fig. 2C). Researchers at the Memorial Sloan Kettering hospital have been pioneers in this field and their recently developed liquid biopsy MSK-ACCESS (Analysis of Circulating Cell- free DNA to Evaluate Somatic Status) assay [32, 82] has been approved by the New York State Department of Health. However, many labs rely on commercial kits to provide in-house end-to-end molecular profiling workflows. Clinics that do not have access to local NGS-based molecular profiling from liquid biopsy through an academic partner may seek solutions from commercial end-to-end providers (Fig. 2C). With this approach, clinicians carry out sample acquisition and shipping in accordance with the vendor’s guidelines and receive a report of molecular findings in return, although reimbursement of such testing approaches is not universal and the service often does not include interpretation or consultation of the findings. Examples for both in-house and service provider solutions include the AVENIO ctDNA panels (Roche; in-house solution), Oncomine™ Pan-Cancer Cell-Free Assay (ThermoFisher; in-house solution), TruSight Oncology 500 ctDNA (Illumina; in-house solution), FoundationOne® Liquid CDx (Foundation Medicine; end-to-end commercial provider) Guardant360™ (Guardant Health; end-to-end commercial provider), Tempus xF Liquid Biopsy Assay (Tempus; end-to-end commercial provider), and elio™ Plasma Resolve (PGDx; end-to-end commercial provider). Many of these CGP solutions offer supplementary and clinically important biomarker information, such as tumor fraction in plasma, estimations of tumor mutational burden (TMB) and microsatellite instability (MSI) status. Industry offerings of CGP solutions for treatment selection in the advanced disease settings are plentiful. However, the SEQC2 Working Group recently reported that when using these kits, the detection of variants had a limited reliability below an allele fraction of 0.5% [83]. Hence, other strategies, such as personalizing assays, are needed to increase sensitivity further.
Personalized, targeted assays
Personalized ctDNA assays (Fig. 2A,B) have found application particularly in the early disease setting. Typically, WES or WGS is employed on a patient’s tumor or a baseline plasma sample to identify somatic variants that were not found in the germline sample (Fig. 2B). These detected variants can then be used to design patient-specific multiplex assays to track the mutations from ctDNA in a personalized fashion, increasing the sensitivity of variant detection [84]. Personalized panels are thus designed to maximize the number of informative reads generated. One of the first studies targeted a median of 18 somatic variants and classified a plasma sample as ctDNA-positive when at least two of these variants were detected [69]. Several studies have described tumor-guided sequencing panels with up to 20 different variants [45, 74, 79, 85,86,87]. However, detection of ctDNA can be vastly enhanced by increasing the number of informative targets in an assay. For example, MRDetect is a tumor-informed detection approach particularly for the minimal residual disease (MRD) setting, which leverages the thousands of somatic mutations typically detectable in solid malignancies to detect tumor fractions with a sensitivity as low as 10− 5 [48]. Similar resolution limits were achieved with the Integration of Variant Reads (INVAR) pipeline, which also targets thousands of informative reads [88]. As both approaches involve sequencing of large regions, which is prone for the accumulation of sequencing errors, a sophisticated custom-made error suppression solution is needed. Importantly, both approaches put the need for high sequencing depth into perspective because of the many targets being analyzed. Hence, the sensitivity of such multi-target approaches is rather determined by the breadth and less by the coverage (Fig. 2B). Both approaches are very sensitive for the detection of the presence of ctDNA, but they are less sensitive for the detection of any specific site, such as a driver mutational event, which could be informative about potential therapies. This is a limiting factor of these approaches if the objective of the analysis is to search for druggable targets.
Several commercial providers have adopted such a “tumor-informed” approach. For example, Natera received a Breakthrough Device designation by the FDA in 2019 for its Signatera™ assay that uses a patient’s own tumor mutation signature to personalize an assay for the detection of molecular residual disease, for which utility was originally demonstrated for disease surveillance for patients with metastatic breast cancer (mBC) [89]. Similarly, Inivata’s RaDaR™ assay, which tracks a set of up to 48 tumor-specific variants, was also granted Breakthrough Device Designation as an assay for the detection of residual disease. The sensitivity of RaDaR™ was evaluated in a study of 90 patients with stage IA-IIIB non-small cell lung cancer (NSCLC) who were undergoing radical treatment with curative intent [90]. In early 2021, Exact Sciences acquired a worldwide exclusive license to the Targeted Digital Screening (TARDIS) assay, which was shown to guide treatment strategies in patients with early-stage breast cancer, albeit that the clinically relevant diagnostic threshold will likely have to be refined in further studies [45].
As a substantial number of resources must be invested to devise and validate targeted liquid biopsy platforms, the trend seems to hold that novel methodologies are first developed in the academic setting (Fig. 2C) and then scaled and offered by companies who focus their efforts on improving their implementation and expanding their adoption. As such, centralized ctDNA testing offered by commercial providers may change the paradigm of tumor molecular profiling and some predict that the decentralized model, i.e. testing performed at local labs, will in the future be limited to single-locus or small gene panels, whereas large-scale ctDNA assays will primarily be outsourced to central labs [52].
However, despite all of these caveats, confident detection of MRD is possible in plasma DNA. Tumor genotype-informed MRD detection approaches can attain LODs of ≤ 0.01% (Fig. 2B), which makes them preferable for detection of minute amounts of MRD [69, 84, 85, 91].
Analysis of SCNAs
For the detection of genome-wide SCNAs from plasma, a shallow coverage of 0.1x suffices to call these aberrations accurately (Fig. 2A), including the identification of focal events [92,93,94]. Additionally, tumor fraction in plasma can be estimated specifically from shallow WGS (sWGS) data, sometimes referred to as low-pass WGS [17], which is pertinent to the downstream interpretation of detected alterations as well as any lack of detection.
Current status of clinical use of ctDNA testing in patients with cancer
The overwhelming technical options of liquid biopsy approaches raise the question as to whether their applications ultimately make a difference for patients’ treatment outcomes. For adoption of a biomarker test in clinical care, three criteria, i.e., analytical validity (measures the accuracy, reliability and reproducibility of a test), clinical validity (assesses the ability of a test to divide a population into separate groups with significantly different clinical outcomes), and clinical utility (evaluates whether outcomes are improved for patients who received the test compared with those who did not), were defined [95]. However, a recent, joint review by the American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP) came to the conclusion that there is insufficient evidence of clinical validity and utility for the majority of ctDNA assays in routine clinical care [96]. The ASCO-CAP panel recommended that ctDNA testing should be used only within clinical trials. However, since the publication of this statement, increasing evidence of clinical validity and clinical utility of ctDNA testing has been reported (Table 1).
In the clinic, ctDNA analysis is emerging as a biomarker for three different scenarios: first, as a prognostic biomarker across multiple cancer types; second, for response assessment; and third, for resistance monitoring, i.e., identification of disease progression during or after systemic therapy ahead of clinical or radiographic indicators.
This development is reflected in key recommendations from the NCCN guidelines, which were recently reviewed [107]. In brief, NSCLC has evolved to the tumor type with the most compelling and comprehensive evidence for ctDNA testing. The NCCN Guidelines for NSCLC (version 7.2021) (https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1450) recommend molecular testing at the time of diagnosis, with repeat biopsy or plasma testing to enable identification of genomic alterations in EGFR, ALK, ROS1, BRAF, MET, and RET to guide the use of US Food and Drug Administration (FDA)-approved targeted therapies in the first-line advanced disease setting. Two recent studies confirmed that plasma ctDNA NGS in advanced NSCLC can increase the positive identification of guideline-recommended genomic biomarkers and actionable alterations [100, 103]. The cobas® EGFR mutation test v2 was the first liquid biopsy assay approved by the FDA [108] as a companion diagnostic test for screening EGFR mutations from plasma cfDNA.
Furthermore, a current challenge is the identification of patients with NSCLC who may achieve durable benefit from immune checkpoint inhibitor (ICI) treatment. On-treatment changes of ctDNA in plasma have been shown to reveal predictive information for long-term clinical benefit in ICI-treated patients and molecular ctDNA responses correlated with radiographic response to ICI [35, 109,110,111,112]. A multi-parameter model that integrates ctDNA levels with circulating immune cells, which mirror the immune milieu, may further improve prediction of tumor response to ICI treatment [113].
Another key cancer type with compelling evidence that ctDNA testing provides clinically relevant information is breast cancer. Approximately 40% of hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative breast cancers carry PIK3CA mutations. Identification of these PIK3CA mutations are informative about treatment with the PI3Kα-specific inhibitor alpelisib in combination with fulvestrant as second-line therapy for advanced disease [114, 115] and, accordingly, the NCCN guidelines for invasive breast cancer (version 1.2022) (https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1419) recommend assessment for PIK3CA mutations using tumor tissue or ctDNA testing, with reflex tumor testing if ctDNA results are negative. Indeed, plasma-based reassessing of PIK3CA status is important, as PIK3CA mutational status can change upon disease recurrence [25, 78]. On the basis of data from the phase III SOLAR-1 trial [114], the therascreen® PIK3CA RGQ PCR kit was granted FDA approval for the detection of PIK3CA mutations in plasma or tumor tissue in patients with advanced-stage (HR)+/HER2 − breast cancer. Further examples are patients with metastatic, ER-positive breast cancer who progressed on endocrine therapies. Variants in ESR1 become much more prevalent in mBC, indicating that the presence of these mutations arise because of the evolving cancer. Importantly, ctDNA analysis can non-invasively detect ESR1 mutations that herald resistance to aromatase inhibitors to tailor adjuvant therapies [116].
