
Abstract
The development of immune checkpoint blockade (ICB)-based immunotherapy has dramatically changed methods of cancer treatment. This approach triggers a durable treatment response and prolongs patients' survival; however, not all patients can benefit. Accumulating evidence demonstrated that the efficacy of ICB is dependent on a robust antitumor immune response that is usually damaged in most tumors. Conventional chemotherapy and targeted therapy promote the antitumor immune response by increasing the immunogenicity of tumor cells, improving CD8+ T cell infiltration, or inhibiting immunosuppressive cells in the tumor microenvironment. Such immunomodulation provides a convincing rationale for the combination therapy of chemotherapeutics and ICBs, and both preclinical and clinical investigations have shown encouraging results. However, the optimal drug combinations, doses, timing, and sequence of administration, all of which affect the immunomodulatory effect of chemotherapeutics, as well as the benefit of combination therapy, are not yet determined. Future studies should focus on these issues and help to develop the optimal combination regimen for each cancer.
Similar content being viewed by others

Introduction
Immune checkpoint blockade (ICB)-based immunotherapy has resulted in a revolutionary shift in cancer treatment. Distinguished from conventional chemotherapy and radiotherapy, which suppress tumors by directly killing malignant cells, ICBs rescue the antitumor activity of T cells through targeted blockade of checkpoints, such as cytotoxic T lymphocyte-associated protein 4 (CTLA-4), programmed cell death 1 (PD-1), and its ligand PD-L1 (also known as CD274), and are superior in establishing immune memory and preventing recurrence [1]. In the past decade, the clinical uses of ICBs have shown promising results in the treatment of many different kinds of malignancies [2,3,4]. To date, several distinct ICBs, including (1) the CTLA-4 antibody ipilimumab (Yervoy); (2) the PD-1 inhibitors: Cemiplimab (Libtayo), nivolumab (Opdivo), and pembrolizumab (Keytruda); and (3) the PD-L1 blockers: Atezolizumab (Tecentriq), avelumab (Bavencio), and durvalumab (Imfinzi), have been approved to treat a variety of advanced cancers, including melanoma, non-small cell lung cancer (NSCLC), hepatocellular carcinoma (HCC), head and neck squamous cell carcinoma (HNSCC), and urothelial carcinoma. Furthermore, these and several other ICBs are under clinical test and are expected to expand the panel of oncological indications.
Despite their increasing varieties and indications, ICBs have been demonstrated to induce an effective and durable antitumor immune response only in a small subset of patients. The response rates to ICBs used as stand-alone therapeutic interventions in unselected patients are mostly less than 30% in a variety of tumor types [5], which is unsatisfactory. Further improving the antitumor efficacy of ICB-based immunotherapy has become one of the main challenges in clinical oncology.
The clinical efficacy of ICBs depends on the pre-existing antitumor immunity. Immune checkpoints are the negative regulators of antitumor immunity. PD-1 and CTLA-4 are expressed on T cells during priming and activation, while in the tumor microenvironment (TME), local interferon-gamma (IFNγ), mainly derived from effector lymphocytes, induces the expression of PD-L1 on cancer cells and intra-tumoral immune cells. Accumulating evidence suggests that tumors infiltrated by CD8+ T cells that can recognize and kill cancer cells are more likely to respond to ICB treatment [6].
Historically, conventional chemotherapy was considered as immunosuppressive because it broadly affects immune cells, in addition to tumor cells, resulting in myelosuppression and leukopenia. However, recent studies have demonstrated that chemotherapy can activate an endogenous antitumor immune response, which partly contributes to their therapeutic effects. Similar immunomodulatory effects were also observed for targeted agents, such as tyrosine kinase inhibitor (TKI), which originally inhibited the proliferation of neoplastic cells with cancer-specific alterations, likely because of the shared signaling pathways between cancer and immune cells. Following these findings, both conventional chemotherapeutics and targeted agents have been suggested to be combined with ICBs to enhance antitumor efficacy. Previous treatment success of the combination therapies confirmed their synergistic effect [7,8,9,10] and encouraged further investigations. Given the clinical momentum in combining these two classes of therapies, it is crucial to understand the actions of chemotherapeutics on the antitumor immune response. Here, we summarize the discovered immunomodulatory chemotherapeutics and targeted agents, discuss how they modify antitumor immunity, and review the possibility of combining these medications with ICBs.