For most other tumor entities, the NCCN guidelines do not directly address plasma ctDNA testing but acknowledge that relevant genomic alterations may be identified by evaluating ctDNA in the blood for a variety of cancers [107]. However, there are compelling near-term emerging applications for ctDNA analysis, in particular for patients with prostate cancer. The PARP inhibitor (PARPi) olaparib was FDA-approved for patients with metastatic, castration-resistant prostate cancer (CRPC) with deleterious or suspected deleterious germline or somatic homologous recombination repair (HRR) gene mutations [117]. For patients with metastatic CRPC (mCRPC) and deleterious germline and/or somatic BRCA mutations, rucaparib received approval [118]. Plasma ctDNA testing will be useful to identify the subset of patients eligible for these treatment approaches. Furthermore, ctDNA testing has been applied to understand primary resistance to abiraterone and enzalutamide [119,120,121] as well as studying molecular alterations involved in neuroendocrine transformation [122].
Importantly, several seminal studies have demonstrated the prognostic value of ctDNA MRD detection and in doing so have proven the clinical validity of these strategies. A recent meta-analysis of these studies came to the conclusion that, following definitive therapy for solid cancers, ctDNA MRD testing is strongly prognostic and has high positive-predictive value for risk of occurrence [84]. The clinical sensitivity, i.e., the percentage of patients with recurrent disease and who were ctDNA-positive after therapy, approached 100% in most studies when a surveillance strategy, i.e., evaluation of multiple posttreatment blood draws during follow-up, was conducted [84]. Another important aspect of ctDNA MRD testing is that residual disease was identified with a lead time of several months earlier than by standard-of-care radiological imaging [84].
However, while the clinical validity of ctDNA MRD testing is clearly established, there is less evidence for its clinical utility, i.e., demonstration of a benefit from early initiation of additional therapy after ctDNA MRD detection. One study conducted with patients with locally advanced NSCLC who were ctDNA MRD-positive after chemoradiation and who received consolidation immunotherapy demonstrated that these patients had significantly better freedom from progression than patients treated with chemoradiation alone [111]. Another study showed that adjuvant atezolizumab in muscle-invasive urothelial cancer might improve survival in ctDNA-positive patients [106]. These two studies provided the first evidence that personalized therapy based on ctDNA MRD status may result in improved patient outcomes. In summary, regarding ctDNA MRD testing, there is clear evidence for clinical validity and emerging signs for clinical utility.
Real-world ctDNA use cases for advanced cancer
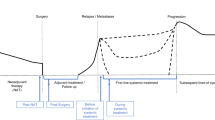
In parallel to the numerous ongoing efforts striving to prove the clinical validity and utility of ctDNA-based testing as described (Table 1), the fact is that molecular profiling of plasma DNA is already a reality for many in academic clinical settings who treat patients with advanced disease. Although tissue biopsy remains the gold standard of cancer diagnosis and represents the primary analyte for guiding treatment decisions, it is not at all uncommon that tumor tissue is simply not available. For example, a patient with clinically confirmed progressive disease who has exhausted all standard lines of treatment may be asked to undergo re-biopsy for retrieval of the latest tumor signal from tissue to derive the next decision for therapy. However, patients may either refuse re-biopsy or may not qualify as candidates for such a procedure. Rather than skipping molecular profiling altogether, here, the analysis of ctDNA may serve as a tumor-agnostic surrogate analyte to detect potential actionable targets. In several cohorts, including prospective studies, large NGS panels have proven advantageous in the detection of actionable targets from ctDNA [19, 30, 98, 99, 102, 123], which is also reflected in the wide assortment of industry offerings of CGP solutions for treatment selection in the advanced disease setting. We have summarized the indication, liquid biopsy rationale, and NGS data from three real-world cases of patients who underwent profiling to identify actionable targets (Fig. 3A). As with any biological data that is unique to the individual, interpretation of the alterations detected and subsequent clinical decisions are not always clear-cut, but may be simplified for exemplary purposes in the form of a decision tree (Fig. 3B). Here, it is important to emphasize that variant interpretation is an intricate process, requiring molecular expertise and at present not entirely standardized [124,125,126]. As such, clinical decisions derived from combined genomic and immunohistochemistry data must be discussed within the framework of an experienced MTB. Similarly, in the advanced disease setting, patients enrolled in liquid biopsy programs may undergo serial sampling to monitor their treatment. In certain scenarios, real-time treatment monitoring via ctDNA analysis may uncover a novel actionable target before it is detected via tissue biopsy or may provide evidence for the development of a novel resistance-related event (Fig. 4A), both which indicate the potential for a change in the course of treatment (Fig. 4B).

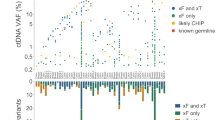
Use cases for ctDNA analysis throughout the cancer patient journey: identification of actionable targets in patients with advanced cancer. A Representation of 3 real-world cases of patients with confirmed progressive disease where liquid biopsy was justified to identify actionable targets. Available clinical patient characteristics and primary tumor biopsy profiling data are displayed in the white box. The last received therapy along with the associated measured radiological response (RECIST 1.1) are in the dark blue box and the specific rationale for ordering a liquid biopsy is listed in the dark green box. Below, a summary of the NGS results from comprehensive genomic profiling (CGP) via the AVENIO ctDNA Expanded Panel are documented, including: tumor fraction in plasma estimated via ichorCNA (%; LOD 3%); clinically relevant and pathogenic somatic copy number alterations (SCNAs) and variants, with variant allele frequency (VAF, %) detected from plasma DNA; non-actionable and variants of unknown significance (VUS). Of particular note is Case 3, which had a relatively high tumor fraction of 21%, but the two pathogenic variants detected had VAFs <1%. As these low allele fractions (KRAS G12A: 0.23%, PTEN N323fs: 0.24%) do not align with the overall tumor content of the sample, these VAFs may indicate subclonality of the alterations or potential sequencing artifacts. For this reason, it would be necessary to confirm their presence with an orthogonal approach using a new blood sample, especially if they were to influence a treatment decision. B Basic decision tree for this use case and the interpretation of detected alterations from liquid biopsy NGS data. The cases in (A) are mapped at the corresponding position that reflects the individual scenario. The critical starting point is the assessment of ctDNA level, i.e. tumor fraction (TF), in plasma, as samples with sufficiently low TF may not yield any detected alterations (Case 2). In such cases, reflex tissue testing is the clinical standard. If the sample has sufficient a ctDNA level, the analyst must rule out potential CHIP or germline variants before moving on to actionability assessment. In some cases, mutations associated with resistance are detected (Case 1), but no therapeutic targets are found. The identification of actionable targets and matching of potential suitable, evidence-based treatments is not a straightforward process and thus should be discussed at a molecular tumor board with oncologists to derive the final treatment decision (Case 3)

Use cases for ctDNA analysis throughout the cancer patient journey: disease monitoring. A Representation of 3 real-world cases of patients who underwent serial liquid biopsy sampling for disease monitoring purposes via shallow whole-genome sequencing (sWGS). Patient age and tumor entity are displayed in the white box. In the green panels, the NGS results from sWGS monitoring are shown in patient timelines. The serial samples are listed in the gray boxes (e.g. S1, S2, etc.). Detected focal somatic copy number alterations (SCNAs) are shown in the green callout boxes at the corresponding time point that they were detected via sWGS.B Basic decision tree for this use case and the interpretation of detected alterations from disease monitoring data via liquid biopsy. The cases in (A) are mapped at the corresponding position that reflects the individual scenario. Again, assessment of the ctDNA level in plasma represents the critical first step, as decreases in ctDNA from the previous sample may indicate a response to therapy, whereas unchanged levels may indicate stable disease. Increases in ctDNA fraction are generally associated with progressive disease. In some cases, novel alterations may be detected via monitoring and may represent novel druggable targets that were not observed from previous profiling (Case 1), known resistance markers (Case 2), or a clonal switch, which demonstrates the adaptive nature of tumors under the selective pressure of targeted therapies (Case 3)
Confounding factors and limitations of ctDNA assays
In order to properly obtain and preserve rare DNA fragments from the circulation, efforts have been dedicated to determining proper pre-analytical workflows crucial to reliable downstream cfDNA analysis, such as blood collection and sample transport, centrifugation, storage and isolation methods [60, 70, 127], as well as technical aspects of the detection of aberrations [55, 128].
To date, a fundamental understanding regarding the biological mechanisms behind release and clearance of cfDNA is still lacking, although this topic is increasingly being explored [129,130,131,132]. Levels of normal cfDNA may also increase due to non-malignant conditions, such as tissue injury or inflammation. In particular, after tumor removal, post-surgical inflammatory changes may cause an increase in cfDNA levels postoperatively for several weeks and hence dilute the allele fraction of ctDNA molecules [133]. Hence, increased plasma DNA levels cannot generally be equated with increased ctDNA levels in patients with cancer.
The low ctDNA fractions pose several challenges and detailed knowledge about the limit of detection (LOD) of the selected assay is vital [84]. The LOD depends on both biological and technical factors. Early landmark studies have described the variable levels of ctDNA in diverse solid tumors and across stages that correlate with tumor burden [24] and demonstrated a half-life of ctDNA ranging from several minutes to several hours [134], although proper pharmacokinetic studies to accurately determine this have not yet been performed. Biological factors determine the tumor DNA shedding rate. Indeed, not every tumor type “sheds” its DNA equally into the bloodstream, which influences the measurable quantity of ctDNA [24, 70]. Biological factors associated with tumor DNA shedding include tumor volume and tumor surface area, vascularization, tumor cell growth and death rates, mitotic and metabolic activity, and cell morphology. Furthermore, cancer signal detection is associated with active proliferation and explains why more aggressive cancers tend to shed more DNA into the bloodstream [135]. In fact, longitudinal follow-up data to evaluate the prognostic significance of a multi-cancer early detection (MCED) test has suggested that this test detected more clinically significant cancers and that detection was prognostic beyond clinical stage and method of clinical diagnosis. Accordingly, cancers not detected by the MCED test tended to be less aggressive [135]. Hence, biological differences in shedding rates may explain the differences in sensitivity between various tumors and high ctDNA levels may be an indication for more aggressive cancers.