Antitumor CD8+ T-cell immunity – the basis for ICB treatment efficacy
CD8+ cytotoxic T lymphocytes (CTLs)-mediated antitumor immunity is the backbone of immune elimination of cancer, as well as the basis for the effectiveness of the ICB. This multistep event is also termed as the cancer-immunity cycle. It starts with the release of neoantigens created by oncogenesis (step 1). Next, antigen-presenting cells (APCs), such as dendritic cells (DCs), will capture these neoantigens and migrate to the draining lymph nodes, where they present the processed peptide to naïve T cells (step 2); leading to the priming and activation of tumor-specific T cells (step 3). During activation, T cells also acquire a chemotactic ability toward the tumor via their expression of C-X-C motif chemokine receptor 3 (CXCR3), a chemokine receptor that can bind to cancer-derived ligands (such as C-X-C motif chemokine ligand, CXCL9, CXCL10, and CXCL11). Then, under the chemokine-receptor interaction, the activated T cells traffic to (step 4) and infiltrate into the tumor bed (step 5), where they specifically recognize (step 6) and eventually kill their target cancer cells (step 7) [11].
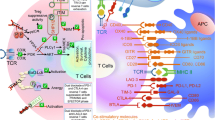
Immune escape from ICB-based immunotherapy has been attributed to failures in the steps of the cancer-immunity cycle, which varies in different tumor types [12]. Various cellular and humoral factors in the TME drive or reduce anticancer immunity to account for these failures. They constitute different tumor immune landscapes and have been demonstrated to be associated with the tumor response to ICBs (Fig. 1).

Failure of cancer-immunity cycles in three different tumor-immune landscapes. a In the immune-desert tumor, the generation of tumor-specific CD8+ T cells is impaired because of low immunogenicity. b In the immune-excluded tumor, CTL infiltration is impaired because the arriving tumor-specific CD8+ T cells are shut out by the aberrant vasculature and stromal barriers. c In the immune-inflamed tumor, the immunosuppressive cells directly impair the cytotoxicity of tumor-specific CD8+ T cells and indirectly inhibit T cell activation by suppressing DCs. Arg1: argase1; Breg: regulatory B cell, CTLA-4: cytotoxic T lymphocyte-associated protein 4, DCs: dendritic cells, IDO1: indoleamine 2,3-dioxygenase 1, IL: interleukin, MHC: major histocompatibility complex, NETs: neutrophil extracellular traps, PD-1/PD-L1: programmed cell death 1 and its ligand 1, TAM: tumor-associated macrophage, TAN: tumor-associated neutrophils, TGFβ: transform grow factor-β, TMB: tumor mutation burden, Treg: regulatory T cell, VEGF-A & VEGFR: vascular endothelial growth factor A and its receptor
Tumor immunogenicity plays a central role in initiating the antitumor immune response [13]. This depends on two key factors: (1) tumor antigenicity, i.e., the tumor neoantigens which can be recognized as non-self compounds; and (2) immune adjuvanticity, i.e., the inflammatory signals that promote the recruitment, maturation, and antigen presentation of immune cells such as DCs. In other words, the tumor immunogenicity favors the generation and recruitment of tumor-specific CTLs, avoiding the immune-desert phenotype which indicates a non-response to ICB treatment. Consistent with these notions, highly-mutated malignancies with an abundance of neoantigens, such as melanoma, NSCLC, and HNSCC, are more sensitive to ICB treatment [14, 15]. Patients with colorectal cancer (CRC) with high microsatellite instability or defects in the mismatch repair system are likely to have improved tumor control after treatment with PD-1/CTLA-4 inhibitors [16, 17].