Somatic variants from non-tumor tissues represent another biological confounding factor and mainly relate to the prevalence of CHIP (clonal hematopoiesis of indeterminate potential) derived mutations in ctDNA, i.e., normal hematopoietic cells accumulating somatic mutations during the aging process in the absence of cancer [136,137,138]. CHIP is highly prevalent in the general population and these mutations from hematopoietic cells may be misinterpreted as tumor-derived in cfDNA analysis. In fact, a majority of cfDNA mutations may be derived from clonal hematopoiesis, making matched cfDNA-white blood cell sequencing mandatory for accurate variant interpretation [27, 35, 82], albeit not yet a universally adopted practice due to the associated increased costs of sequencing an additional sample per patient.
Importantly, commercial products should not be considered a panacea, as their actual performance capabilities have yet to be critically tested in large, multicenter studies. As mentioned above, their reliability below an allele fraction of 0.5% is likely limited [83]. Furthermore, numerous published studies involve black box chemistry and complex bioinformatics, which are hard to comprehend or to verify, even for experts in the field. As a consequence, assessment of reproducibility and sensitivity by second non-profit-based parties is often lacking. At the same time, the innovative bioinformatics algorithms developed for WGS analysis are by no means self-explanatory and require a deeper intersection of basic biological and technical knowledge. Some studies, which are based on use of machine learning classifiers, may be prone to over-performance, even if they used separate training and validation cohorts, such that many of the published models require validation in prospective, multi-center studies.
A detailed discussion of technical errors, which occur ex vivo during the various molecular biology steps, leading to artificial mutations, is beyond the scope of this review and has been reviewed previously [50, 84].
Breakdown of applications in the pipeline
So far, the ctDNA assays described herein have focused on somatic tumor-associated mutations and copy number alterations. However, epigenetic alterations, such as methylation markers, cfDNA fragment length, and open chromatin regions, will increasingly take on an important role [139, 140]. Tissue-specific, stable, and universal methylation patterns can be used to detect cell death and to monitor even common diseases, such as inflammation, cardiomyocyte cell death or pancreatitis with cfDNA testing (Fig. 5A). These epigenetic differences can be leveraged in cfDNA analysis to determine the exact origin of cfDNA, which is referred to as plasma DNA tissue mapping or plasma DNA tissue deconvolution (Fig. 5A). To date, most plasma DNA tissue mapping studies have been conducted using methylation markers [11, 141,142,143,144,145]. Furthermore, the MCED tests mentioned above have been based on such methylation markers and have described the option of detecting more than 50 different cancer types [135, 146]. Early detection of cancer is at present a very active area of research and other efforts have used a combination of mutations and circulating proteins [147, 148]. These tests offer the option of detecting a range of cancers early, which may reduce cancer-related death. At present, their feasibility as a screening test in healthy populations is being tested with large-scale prospective studies, such as Grail’s The Circulating Cell-free Genome Atlas Study (CCGA; NCT02889978) or STRIVE trial (NCT03085888) as well as Thrive’s ASCEND trial (NCT04213326), all of which already began before 2020. Several of such early detection efforts have recently been reviewed [149].

Methylation analysis and whole-genome sequencing of cfDNA enables applications that extend beyond DNA sequence and copy number. A (Left) Methylation patterns of DNA in tumor cells may look different from their normal, healthy counterparts. Generally, CpG islands are associated with promoter regions of genes and these regions are prone to hypermethylation, i.e., gain of methylation, in tumor cells, leading to a block of gene transcription, as the bulky transcription machinery is prohibited from binding to the hypermethylated site. Conversely, tumor cells exhibit a general trend of global hypomethylation, i.e., loss of methylation, throughout the genome, which is frequently observed at repetitive sequences. The lollipops represent CpG sites, with white lollipops indicating no methylation at this particular cytosine and dark blue representing methylation at the cytosine. (Right) Typically, beadchip array data can be harvested to perform differential methylation analysis between various tissue types of interest, e.g. comparing normal breast tissue and malignant breast tissue or identifying differences in methylation between healthy colon or lung tissue. Differential individual CpGs or regions of differential methylation can be identified for use as a tissue-specific marker for downstream purposes. CpGs or regions of CpGs that do not confer a highly differential methylation signal from other analyzed tissues will not constitute a robust tissue-specific marker. B Apoptotic death of cells results in the digestion of open chromatin, i.e. regions of DNA not bound to and protected by nucleosomes. Naked DNA not associated with proteins, e.g., histones or TFs, will be digested and not detected in the circulation. C The majority of cfDNA is thus mononucloeosomal DNA. However, longer fragments of DNA may be protected by two nucleosomes, i.e. dinucleosome. D The coverage patterns of where the reads align in the genome reflect the biology of that particular region. The coverage patterns at regions of interest (ROI) reflect the original positioning of nucleosomes in cells. Generally, well-defined nucleosome organization and positioning in cancer cells may indicate that the ROI is “open” or accessible. This is accompanied by a drop in coverage at the ROI, where no nucleosomes were positioned, resulting in what is referred to as the nucleosome-depleted region (NDR). Densely packed nucleosomes with less defined positioning reflects that the region is not accessible or “closed”, with no drop in coverage at the NDR and no oscillation of coverage upstream or downstream to the ROI. Example ROIs are transcription start sites (TSS), transcription factor binding sites (TFBSs), or DNase hypersensitivity sites (DHS). E The types of fragment features that can be observed are diverse, such that there is no one-size-fits-all approach to applying fragmentomics to cfDNA. Exemplary features that can be harvested for analysis are illustrated on this DNA strand, including fragment length. Green stars represent that plasma DNA ends show prevalence of certain nucleotide contexts, i.e., preferred fragment end motifs, which are defined as a few nucleotides at plasma DNA ends regardless of the site of origin within the genome. Detection of double-stranded plasma molecules carrying single-stranded protruding ends are termed jagged ends, which may be harnessed to assess the jaggedness across varying plasma DNA fragment sizes and their association with nucleosomal patterns. F Because ctDNA has a modal size profile shorter than that of the background cfDNA originating from non-cancerous cells, this fragment size feature can be used to enhance detection of tumor-associated alterations. Shorter fragments of cfDNA can be harvested either through specialized library preparation approaches that enrich for short cfDNA molecules, through in silico size selection approaches, or a combination of both. G Fragment size differences have also been shown to differentiate between mutations stemming from CHIP and those originating from the tumor. CHIP-associated mutations are associated with fragment size distributions of wildtype molecules (black distribution), whereas tumor-associated mutations typically reside on short cfDNA fragments (green distribution). H Using WGS data, global fragmentation patterns can be observed. By establishing coverage and size distribution references of cfDNA fragments in defined genomic windows in both healthy and cancer populations, it can be determined whether an individual’s cfDNA distribution is likely to be healthy (blue signal) or cancer-derived (red signal). By comparing genome-wide profiles between various tissues, these patterns may also be used for tissue deconvolution purposes. I Nucleosomes (purple circles) are shown in the form of heterochromatin or open chromatin regions along a length of DNA. Open chromatin consists of regulatory regions within the genome, such as enhancers, transcription factor binding sites (TFBS), promoters, and transcription start sites (TSSs), to which proteins may bind. These are highlighted in green and collectively represent DNase hypersensitivity sites (DHS). When a canonical nucleosome is supplanted, the underlying DNA is rendered accessible to nucleases and other protein factors
In general, typical cfDNA fragment lengths after enzymatic processing in apoptotic cells have a modal distribution of 166 bp, a size that corresponds approximately to the length of DNA wrapped around a nucleosome (∼147 bp) and a linker fragment (∼20 bp) [150,151,152]. The nucleosome protects the DNA from enzymatic digestion in apoptotic cells so that DNA is mainly degraded in the intervening linker fragments (Fig. 5B). As a result, cfDNA consists mostly of mononucleosomal DNA, but dinucleosomal cfDNA fragments may also be observed (Fig. 5C). Several studies have provided evidence that cfDNA indeed reflects such nucleosome footprints [122, 153, 154]. Coverage-based analysis can be used for nucleosome position mapping (Fig. 5D), but other approaches for nucleosome position mapping have also been described [122, 145, 153]. The strong impact of the cellular nucleosomal organization on the DNA fragmentation patterns [29, 155,156,157,158] results in characteristic signatures regarding fragment size and nucleotide motifs including—in addition to the fragment length—end-motif frequency or jagged ends (Fig. 5E). Applications of special protocols may reveal the presence of cfDNA fragments that deviate from the canonical cfDNA sizes. Single-stranded DNA sequencing revealed the existence of ultrashort cfDNA (~ 50 bp) [159], whereas single-molecule sequencing found a population of long (up to ~ 23,000 bp) cfDNA molecules [160]. Such molecules at the extreme spectrum cfDNA fragment lengths may open up new clinical applications. Altogether, this novel and very evolving area in liquid biopsy research is referred to as “fragmentomics” [161, 162] (Table 2).
Importantly, several studies have demonstrated that ctDNA has a modal size profile shorter than that of the background cfDNA originating from non-cancerous cells [156, 168]. To date, there are many efforts to explore how this knowledge can be leveraged to employ fragment size analysis to enhance detection of ctDNA [156]. For example, it has been shown that focusing on shorter cfDNA fragments enhances the detection of ctDNA [156] (Fig. 5F). Another important application is to use these size differences to distinguish between CHIP-associated mutations, which usually reside on cfDNA fragments with a size distribution of non-cancerous molecules, whereas mutations present in matched tumor specimen are more frequently on significantly shorter cfDNA molecules (Fig. 5G) [27]. Furthermore, machine learning models have been developed for detecting tumor-derived cfDNA through genome-wide analyses of cfDNA fragmentation in individuals at risk for lung cancer, suggesting that global fragmentation profiles may emerge as a tool for non-invasive detection of lung cancer [29, 157] (Fig. 5H). Importantly, as in any cfDNA application, pre-analytical processing methods must be carefully considered when performing fragmentomics-based analyses [159].
Open chromatin regions refer to regulatory regions within the genome, such as promoters, enhancers, or silencers (Fig. 5I), to which proteins bind, supplanting a canonical nucleosome and rendering the underlying DNA accessible to nucleases and other protein factors (Fig. 5I). As these are often regulatory regions with a biological role in tissue differentiation [169], nucleosome patterns and open chromatin regions are—similar to methylation markers—highly tissue-specific and can be also be used for plasma DNA tissue mapping and even tumor subtyping [122, 153]. Furthermore, plasma DNA-deduced nucleosome maps have been shown to result in characteristic coverage densities at transcription start sites, which correlate with gene expression from cells releasing their DNA into the circulation [154]. Similar patterns can also be observed at transcription factor binding sites (TFBS) and correlate with the accessibility of these transcription factors such that deregulation of transcription factors, an important driver in tumorigenesis, can be inferred from cfDNA [122]. Another application demonstrated that tissue-specific cfDNA degradation and nucleosome patterns enable the quantification of ctDNA burden [170] as well as for the detection and classification of pediatric solid tumors [167].
In summary, the detailed analyses of these epigenetic cfDNA features represents a novel, emerging area that will enable vital biological insights into the process of DNA release into the circulation [162] as well as more detailed clinically relevant characteristics about the tumor genome that are not attainable by the mere investigation of mutations or SCNAs.
From these recent developments, we dare to provide a personal view as to how cfDNA will be analyzed in the future (Fig. 6). We envision that instead of panels, only WGS will be conducted with a moderate sequencing depth of 30-35x. Such a sequencing depth, using appropriate bioinformatics tools, will be sufficient to track hundreds to thousands of somatic tumor-associated mutations and copy number alterations in a personalized setting [48]. Fragmentation patterns, i.e., variability of cfDNA lengths, can be investigated for further evidence for the presence of ctDNA. From nucleosome position mapping, essential biological information can be inferred, such as which genes are expressed or which transcription factors are active in the tumor cells. These features will be fed into multi-parameter classification models to enable cfDNA analyses with unprecedented resolution. With the wealth of information that can be extracted from cfDNA WGS at 30-35x coverage, this cfDNA analysis strategy will be applicable to both early-stage and advanced-stage cancer diseases. Importantly, such cfDNA evaluation strategies may well extend beyond applications in cancer to a more general use, for example, for evaluating chronic diseases or the health status of an individual.

A personal viewpoint on future plasma DNA analyses: one single dataset, an impressive number of analysis opportunities from cfDNA. In the future, there is little doubt that we will be sequencing whole patient genomes. Although current precision cancer medicine programs predominantly rely on gene panel sequencing, the decreasing cost of WGS will soon provide an attractive alternative, replacing stand-alone cancer diagnostics tests that require separate validation and standardization procedures. From a moderate sequencing coverage of 30-35x, we will be able to harvest cfDNA information from a single dataset, encompassing analysis possibilities ranging from personalized mutation tracking to tissue deconvolution. However, as methylation markers serve as the current predominant tissue-specific identifiers, it may be that whole-genome bisulfite sequencing (WGBS) provides an alternative to WGS for tissue deconvolution purposes. The complex array of data that can be obtained from diverse WGS analyses will represent multi-dimensional data to be subjected to feature extraction and various machine learning approaches. Ultimately, it will be possible to develop a model capable of distinguishing cfDNA that was derived from blood of a healthy individual and cfDNA derived from a patient with cancer. In the latter case, appropriate models may allow for tumor classification/subtyping, assessment of tumor evolution and identification of druggable targets or resistance markers. Furthermore, this would also open up exciting avenues for the analysis of cfDNA that extend beyond application in oncology
Concluding remarks
Owing to its diverse non-invasive application, analysis of ctDNA represents a future standard practice of clinical oncology. Herein, we hope to have summarized the most current applications, particularly distinguishing between established use cases and those with promising future potential, in turn supporting clinicians who may order and rely on these molecular testing modalities to guide their patient care in real-time (for a summary of open issues and potential solutions, see the summary box in the Appendix). However, as the number and types of liquid biopsy testing grow, so, too, does the requirement for information across multiple, complex knowledge domains in order to understand these novel approaches. Although liquid biopsy is just one tool in the precision oncology arsenal, the general growing and overwhelming amount of “omics” data coupled with novel therapy indications has rendered critical point-of-care decisions for oncologists and other precision medicine players challenging. Such complexity creates a disconnect between the researchers who develop assays and associated bioinformatics analyses, the clinical practitioners who must keep up to date with validated testing strategies, payer institutions who must determine which tests represent medical necessity and thus justify reimbursement, and patients and their families who may struggle to understand how treatment decisions are made throughout their journey with cancer. In this regard, interdisciplinary solutions must be established to facilitate ongoing exchange between the various experts of precision medicine. Molecular tumor boards represent critical instruments for bringing together these decoupled knowledge domains to inform clinical decisions that ultimately benefit the patient and relieve healthcare systems [33, 67, 171,172,173]. Interestingly, although its importance is frequently emphasized at conferences and in other media [52, 174, 175], further education platforms for clinicians without access to MTBs are not prevalent, although young oncologists in particular would benefit immensely from learning about the fundamentals of genomics and liquid biopsy profiling early on in their training. We have recently begun one such initiative to generate high-quality content tailored to clinicians (https://www.vesseldna.com/a-clinicians-handbook-for-ctdna/). Similarly, most current oncology workflows overlook direct incorporation of the patient into critical decision-making processes, as most patients do not possess the necessary capacity to understand such decisions based on genomics data. It would be worthwhile to evaluate workflows geared towards patient education and personalized consultation such that the patient may better understand the interpretation of his or her molecular testing and liquid biopsy results which guide treatment decisions and potentially influence his or her clinical outcome. As the field of precision oncology and liquid biopsy methodology begin to mature, researchers, clinicians and patients may look forward to innovative opportunities in personalized cancer care.
Availability of data and materials
Not applicable.
Abbreviations
- ASCO:
-
American Society of Clinical Oncology
- CAPP-Seq:
-
CAncer Personalized Profiling by deep Sequencing
- cfDNA:
-
Cell-free DNA
- CGP:
-
Comprehensive genomic profiling
- CHIP:
-
Clonal hematopoiesis of indeterminate potential
- CRPC:
-
Castration-resistant prostate cancer
- ctDNA:
-
Circulating tumor DNA
- DHS:
-
DNase hypersensitivity site
- DNA:
-
Deoxyribonucleic acid
- dPCR:
-
Digital PCR
- ER:
-
Estrogen receptor
- FDA:
-
Food and Drug Administration
- HR:
-
Hormone receptor
- HRR:
-
Homologous recombination repair
- ICI:
-
Immune checkpoint inhibitor
- LOD:
-
Limit of detection
- mBC:
-
Metastatic breast cancer
- MCED:
-
Multi-cancer early detection
- mCRPC:
-
Metastatic castration-resistant prostate cancer
- MRD:
-
Minimal residual disease
- MTB:
-
Molecular tumor board
- NCCN:
-
National Comprehensive Cancer Network
- NDR:
-
Nucleosome-depleted region
- NGS:
-
Next-generation sequencing
- NSCLC:
-
Non-small cell lung cancer
- PARPi:
-
Poly adenosine diphosphate-ribose polymerase inhibitor
- RNA:
-
Ribonucleic acid
- ROI:
-
Region of interest
- Safe-SeqS:
-
Safe Sequencing System
- SBS:
-
Sequencing-by-synthesis
- SCNA:
-
Somatic copy number alteration
- SiMSen-Seq:
-
Simple, multiplexed, PCR-based barcoding of DNA for sensitive mutation detection using sequencing
- sWGS:
-
Shallow whole-genome sequencing
- TAm-Seq:
-
Tagged Amplicon Sequencing
- TARDIS:
-
Targeted Digital Sequencing
- TFBS:
-
Transcription factor binding site
- TKI:
-
Tyrosine kinase inhibitor.
- TSS:
-
Transcription start site
- VAF:
-
Variant allele frequency
- WGS:
-
Whole-genome sequencing
References
Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76.
Stockley TL, Oza AM, Berman HK, Leighl NB, Knox JJ, Shepherd FA, et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med. 2016;8(1):109–016–0364–2.
Hyman DM, Taylor BS, Baselga J. Implementing Genome-Driven Oncology. Cell. 2017;168(4):584–99.
Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol. 2020;38(1):1–10.
Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977;37(3):646–50.
Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11(6):426–37.
Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32(6):579–86.
Duvvuri B, Lood C. Cell-Free DNA as a Biomarker in Autoimmune Rheumatic Diseases. Front Immunol. 2019;10:502.
Lehmann-Werman R, Magenheim J, Moss J, Neiman D, Abraham O, Piyanzin S, et al. Monitoring liver damage using hepatocyte-specific methylation markers in cell-free circulating DNA. JCI Insight. 2018;3(12). https://doi.org/10.1172/jci.insight.120687. eCollection 2018 Jun 21.
Zemmour H, Planer D, Magenheim J, Moss J, Neiman D, Gilon D, et al. Non-invasive detection of human cardiomyocyte death using methylation patterns of circulating DNA. Nat Commun. 2018;9(1):1443–018–03961–y.
Lui YY, Chik KW, Chiu RW, Ho CY, Lam CW, Lo YM. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin Chem. 2002;48(3):421–7.
Zheng YW, Chan KC, Sun H, Jiang P, Su X, Chen EZ, et al. Nonhematopoietically derived DNA is shorter than hematopoietically derived DNA in plasma: a transplantation model. Clin Chem. 2012;58(3):549–58.
Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–4.
Ramalingam N, Jeffrey SS. Future of Liquid Biopsies With Growing Technological and Bioinformatics Studies: Opportunities and Challenges in Discovering Tumor Heterogeneity With Single-Cell Level Analysis. Cancer J. 2018;24(2):104–8.
Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, et al. Genomic characterization of metastatic breast cancers. Nature. 2019;569(7757):560–4.
Reiter JG, Baretti M, Gerold JM, Makohon-Moore AP, Daud A, Iacobuzio-Donahue CA, et al. An analysis of genetic heterogeneity in untreated cancers. Nat Rev Cancer. 2019;19(11):639–50.