Another prerequisite for a successful ICB response is the infiltration of activated tumor-specific CTLs. High intra-tumoral CTL levels are recognized as a predictor of improved response and treatment outcome of ICB therapy, while immune-excluded tumors, with T cells present at the invasive margin, usually respond less to ICBs. Accumulating evidence suggests that the desmoplastic stroma and disorganized tumor vasculature of the TME are the main reasons for the immune-excluded phenotype. In line with this notion, single-agent ipilimumab has been demonstrated to be ineffective to treat advanced pancreatic ductal adenocarcinoma (PDAC), a tumor characterized by fibrotic stroma [18]. Furthermore, vascular endothelial growth factor A (VEGF-A), which promotes the tumor vasculature, is associated with treatment resistance to anti-CTLA-4 antibodies in patients with melanoma [19], likely because it impairs endothelial-T cell adhesion and subsequent T-cell infiltration by reducing levels of intercellular adhesion molecule–1 (ICAM-1), as well as vascular cell adhesion molecule–1 (VCAM-1) [20], and triggers CD8+ T cell apoptosis by inducing FASL on endothelial cells [21].
Immunosuppressive cells are the most important suppressors for the antitumor immune response. Previous studies have confirmed the immunosuppressive roles of regulatory T cells and B cells (Tregs and Bregs), anti-inflammatory tumor-associated macrophages (M2-TAMs), tumor-associated neutrophils (TANs), and myeloid-derived suppressor cells (MDSCs) in various kinds of cancers. These populations are selectively accumulated and activated in tumor sites by either cancer-cell secreted chemokines or chronic inflammatory signals. Cellular and humoral factors have been exploited to suppress CTL-mediated antitumor response, including (1) immunosuppressive cytokines, such as interleukin (IL)-10, IL-35, and transforming growth factor-beta (TGF-β), which inhibit DC maturation and antigen presentation; T-cell activation; and the priming and cytotoxicity of CTLs; (2) immunosuppressive metabolites, including adenosine, kynurenine, reactive oxygen species, lactic acid, and nitric oxide are generally derived from MDSCs, M2-TAMs, Tregs, and tumor-associated neutrophils. These molecules not only directly impaired CTL antitumor capacity by decreasing effective molecules like IFNγ and perforin, and upregulating co-inhibitory receptors such as PD-1 and TIM3, but also positively increased the recruitment of suppressor cells, thereby exacerbating immunosuppression; (3) immunosuppressive ligands: M2-TAMs, TANs, Bregs, and MDSCs usually have high levels of surface inhibitors, such as PD-L1, while Tregs can also inactivate DCs via the expression of CTLA-4, which binds to CD80/CD86 on DCs and transduces suppressive signals to interrupt DC activation [22]; (4) consumption of key nutrients and grow factors: Tregs competitively consume IL-2, while M2-TAMs and MDSCs can reduce local L-cysteine, L-arginine, and tryptophan, thereby restricting the activation and proliferation of CTLs; (5) vascularization and stroma promotion: both M2-TAMs and MDSCs are important sources of VEGF-A and they prevent CTL infiltration by promoting the proliferation of cancer-associated fibroblasts [23, 24]; and (6) cancer cell protection: Neutrophil extracellular traps wrap and coat tumor cells and thus protect them from CTL-mediated killing [25].
In addition to immunosuppression, these populations, especially M2-TAMs, play important roles in tumorigenesis. Tissue chronic inflammation mediated by macrophages was thought to be mutagenic and growth-promoting. With the expression of pro-tumoral cytokines, such as EGF and CCL18, M2-TAMs, MDSCs, and TANs directly activate the migration of cancer cells [26,27,28]. M2-TAM and MDSCs induce the rapid generation of immature vascular networks providing the nutrients and oxygen for tumor proliferation [29]. M2-TAMs can also enhance tumor invasion directly [30], and induce the entrance of tumor cells into circulation [31]. In the metastatic tumor microenvironment, M2 macrophage assist tumor cell extravasation from blood vessels [32] and supported the seeding, survival, and prospering of tumor cells through the formation of a nurturing niche [33, 34]. Consistent with their tumor promotion and immune suppression effects, accumulating evidence suggests that the high accumulation of Tregs, Bregs, M2-TAMs, TAN, or MDSCs is associated with a poor response to ICB treatment in a variety of cancers [35, 36].