Adalsteinsson VA, Ha G, Freeman SS, Choudhury AD, Stover DG, Parsons HA, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun. 2017;8(1):1324–017–00965–y.
Odegaard JI, Vincent JJ, Mortimer S, Vowles JV, Ulrich BC, Banks KC, et al. Validation of a Plasma-Based Comprehensive Cancer Genotyping Assay Utilizing Orthogonal Tissue- and Plasma-Based Methodologies. Clin Cancer Res. 2018;24(15):3539–49.
Rothwell DG, Ayub M, Cook N, Thistlethwaite F, Carter L, Dean E, et al. Utility of ctDNA to support patient selection for early phase clinical trials: the TARGET study. Nat Med. 2019;25(5):738–43.
Bieg-Bourne CC, Okamura R, Kurzrock R. Concordance between TP53 alterations in blood and tissue: impact of time interval, biopsy site, cancer type and circulating tumor DNA burden. Mol Oncol. 2020;14(6):1242–51.
Park S, Olsen S, Ku BM, Lee MS, Jung HA, Sun JM, et al. High concordance of actionable genomic alterations identified between circulating tumor DNA-based and tissue-based next-generation sequencing testing in advanced non-small cell lung cancer: The Korean Lung Liquid Versus Invasive Biopsy Program. Cancer. 2021;127(16):3019–28.
Zardavas D, Irrthum A, Swanton C, Piccart M. Clinical management of breast cancer heterogeneity. Nat Rev Clin Oncol. 2015;12(7):381–94.
De Mattos-Arruda L, Weigelt B, Cortes J, Ng CKY, Nuciforo P, Bidard FC, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol. 2018;29(11):2268.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24.
Higgins MJ, Jelovac D, Barnathan E, Blair B, Slater S, Powers P, et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res. 2012;18(12):3462–9.
Barata PC, Koshkin VS, Funchain P, Sohal D, Pritchard A, Klek S, et al. Next-generation sequencing (NGS) of cell-free circulating tumor DNA and tumor tissue in patients with advanced urothelial cancer: a pilot assessment of concordance. Ann Oncol. 2017;28(10):2458–63.
Chabon JJ, Hamilton EG, Kurtz DM, Esfahani MS, Moding EJ, Stehr H, et al. Integrating genomic features for non-invasive early lung cancer detection. Nature. 2020;580(7802):245–51.
Chen X, Gole J, Gore A, He Q, Lu M, Min J, et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat Commun. 2020;11(1):3475–020–17316–z.
Mathios D, Johansen JS, Cristiano S, Medina JE, Phallen J, Larsen KR, et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat Commun. 2021;12(1):5060–021–24994–w.
Aggarwal C, Thompson JC, Black TA, Katz SI, Fan R, Yee SS, et al. Clinical Implications of Plasma-Based Genotyping With the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol. 2019;5(2):173–80.
Mack PC, Banks KC, Espenschied CR, Burich RA, Zill OA, Lee CE, et al. Spectrum of driver mutations and clinical impact of circulating tumor DNA analysis in non-small cell lung cancer: Analysis of over 8000 cases. Cancer. 2020;126(14):3219–28.
Rose Brannon A, Jayakumaran G, Diosdado M, Patel J, Razumova A, Hu Y, et al. Enhanced specificity of clinical high-sensitivity tumor mutation profiling in cell-free DNA via paired normal sequencing using MSK-ACCESS. Nat Commun. 2021;12(1):3770–021–24109–5.
Sicklick JK, Kato S, Okamura R, Schwaederle M, Hahn ME, Williams CB, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med. 2019;25(5):744–50.
Moser T, Waldispuehl-Geigl J, Belic J, Weber S, Zhou Q, Hasenleithner SO, et al. On-treatment measurements of circulating tumor DNA during FOLFOX therapy in patients with colorectal cancer. NPJ Precis Oncol. 2020;4(1):30–020–00134–3.
Weber S, van der Leest P, Donker HC, Schlange T, Timens W, Tamminga M, et al. Dynamic Changes of Circulating Tumor DNA Predict Clinical Outcome in Patients With Advanced Non–Small-Cell Lung Cancer Treated With Immune Checkpoint Inhibitors. JCO Precision Oncology. 2021;12(5):1540–53.
Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815.
Zhou Q, Perakis SO, Ulz P, Mohan S, Riedl JM, Talakic E, et al. Cell-free DNA analysis reveals POLR1D-mediated resistance to bevacizumab in colorectal cancer. Genome Med. 2020;12(1):20–020–0719–6.
Dagogo-Jack I, Brannon AR, Ferris LA, Campbell CD, Lin JJ, Schultz KR, et al. Tracking the Evolution of Resistance to ALK Tyrosine Kinase Inhibitors through Longitudinal Analysis of Circulating Tumor DNA. JCO Precis Oncol. 2018;2018:https://doi.org/10.1200/PO.17.00160. Epub 23 Jan 2018.
Dagogo-Jack I, Rooney M, Nagy RJ, Lin JJ, Chin E, Ferris LA, et al. Molecular Analysis of Plasma From Patients With ROS1-Positive NSCLC. J Thorac Oncol. 2019;14(5):816–24.
Dagogo-Jack I, Rooney M, Lin JJ, Nagy RJ, Yeap BY, Hubbeling H, et al. Treatment with Next-Generation ALK Inhibitors Fuels Plasma ALK Mutation Diversity. Clin Cancer Res. 2019;25(22):6662–70.
Recondo G, Bahcall M, Spurr LF, Che J, Ricciuti B, Leonardi GC, et al. Molecular Mechanisms of Acquired Resistance to MET Tyrosine Kinase Inhibitors in Patients with MET Exon 14-Mutant NSCLC. Clin Cancer Res. 2020;26(11):2615–25.
Azad TD, Chaudhuri AA, Fang P, Qiao Y, Esfahani MS, Chabon JJ, et al. Circulating Tumor DNA Analysis for Detection of Minimal Residual Disease After Chemoradiotherapy for Localized Esophageal Cancer. Gastroenterology. 2020;158(3):494-505.e6.
Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 2017;7(12):1394–403.
Chen K, Shields MD, Chauhan PS, Ramirez RJ, Harris PK, Reimers MA, et al. Commercial ctDNA Assays for Minimal Residual Disease Detection of Solid Tumors. Mol Diagn Ther. 2021;25(6):757–74.
McDonald BR, Contente-Cuomo T, Sammut SJ, Odenheimer-Bergman A, Ernst B, Perdigones N, et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci Transl Med. 2019;11(504). https://doi.org/10.1126/scitranslmed.aax7392.
Parsons HA, Rhoades J, Reed SC, Gydush G, Ram P, Exman P, et al. Sensitive Detection of Minimal Residual Disease in Patients Treated for Early-Stage Breast Cancer. Clin Cancer Res. 2020;26(11):2556–64.
Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8(346):346ra92.
Zviran A, Schulman RC, Shah M, Hill STK, Deochand S, Khamnei CC, et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat Med. 2020;26(7):1114–24.
Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17(4):223–38.
Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. 2019;20(2):71–88.
Cescon DW, Bratman SV, Chan SM, Siu LL. Circulating tumor DNA and liquid biopsy in oncology. Nature Cancer. 2020;1(3):276–90.
Ignatiadis M, Sledge GW, Jeffrey SS. Liquid biopsy enters the clinic - implementation issues and future challenges. Nat Rev Clin Oncol. 2021;18(5):297–312.
Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol. 2017;14(9):531–48.
Bohers E, Viailly PJ, Jardin F. cfDNA Sequencing: Technological Approaches and Bioinformatic Issues. Pharmaceuticals (Basel). 2021;14(6). https://doi.org/10.3390/ph14060596.
Keller L, Belloum Y, Wikman H, Pantel K. Clinical relevance of blood-based ctDNA analysis: mutation detection and beyond. Br J Cancer. 2021;124(2):345–58.
Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015;61(1):112–23.
Chan KC, Yeung AU- Lui, Wing-Bong, Lui WB, Rainer TH, Lo YM. Effects of preanalytical factors on the molecular size of cell-free DNA in blood. Clin Chem. 2005;51(4):781–4.
Chiu RW, Poon LL, Lau TK, Leung TN, Wong EM, Lo YM. Effects of blood-processing protocols on fetal and total DNA quantification in maternal plasma. Clin Chem. 2001;47(9):1607–13.
Jung M, Klotzek S, Lewandowski M, Fleischhacker M, Jung K. Changes in concentration of DNA in serum and plasma during storage of blood samples. Clin Chem. 2003;49(6 Pt 1):1028–9.
Lampignano R, Neumann MHD, Weber S, Kloten V, Herdean A, Voss T, et al. Multicenter Evaluation of Circulating Cell-Free DNA Extraction and Downstream Analyses for the Development of Standardized (Pre)analytical Work Flows. Clin Chem. 2020;66(1):149–60.
Parpart-Li S, Bartlett B, Popoli M, Adleff V, Tucker L, Steinberg R, et al. The Effect of Preservative and Temperature on the Analysis of Circulating Tumor DNA. Clin Cancer Res. 2017;23(10):2471–7.
van Dessel LF, Beije N, Helmijr JC, Vitale SR, Kraan J, Look MP, et al. Application of circulating tumor DNA in prospective clinical oncology trials - standardization of preanalytical conditions. Mol Oncol. 2017;11(3):295–304.
Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333–51.
Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11(1):31–46.
Jain NM, Schmalz L, Cann C, Holland A, Osterman T, Lang K, et al. Framework for Implementing and Tracking a Molecular Tumor Board at a National Cancer Institute-Designated Comprehensive Cancer Center. Oncologist. 2021;26(11):e1962–70.
Knepper TC, Bell GC, Hicks JK, Padron E, Teer JK, Vo TT, et al. Key Lessons Learned from Moffitt’s Molecular Tumor Board: The Clinical Genomics Action Committee Experience. Oncologist. 2017;22(2):144–51.