In contrast to suppressive immune populations, CTL-promoting cells are also present in the TME and are usually related to a better response to ICB therapy. Natural killer (NK) cells and gamma/delta T cells (γ/δ T cells) are the important lymphocytes in the innate immune system. In an MHC-independent cytolytic manner, these two populations can effectively deplete tumor cells with antigen-presentation deficiency, and thus serve as allies of antitumor CTLs. Besides, previous studies have found that both NK and γ/δ T cells can promote the generation of CTLs. NK cells can recruit conventional DCs to the tumor bed via chemo-attractants, such as XCL1, CCL5, and Fms-related tyrosine kinase 3 ligand (FLT3LG) [37, 38], while γ/δ T cells can directly act as professional APCs [39]. B cells are also key professional APCs in cancer. Recent studies have demonstrated that they participate in the formation of tertiary lymphoid structures (TLS), allowing the generation of tumor-specific CTLs, and eventually driving the tumor response to ICB treatments [40,41,42].
Chemotherapeutics promote antitumor immunity (Fig. 2)
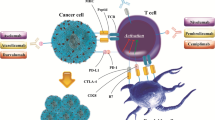
Chemotherapeutics originally direct inhibited or killed malignant cells to achieve their therapeutic effects. Recently, some frontline drugs have been found to additionally promote antitumor immunity by increasing tumor immunogenicity, improving T cell infiltration, or depleting the immunosuppressive populations. Therefore, it is reasonable to hypothesize that chemotherapeutics specifically remove the constraints of the antitumor immune response in different TMEs, making them the first-line option for these tumors. The immunomodulatory effects of some popular chemotherapeutics are summarized in Table 1. Some of them have been well-reviewed previously [85]; therefore, the current review mainly focuses on recent findings and their role in combination therapy with ICBs.

Chemotherapeutics drive cancer-immunity cycles. Chemotherapeutics induce immunogenic cell death (a), promote CD8+ T cell infiltration (b), and inhibit immunosuppressive cells (c). ATP: adenosine-triphosphate, BRAF: B-Raf proto-oncogene, serine/threonine kinase, CTL: Cytotoxic T lymphocytes, CXCL10: C-X-C motif chemokine ligand 10, DC: dendritic cells, HMGB1: high-mobility group box1, ICAM-1: intercellular adhesion molecule, IFN-γ: interferon-gamma, MDSC: myeloid-derived suppressor cell, NK cells: natural killer cells, PARP: Poly (ADP-ribose) polymerase, TAM: tumor-associated macrophage, TKI: tyrosine kinase inhibitor, TLS: Tertiary lymphoid structures, TNF: tumor necrosis factor, Treg: regulatory T cell, VCAM: vascular cell adhesion molecule
Chemotherapeutic and targeted agents activate antitumor CD8+ T cell immunity
Decreased immunogenicity is one of the most important characteristics of malignancies, leading to immune ignorance. Besides, in most cases, chemotherapeutics induce apoptosis and necrosis of cancer cells, which is incapable of inducing adaptive immunity. By contrast, immunogenic cell death (ICD) is characterized by its potential to increase tumor immunogenicity and thus establish long-lasting antitumor immunity. To date, several kinds of chemotherapeutics that have been commonly used in clinical practice were found to mediate their antineoplastic activity by inducing ICD of malignant cells [86].
Anthracyclines, oxaliplatin, and paclitaxel are well-recognized as ICD inducers. Preclinical and clinical histological observations have demonstrated that these agents significantly increase the abundance of intra-tumoral CD8+ T cells, which favors their antineoplastic efficacy and is associated with better patient outcomes [85, 87]. The mechanisms through which they induce ICD have been determined [13]. Traditionally, several processes, such as the unfolded protein response, autophagy, inflammasome signaling, Toll-like receptor 3 (TLR3) signaling, and type 1 interferon response are related to ICD. During these processes, damage-associated molecular patterns (DAMPs) including adenosine triphosphate (ATP), the surface-exposure of calreticulin, high-mobility group box1 (HMGB1) promoted DC-mediated CTL activation; and chemokines such as CXCL10 enhanced CTL recruitment. Moreover, recent studies have found that ICD-inducing drugs can also modulate antitumor CTL immunity through tumor-infiltrating NK cells and B cells. In human ovarian cancer, platinum and taxane chemotherapy significantly increase NK cell infiltration and local T cell oligoclonal expansion [47]. While in human breast cancer, the neoadjuvant doxorubicin, cyclophosphamide, and paclitaxel combination regimen switched the tumor-infiltrating B cells to a new ICOSL+ phenotype. These newly emerging B cells are involved in the formation of the TLS, and significantly increased the numbers and the cytotoxicity of tumor-specific CD8+T cells [52] (Fig. 1a).