Koopman B, Groen HJM, Ligtenberg MJL, Grunberg K, Monkhorst K, de Langen AJ, et al. Multicenter Comparison of Molecular Tumor Boards in The Netherlands: Definition, Composition, Methods, and Targeted Therapy Recommendations. Oncologist. 2021;26(8):e1347–58.
Mosele F, Remon J, Mateo J, Westphalen CB, Barlesi F, Lolkema MP, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO Precision Medicine Working Group. Ann Oncol. 2020;31(11):1491–505.
Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545(7655):446–51.
van der Pol Y, Mouliere F. Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA. Cancer Cell. 2019;36(4):350–68.
Finan C, Gaulton A, Kruger FA, Lumbers RT, Shah T, Engmann J, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med. 2017;9(383). https://doi.org/10.1126/scitranslmed.aag1166.
Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, et al. Target-enrichment strategies for next-generation sequencing. Nat Methods. 2010;7(2):111–8.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47(D1):D941–7.
Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4(136):136ra68.
Gale D, Lawson ARJ, Howarth K, Madi M, Durham B, Smalley S, et al. Development of a highly sensitive liquid biopsy platform to detect clinically-relevant cancer mutations at low allele fractions in cell-free DNA. PLoS One. 2018;13(3).
Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108(23):9530–5.
Stahlberg A, Krzyzanowski PM, Egyud M, Filges S, Stein L, Godfrey TE. Simple multiplexed PCR-based barcoding of DNA for ultrasensitive mutation detection by next-generation sequencing. Nat Protoc. 2017;12(4):664–82.
Suppan C, Graf R, Jahn S, Zhou Q, Klocker EV, Bartsch R, et al. Sensitive and robust liquid biopsy-based detection of PIK3CA mutations in hormone-receptor-positive metastatic breast cancer patients. Br J Cancer. 2021.
Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–54.
Dawson SJ, Rosenfeld N, Caldas C. Circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;369(1):93–4.
Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497(7447):108–12.
Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med. 2019;25(12):1928–37.
Deveson IW, Gong B, Lai K, LoCoco JS, Richmond TA, Schageman J, et al. Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology. Nat Biotechnol. 2021;39(9):1115–28.
Moding EJ, Nabet BY, Alizadeh AA, Diehn M. Detecting Liquid Remnants of Solid Tumors: Circulating Tumor DNA Minimal Residual Disease. Cancer Discov. 2021.
Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34(5):547–55.
Reinert T, Henriksen TV, Christensen E, Sharma S, Salari R, Sethi H, et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019;5(8):1124–31.
Christensen E, Birkenkamp-Demtroder K, Sethi H, Shchegrova S, Salari R, Nordentoft I, et al. Early Detection of Metastatic Relapse and Monitoring of Therapeutic Efficacy by Ultra-Deep Sequencing of Plasma Cell-Free DNA in Patients With Urothelial Bladder Carcinoma. J Clin Oncol. 2019;37(18):1547–57.
Wan JCM, Heider K, Gale D, Murphy S, Fisher E, Mouliere F, et al. ctDNA monitoring using patient-specific sequencing and integration of variant reads. Sci Transl Med. 2020;12(548). https://doi.org/10.1126/scitranslmed.aaz8084.
Coombes RC, Page K, Salari R, Hastings RK, Armstrong A, Ahmed S, et al. Personalized Detection of Circulating Tumor DNA Antedates Breast Cancer Metastatic Recurrence. Clin Cancer Res. 2019;25(14):4255–63.
Heider K, Gale DG, Marsico G, Ruiz-Valdepeñas A, Sharma G, Perry M, et al. Detection of residual disease and recurrence in early-stage non-small cell lung cancer (NSCLC) patients using sensitive personalized ctDNA sequencing assays. JCO. 2020 05/20; 2021/12;38(15):e15560–e15560.
Kurtz DM, Soo J, Co Ting Keh L, Alig S, Chabon JJ, Sworder BJ, et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA. Nat Biotechnol. 2021.
Heitzer E, Ulz P, Belic J, Gutschi S, Quehenberger F, Fischereder K, et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013;5(4):30.
Ulz P, Heitzer E, Speicher MR. Co-occurrence of MYC amplification and TP53 mutations in human cancer. Nat Genet. 2016;48(2):104–6.
Ulz P, Belic J, Graf R, Auer M, Lafer I, Fischereder K, et al. Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer. Nat Commun. 2016;7:12008.
Teutsch SM, Bradley LA, Palomaki GE, Haddow JE, Piper M, Calonge N, et al. The Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Initiative: methods of the EGAPP Working Group. Genet Med. 2009;11(1):3–14.
Merker JD, Oxnard GR, Compton C, Diehn M, Hurley P, Lazar AJ, et al. Circulating Tumor DNA Analysis in Patients With Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J Clin Oncol. 2018;36(16):1631–41.
Nakamura Y, Taniguchi H, Ikeda M, Bando H, Kato K, Morizane C, et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat Med. 2020;26(12):1859–64.
Palmero R, Taus A, Viteri S, Majem M, Carcereny E, Garde-Noguera J, et al. Biomarker Discovery and Outcomes for Comprehensive Cell-Free Circulating Tumor DNA Versus Standard-of-Care Tissue Testing in Advanced Non–Small-Cell Lung Cancer. JCO Precision Oncol. 2021;12(5):93–102.
Turner NC, Kingston B, Kilburn LS, Kernaghan S, Wardley AM, Macpherson IR, et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 2020;21(10):1296–308.
Leighl NB, Page RD, Raymond VM, Daniel DB, Divers SG, Reckamp KL, et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non-small Cell Lung Cancer. Clin Cancer Res. 2019;25(15):4691–700.
Parsons HA, Beaver JA, Cimino-Mathews A, Ali SM, Axilbund J, Chu D, et al. Individualized Molecular Analyses Guide Efforts (IMAGE): A Prospective Study of Molecular Profiling of Tissue and Blood in Metastatic Triple-Negative Breast Cancer. Clin Cancer Res. 2017;23(2):379–86.
Riedl JM, Hasenleithner SO, Pregartner G, Scheipner L, Posch F, Groller K, et al. Profiling of circulating tumor DNA and tumor tissue for treatment selection in patients with advanced and refractory carcinoma: a prospective, two-stage phase II Individualized Cancer Treatment trial. Ther Adv Med Oncol. 2021;13:1758835920987658.
Pritchett MA, Camidge DR, Patel M, Khatri J, Boniol S, Friedman EK, et al. Prospective Clinical Validation of the InVisionFirst-Lung Circulating Tumor DNA Assay for Molecular Profiling of Patients With Advanced Nonsquamous Non-Small-Cell Lung Cancer. JCO Precis Oncol. 2019;3. https://doi.org/10.1200/PO.18.00299. eCollection 2019.
O’Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov. 2018;8(11):1390–403.
Siravegna G, Lazzari L, Crisafulli G, Sartore-Bianchi A, Mussolin B, Cassingena A, et al. Radiologic and Genomic Evolution of Individual Metastases during HER2 Blockade in Colorectal Cancer. Cancer Cell. 2018;34(1):148–162.e7.
Powles T, Assaf ZJ, Davarpanah N, Banchereau R, Szabados BE, Yuen KC, et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature. 2021;595(7867):432–7.
Cheng ML, Pectasides E, Hanna GJ, Parsons HA, Choudhury AD, Oxnard GR. Circulating tumor DNA in advanced solid tumors: Clinical relevance and future directions. CA Cancer J Clin. 2021;71(2):176–90.
US Food and Drug Administration. cobas® EGFR Mutation Test V. 2016; Available at: https://www.accessdata.fda.gov/cdrh_docs/pdf12/P120019S007c.pdf. Accessed 3 Dec 2021.
Anagnostou V, Forde PM, White JR, Niknafs N, Hruban C, Naidoo J, et al. Dynamics of Tumor and Immune Responses during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Res. 2019;79(6):1214–25.
Goldberg SB, Narayan A, Kole AJ, Decker RH, Teysir J, Carriero NJ, et al. Early Assessment of Lung Cancer Immunotherapy Response via Circulating Tumor DNA. Clin Cancer Res. 2018;24(8):1872–80.
Moding EJ, Liu Y, Nabet BY, Chabon JJ, Chaudhuri AA, Hui AB, et al. Circulating Tumor DNA Dynamics Predict Benefit from Consolidation Immunotherapy in Locally Advanced Non-Small Cell Lung Cancer. Nat Cancer. 2020;1(2):176–83.
Raja R, Kuziora M, Brohawn PZ, Higgs BW, Gupta A, Dennis PA, et al. Early Reduction in ctDNA Predicts Survival in Patients with Lung and Bladder Cancer Treated with Durvalumab. Clin Cancer Res. 2018;24(24):6212–22.
Nabet BY, Esfahani MS, Moding EJ, Hamilton EG, Chabon JJ, Rizvi H, et al. Noninvasive Early Identification of Therapeutic Benefit from Immune Checkpoint Inhibition. Cell. 2020;183(2):363–76 e13.
Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2019;380(20):1929–40.
Rugo HS, Lerebours F, Ciruelos E, Drullinsky P, Ruiz-Borrego M, Neven P, et al. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): one cohort of a phase 2, multicentre, open-label, non-comparative study. Lancet Oncol. 2021;22(4):489–98.
Page K, Martinson LJ, Fernandez-Garcia D, Hills A, Gleason KLT, Gray MC, et al. Circulating Tumor DNA Profiling From Breast Cancer Screening Through to Metastatic Disease. JCO Precis Oncol. 2021;5. https://doi.org/10.1200/PO.20.00522. eCollection 2021.
de Bono J, Kang J, Hussain M. Olaparib for Metastatic Castration-Resistant Prostate Cancer. Reply N Engl J Med. 2020;383(9):891.
Abida W, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J Clin Oncol. 2020;38(32):3763–72.
Annala M, Vandekerkhove G, Khalaf D, Taavitsainen S, Beja K, Warner EW, et al. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov. 2018;8(4):444–57.
Belic J, Graf R, Bauernhofer T, Cherkas Y, Ulz P, Waldispuehl-Geigl J, et al. Genomic alterations in plasma DNA from patients with metastasized prostate cancer receiving abiraterone or enzalutamide. Int J Cancer. 2018;143(5):1236–48.
Conteduca V, Wetterskog D, Sharabiani MTA, Grande E, Fernandez-Perez MP, Jayaram A, et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: a multi-institution correlative biomarker study. Ann Oncol. 2017;28(7):1508–16.
Ulz P, Perakis S, Zhou Q, Moser T, Belic J, Lazzeri I, et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat Commun. 2019;10(1):4666–019–12714–4.
Sicklick JK, Kato S, Okamura R, Schwaederle M, Hahn ME, Williams CB, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med. 2019;25(5):744–50.
Katsoulakis E, Duffy JE, Hintze B, Spector NL, Kelley MJ. Comparison of Annotation Services for Next-Generation Sequencing in a Large-Scale Precision Oncology Program. JCO Precis Oncol. 2020;4. https://doi.org/10.1200/PO.19.00118. eCollection 2020.
Perakis SO, Weber S, Zhou Q, Graf R, Hojas S, Riedl JM, et al. Comparison of three commercial decision support platforms for matching of next-generation sequencing results with therapies in patients with cancer. ESMO Open. 2020;5(5):e000872–2020–000872.
Wagner AH, Walsh B, Mayfield G, Tamborero D, Sonkin D, Krysiak K, et al. A harmonized meta-knowledgebase of clinical interpretations of somatic genomic variants in cancer. Nat Genet. 2020;52(4):448–57.
Maass KK, Schad PS, Finster AME, Puranachot P, Rosing F, Wedig T, et al. From Sampling to Sequencing: A Liquid Biopsy Pre-Analytic Workflow to Maximize Multi-Layer Genomic Information from a Single Tube. Cancers (Basel). 2021;13(12): https://doi.org/10.3390/cancers13123002.
Weber S, Spiegl B, Perakis SO, Ulz CM, Abuja PM, Kashofer K, et al. Technical Evaluation of Commercial Mutation Analysis Platforms and Reference Materials for Liquid Biopsy Profiling. Cancers (Basel). 2020;12(6). https://doi.org/10.3390/cancers12061588.
Thierry AR, El Messaoudi S, Gahan PB, Anker P, Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016;35(3):347–76.
Bardelli A, Pantel K. Liquid Biopsies, What We Do Not Know (Yet). Cancer Cell. 2017;31(2):172–9.
Heitzer E, Auinger L, Speicher MR. Cell-Free DNA and Apoptosis: How Dead Cells Inform About the Living. Trends Mol Med. 2020;26(5):519–28.
Fox-Fisher I, Piyanzin S, Ochana BL, Klochendler A, Magenheim J, Peretz A, et al. Remote immune processes revealed by immune-derived circulating cell-free DNA. Elife. 2021;10. https://doi.org/10.7554/eLife.70520.
Henriksen TV, Reinert T, Christensen E, Sethi H, Birkenkamp-Demtroder K, Gogenur M, et al. The effect of surgical trauma on circulating free DNA levels in cancer patients-implications for studies of circulating tumor DNA. Mol Oncol. 2020;14(8):1670–9.
Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985–90.
Chen X, Dong Z, Hubbell E, Kurtzman KN, Oxnard GR, Venn O, et al. Prognostic Significance of Blood-Based Multi-cancer Detection in Plasma Cell-Free DNA. Clin Cancer Res. 2021;27(15):4221–9.
Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–87.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–98.
Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16.
Dor Y, Cedar H. Principles of DNA methylation and their implications for biology and medicine. Lancet. 2018;392(10149):777–86.
Leypold NA, Speicher MR. Evolutionary conservation in noncoding genomic regions. Trends Genet. 2021;37(10):903–18.
Erger F, Norling D, Borchert D, Leenen E, Habbig S, Wiesener MS, et al. cfNOMe - A single assay for comprehensive epigenetic analyses of cell-free DNA. Genome Med. 2020;12(1):54–020–00750–5.
Lam WKJ, Gai W, Sun K, Wong RSM, Chan RWY, Jiang P, et al. DNA of Erythroid Origin Is Present in Human Plasma and Informs the Types of Anemia. Clin Chem. 2017;63(10):1614–23.
Sadeh R, Sharkia I, Fialkoff G, Rahat A, Gutin J, Chappleboim A, et al. ChIP-seq of plasma cell-free nucleosomes identifies gene expression programs of the cells of origin. Nat Biotechnol. 2021;39(5):586–98.
Sun K, Jiang P, Chan KC, Wong J, Cheng YK, Liang RH, et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci U S A. 2015;112(40):E5503–12.
Sun K, Jiang P, Cheng SH, Cheng THT, Wong J, Wong VWS, et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res. 2019;29(3):418–27.
Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV, CCGA Consortium. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. 2020;31(6):745–59.
Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359(6378):926–30.
Lennon AM, Buchanan AH, Kinde I, Warren A, Honushefsky A, Cohain AT, et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science. 2020;369(6499). https://doi.org/10.1126/science.abb9601. Epub 28 Apr 2020.
Cisneros-Villanueva M, Hidalgo-Perez L, Rios-Romero M, Cedro-Tanda A, Ruiz-Villavicencio CA, Page K, et al. Cell-free DNA analysis in current cancer clinical trials: a review. Br J Cancer. 2022;126(3):391–400.
Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005;102(45):16368–73.
Lo YM, Chan KC, Sun H, Chen EZ, Jiang P, Lun FM, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med. 2010;2(61):61ra91.
Ramachandran S, Henikoff S. Replicating Nucleosomes. Sci Adv. 2015;1(7). https://doi.org/10.1126/sciadv.1500587.
Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell. 2016;164(1–2):57–68.
Ulz P, Thallinger GG, Auer M, Graf R, Kashofer K, Jahn SW, et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat Genet. 2016;48(10):1273–8.
Underhill HR, Kitzman JO, Hellwig S, Welker NC, Daza R, Baker DN, et al. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016;12(7):e1006162.
Mouliere F, Chandrananda D, Piskorz AM, Moore EK, Morris J, Ahlborn LB, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med. 2018;10(466). https://doi.org/10.1126/scitranslmed.aat4921.
Cristiano S, Leal A, Phallen J, Fiksel J, Adleff V, Bruhm DC, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570(7761):385–9.
Mouliere F, Smith CG, Heider K, Su J, van der Pol Y, Thompson M, et al. Fragmentation patterns and personalized sequencing of cell-free DNA in urine and plasma of glioma patients. EMBO Mol Med. 2021;13(8).
Hudecova I, Smith CG, Hansel-Hertsch R, Chilamakuri CS, Morris JA, Vijayaraghavan A, et al. Characteristics, origin, and potential for cancer diagnostics of ultrashort plasma cell-free DNA. Genome Res. 2021.
Yu SCY, Jiang P, Peng W, Cheng SH, Cheung YTT, Tse OYO, et al. Single-molecule sequencing reveals a large population of long cell-free DNA molecules in maternal plasma. Proc Natl Acad Sci U S A. 2021;118(50). https://doi.org/10.1073/pnas.2114937118.
Ivanov M, Baranova A, Butler T, Spellman P, Mileyko V. Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genomics. 2015;16 Suppl 13:S1–2164–16–S13–S1. Epub 16 Dec 2015 .
Lo YMD, Han DSC, Jiang P, Chiu RWK. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 2021;372(6538):https://doi.org/10.1126/science.aaw3616.
Chan KC, Jiang P, Sun K, Cheng YK, Tong YK, Cheng SH, et al. Second generation noninvasive fetal genome analysis reveals de novo mutations, single-base parental inheritance, and preferred DNA ends. Proc Natl Acad Sci U S A. 2016;113(50):E8159–68.
Jiang P, Sun K, Tong YK, Cheng SH, Cheng THT, Heung MMS, et al. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2018;115(46):E10925–33.
Jiang P, Sun K, Peng W, Cheng SH, Ni M, Yeung PC, et al. Plasma DNA End-Motif Profiling as a Fragmentomic Marker in Cancer, Pregnancy, and Transplantation. Cancer Discov. 2020;10(5):664–73.
Jiang P, Xie T, Ding SC, Zhou Z, Cheng SH, Chan RWY, et al. Detection and characterization of jagged ends of double-stranded DNA in plasma. Genome Res. 2020;30(8):1144–53.
Peneder P, Stutz AM, Surdez D, Krumbholz M, Semper S, Chicard M, et al. Multimodal analysis of cell-free DNA whole-genome sequencing for pediatric cancers with low mutational burden. Nat Commun. 2021;12(1):3230–021–23445–w.
Jiang P, Chan CW, Chan KC, Cheng SH, Wong J, Wong VW, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci U S A. 2015;17(11):E1317-25.
Meuleman W, Muratov A, Rynes E, Halow J, Lee K, Bates D, et al. Index and biological spectrum of human DNase I hypersensitive sites. Nature. 2020;584(7820):244–51.
Zhu G, Guo YA, Ho D, Poon P, Poh ZW, Wong PM, et al. Tissue-specific cell-free DNA degradation quantifies circulating tumor DNA burden. Nat Commun. 2021;12(1):2229–021–22463–y.
Kato S, Kim KH, Lim HJ, Boichard A, Nikanjam M, Weihe E, et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat Commun. 2020;11(1):4965–020–18613–3.
Larson KL, Huang B, Weiss HL, Hull P, Westgate PM, Miller RW, et al. Clinical Outcomes of Molecular Tumor Boards: A Systematic Review. JCO Precis Oncol. 2021;5: https://doi.org/10.1200/PO.20.00495. eCollection 2021 Jul.
Sicklick JK, Kato S, Okamura R, Patel H, Nikanjam M, Fanta PT, et al. Molecular profiling of advanced malignancies guides first-line N-of-1 treatments in the I-PREDICT treatment-naive study. Genome Med. 2021;13(1):155–021–00969–w.