Given their ability to activate the antitumor CTL response, ICD-inducing chemotherapeutics are believed to combine with and enhance the therapeutic efficacy of ICBs. Doxorubicin plus PD-1 or PD-L1 antibodies showed a significantly improved antitumor effect in various murine cancers, such as melanoma and breast cancer [88, 89]. In human metastatic triple-negative breast cancer (TNBC), short-term doxorubicin induction sensitized the tumor to PD-1 blockade [90]. Similarly, oxaliplatin was demonstrated to boost the efficacy of anti-PD-L1 therapy in murine colorectal cancer [91]. Paclitaxel and ICB combination therapy elicited a superior tumor-suppression effect in nonimmunogenic squamous NSCLC [8].
In addition to anthracyclines, teniposide, another topoisomerase II inhibitor, was reported recently to induce ICD; however, it acts via a different mechanism to that of anthracyclines. Topoisomerase II inhibitors induced the proliferation-arrest or demise of neoplastic cells by increasing DNA double-strand breaks [92]. It has been suggested that damaged DNA fragments in the nucleus could be actively exported to the cytoplasm, possibly to prevent misincorporation into genomic DNA, and trigger the innate immune response mediated by the cGAS-STING (cyclic GMP–AMP synthase/stimulators of interferon gene) pathway [93,94,95]. In line with this notion, teniposide activated the tumor-cell intrinsic type-I interferon (IFN) response and upregulated features of ICD. Besides, teniposide strengthened the tumor cell antigen presentation machinery, which augmented T-cell recognition. Consequently, in rodent colon cancer, teniposide induced robust antitumor CD8+ T-cell immunity and remarkable tumor suppression. Vaccination with teniposide-treated dead tumor cells effectively prevented tumor redevelopment. Furthermore, the administration of teniposide successfully reversed the insensitivity to the PD-1 inhibitor of KRAS mutant CT26 colon cancer [62]. Despite its positive immunomodulatory effect in murine tumors, whether teniposide acts as an ICD inducer in human cancers remains elusive.
Poly (ADP-ribose) polymerase inhibitors (PARPi), including olaparib and niraparib, inhibit DNA repair in homologous-recombination-deficient malignant cells, leading to synthetic lethality [96]. Such retention and accumulation of DNA damage can activate the cGAS-STING pathway and the subsequent type-I IFN response, as mentioned above. In line with this notion, the administration of olaparib to murine BRCA (encoding breast cancer type 1 susceptibility protein) -deficient TNBCs increased the CD8+ T cell abundance and activated antitumor immunity [72]. Despite PARPis generally eliciting antitumor efficacy in BRCA-mutant cancers, clinical investigations have demonstrated the unexpected treatment benefits of niraparib in patients with BRCA-proficient ovarian cancer [97, 98]. A recent preclinical study found that in ovarian cancer, PARPi triggered the STING-dependent immunogenic response, regardless of DNA repair deficiency [74]. A similar observation was also found in small cell lung cancer (SCLC) [73]. In addition to the increased intra-tumoral CTLs, PARPi could upregulate PD-L1 expression in malignant cells in breast cancer, SCLC, and ovarian cancer, regardless of the BRCA mutation status. Such increasing CTL abundance and intra-tumoral PD-L1 level potentiate the combined therapy of PARPi and ICBs [99]. As expected, a combination of niraparib plus pembrolizumab therapy showed promising synergistic antitumor activity in patients with TNBC or ovarian cancer [100, 101], despite the best treatment efficacy still being observed in patients with BRCA-mutant [102]. Although the combination of a PARPi and an ICB (olaparib plus durvalumab) did not show satisfactory therapeutic efficacy in SCLC, it is worth noting that the addition of olaparib might be capable of reversing the ICB resistance of SCLC, because some tumors that progressed in previous ICB treatment maintained stable disease under the combined regimen [103].