The American Journal of Managed Care. Evidence-Based Oncology. 2021;27(7).
The ASCO Post. The Evolution of Liquid Biopsy in Cancer Care. 2021. Available at: https://ascopost.com/issues/october-10-2021/the-evolution-of-liquid-biopsy-in-cancer-care/. Accessed 12 Dec 2021.
Rolfo C, Cardona AF, Cristofanilli M, Paz-Ares L, Diaz Mochon JJ, Duran I, et al. Challenges and opportunities of cfDNA analysis implementation in clinical practice: Perspective of the International Society of Liquid Biopsy (ISLB). Crit Rev Oncol Hematol. 2020;151:102978.
Deans ZC, Butler R, Cheetham M, Dequeker EMC, Fairley JA, Fenizia F, et al. IQN path ASBL report from the first European cfDNA consensus meeting: expert opinion on the minimal requirements for clinical ctDNA testing. Virchows Arch. 2019;474(6):681–9.
Martignano F. Cell-Free DNA. An Overview of Sample Types and Isolation Procedures. Methods Mol Biol. 2019;1909:13–27.
Sorber L, Zwaenepoel K, De Winne K, Van Casteren K, Augustus E, Jacobs J, et al. A Multicenter Study to Assess EGFR Mutational Status in Plasma: Focus on an Optimized Workflow for Liquid Biopsy in a Clinical Setting. Cancers (Basel). 2018;10(9). https://doi.org/10.3390/cancers10090290.
Sorber L, Zwaenepoel K, Jacobs J, De Winne K, Goethals S, Reclusa P, et al. Circulating Cell-Free DNA and RNA Analysis as Liquid Biopsy: Optimal Centrifugation Protocol. Cancers (Basel). 2019;11(4). https://doi.org/10.3390/cancers11040458.
van Ginkel JH, van den Broek DA, van Kuik J, Linders D, de Weger R, Willems SM, et al. Preanalytical blood sample workup for cell-free DNA analysis using Droplet Digital PCR for future molecular cancer diagnostics. Cancer Med. 2017;6(10):2297–307.
Warton K, Graham LJ, Yuwono N, Samimi G. Comparison of 4 commercial kits for the extraction of circulating DNA from plasma. Cancer Genet. 2018;228–229:143–50.
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Non-Small Cell Lung Cancer. Version: 7.2021. Available at: https://www.nccn.org/guidelines/category_1.
Rolfo C, Mack P, Scagliotti GV, Aggarwal C, Arcila ME, Barlesi F, et al. Liquid Biopsy for Advanced NSCLC: A Consensus Statement From the International Association for the Study of Lung Cancer. J Thorac Oncol. 2021;16(10):1647–62.
Boichard A, Richard SB, Kurzrock R. The Crossroads of Precision Medicine and Therapeutic Decision-Making: Use of an Analytical Computational Platform to Predict Response to Cancer Treatments. Cancers (Basel). 2020;12(1). https://doi.org/10.3390/cancers12010166.
Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol. 2017;2017. https://doi.org/10.1200/PO.17.00011. Epub 16 May 2017.
Mateo J, Chakravarty D, Dienstmann R, Jezdic S, Gonzalez-Perez A, Lopez-Bigas N, et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann Oncol. 2018;29(9):1895–902.
Rolfo C, Manca P, Salgado R, Van Dam P, Dendooven A, Ferri Gandia J, et al. Multidisciplinary molecular tumour board: a tool to improve clinical practice and selection accrual for clinical trials in patients with cancer. ESMO Open. 2018;3(5):e000398–2018–000398. eCollection 2018.
Acknowledgements
The majority of the figure illustrations were created with BioRender.com.
Funding
Research in our laboratory is supported by the Austrian Science Fund (FWF; KLI 764 and TCS 101). The Austrian Science Fund had no role in the design of the review and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
S.O.H. and M.R.S. contributed equally to this work. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Summary box
Summary box
Why is analysis of ctDNA not yet a clinical gold standard? Open issues and potential solutions
Type of issue | Bottleneck | Potential workaround/solution |
Logistical issues | Many hospitals face reimbursement issues when it comes to liquid biopsy testing and costs may be difficult to justify and thus are not covered for every ordered test. | This will hopefully change when significant progress is made in the analytical and clinical validity as well as clinical utility of plasma-based assays, especially as evidence from appropriately designed clinical trials accumulates. To date, there are no cost-effectiveness data on cfDNA assays and models for evaluating the economic impact on health must be considered in order for healthcare payers to cover such liquid biopsy solutions [176] |
Access to testing | Those who face reimbursement issues from large, commercial providers may find a solution in partnering with academic labs who perform validated assays routinely | |
Pre-analytical/analytical factors | Despite advances in liquid biopsy technology, pre-analytical and analytical standards are still lacking and may vary between testing labs. The 2018 joint review by ASCO and the College of American Pathologists determined that data on pre-analytic variables, analytical validity, interpretation and reporting are still insufficient to justify widespread adoption for the majority of ctDNA solid tumor assays [96]. | Previous works [60, 62, 128, 177,178,179,180,181,182] have detailed the influence of pre-analytical factors such as blood collection tubes, plasma versus serum, transit time, centrifugation protocols, storage conditions, cfDNA purification, quantification, characterization and analysis platforms. Several collaborative efforts are currently underway to evaluate these findings, devise the criteria for standardizing liquid biopsy workflows, put these into context for providers and end users of cfDNA assays, as well as to bridge the gap between academia and industry to enable a transition of research findings into clinical implementation: The European Liquid Biopsy Society (ELBS); European Liquid Biopsy Academy (ELBA); International Society of Liquid Biopsy (ISLB); BloodPac |
Negative results | According to the ASCO Post, the 2021 National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for molecular and biomarker analysis of NSCLC advises that cfDNA/ctDNA testing should not replace standard histologic diagnosis via tissue testing, unless the patient is unable to undergo invasive tissue sampling [183]. | A recent consensus statement released by the International Association for the Study of Lung Cancer (IASLC) concluded that ctDNA analysis is an acceptable initial approach detection of biomarkers at diagnosis as well as for monitoring targeted therapies [184]. |
Both technical and biological factors can influence the concordance between tumor tissue results and plasma DNA analysis. | The risk of false-positive and false-negative results from liquid biopsy testing have not completely eradicated the necessity of reflex tissue testing, such that the FDA recommends follow-up testing after receiving negative results, including those from commercial providers such as Guardant360 CDx and FoundationOne Liquid CDx | |
Diversity of NGS panels | Generally, there are no harmonized minimal requirements for NGS panels, although most harbor biomarkers within guidelines of national consortia, e.g. NCCN. | Implementation of broader NGS panels, e.g. those that evaluate mutations outside of the canonical hotspots, as these may reflect emerging biomarkers or inclusion criteria for recruiting/ongoing clinical trials |
Clinical validity | Determination of clinical validity for broad NGS panels is challenging, as they are applied in multiple tumor types. For both of the use cases of therapy monitoring and early-stage cancer, evidence of clinical validity is still emerging, whereas there is still no evidence available for cancer screening purposes. | As summarized by Ignatiadis et al., further clinical validation of ctDNA assays will be conducted in the form of large-cohort phase III trials, which should include mandatory blood sampling and emphasize the need to report the results of liquid biopsy assays as official trial results [52]. |
Clinical utility | Currently, liquid biopsy assays lack consistency and precision. In fact, clinical validity and clinical utility have not yet been shown for the vast majority of assays. | Sophisticated multicenter clinical validation studies and regulatory guidelines are lacking but must be established to ensure responsible future application of liquid biopsies in precision oncology. |
It is not yet known whether interventional treatment based on detection of ctDNA relapse via liquid biopsy will actually improve patient outcome, i.e. cure, or whether systemic treatment will simply delay onset of metastatic disease [52]. | Design and conducting of suitable clinical trials with the goal of improving outcomes of patients with detectable ctDNA | |
Bioinformatics | Increasing complexity of bioinformatics algorithms | Need for increased usability, e.g. through user-friendly graphical user interfaces with clear and concise instruction for use and interpretation of results |
Education | Lack of structured, formal training available for clinicians regarding the implementation of genomic medicine: which tests to order for which patients; understanding the benefits and limitations of NGS assays; when to order liquid biopsy; how to interpret the clinical reports, prioritize variants and evidence and convert potential actionable insight into clinical care. | Such questions may be answered within the context of an appropriately set up molecular tumor board (MTB), where a panel of diverse experts, e.g. oncologists, pathologists, clinical geneticists and bioinformaticians meet on an ongoing basis to deliver the most suitable precision oncology approaches. Additionally, innovative education platforms, particularly geared towards young oncologists, may provide the fundamental awareness of such testing modalities early on in a clinician’s training. Molecular profiling and liquid biopsy education should be integrated into university training programs. |
Interpretation bottlenecks | Lack of MTB infrastructure within hospital | With the advent of virtual molecular tumor boards, it may be possible to partner with experienced MTBs to discuss difficult patient cases |
Clinical decision support tools are still in their infancy and harmonization efforts regarding the interpretation of genomic variants have only just begun [124,125,126, 185] . | Various strategies of decision support software may lead to discrepancies in pathogenicity, actionability and treatment matching when interpreting genomic variants. For this reason, data must be evaluated and prioritized at a multidisciplinary MTB to derive the most suitable treatment decision. | |
The interpretation and prioritization of variants at an MTB may vary from clinic to clinic and some MTBs may still be unfamiliar with data derived from ctDNA assays. | Clinics should strictly employ standardized criteria to define actionability and must rely on validated evidence-based scales, such as the FDA-approved content of OncoKB and European ESCAT guidelines [186,187,188] |
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hasenleithner, S.O., Speicher, M.R. A clinician’s handbook for using ctDNA throughout the patient journey. Mol Cancer 21, 81 (2022). https://doi.org/10.1186/s12943-022-01551-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-022-01551-7