Pyroptosis is a new pattern of cell death, which is mediated by gasdermin (GSDM) proteins. GSDMs, mainly GSDMD and GSDME, are activated after the cleavage of their autoinhibitory N domains by caspases. They translocate to, and form pores on, the cytomembrane, resulting in cell swelling, membrane rupture, and the release of cytosolic contents including DAMPs, such as HMGB1 and ATP [104, 105]. Some conventional chemotherapeutic agents, like cisplatin and etoposide, can induce pyroptosis. However, GSDME is silenced in most cancer cells, but is expressed in many normal cells, including lymphocytes; therefore, these medications were traditionally supposed to impair, rather than promote, antitumor immunity [106, 107]. Intriguingly, a recent study showed that GSDME-mediated pyroptosis acts as a form of ICD and effectively activated antitumor CD8+ T-cell immunity in murine melanoma [108]. The combination of B-Raf proto-oncogene, serine/threonine kinase (BRAF) and MAPK/ERK kinase (MEK) inhibitors, the frontline care for BRAFV600E-mutant melanoma, was found to induce the pyroptosis of melanoma cells by blocking extracellular regulated kinase (ERK)1/2 signaling and subsequently activating the GSDME cleaver, caspase-3. These dual inhibitions significantly increased the intra-tumoral abundance of DCs, as well as CTLs, contributing to durable tumor regression [65]. Besides, BRAF inhibition alone has been demonstrated to increase CD8+ T cells, while MEK inhibitors potentiated anti-tumor T cells by preventing T-cell receptor (TCR)-driven apoptosis [70]. The potential of BRAF and MEK inhibitors to synergize the effects of anti-PD-1 antibodies has been observed in mouse melanoma [109]. In human BRAFV600E-mutant melanoma, such a triplet therapy facilitated a remarkable antitumor response and prolonged the progression-free survival of patients [110, 111]. Similar to BRAF and MEK inhibitors, crizotinib, which is used to treat NSCLC carrying activated anaplastic lymphoma kinase (ALK) and ROS proto-oncogene 1, receptor tyrosine kinase (ROS1), favored the ICD, likely because it triggered the pyroptosis of lung cancer cells in which GSDME is expressed ubiquitously [112]. The administration of crizotinib increased CTL accumulation in murine NSCLC and remarkably sensitized the tumor to PD-1 blockade [69].
Chemotherapeutic and targeted agents enhance CD8+ T-cell infiltration
In addition to promoting the generation of CTLs, chemotherapeutics can enhance their entry into the tumor center. Antiangiogenic molecules that target the VEGF/VEGFR axis are expected to starve the tumor, thus suppressing tumor progression and improving patient survival. However, recent research has found that, instead of simply starving tumor cells to death, antiangiogenic agents also promote immune attack by markedly increasing the infiltration of tumor-specific CTLs after normalizing the immature vessels (Fig. 1b).
In a variety of rodent and human malignancies, both anti-VEGF-A and anti-VEGFR2 agents have been demonstrated to increase T cell infiltration [64, 113], and dual inhibition of the VEGF/VEGFR axis, and other antiangiogenic factors, such as angiopoietin-2 (ANGPT2) and prostaglandin E2 (PGE2), further improved this increment [21, 114]. After infiltration, CTLs recognize the tumor cells and secrete many cytotoxic cytokines, such as IFN-γ, resulting in the induction of PD-L1 expression by tumor cells. Thus, it provides a convincing rationale for the development of the combination of antiangiogenic therapy and immunotherapy. As expected, different combinatorial treatments of antiangiogenic agents and ICBs have shown a higher synergistic effect in tumor control compared with that achieved by monotherapy in various rodent cancers, including breast cancer, pancreatic neuroendocrine tumor, and HCC [84, 113]. Histological examinations showed that the combination of ICB and antiangiogenic agents further promoted vessel normalization, even toward high endothelial venules [84]. Such normalized vessels permitted not only CTLs, but also DCs and B cells, infiltration and accumulation [114], which implied the formation of TLSs allowing local generation and expansion of tumor-destroying CTLs [84]. In human HCC, CD8+ T cells are usually presented at the peritumoral, rather than intra-tumoral, areas [115]. The combination of the anti-VEGF-A antibody bevacizumab with the anti-PD-L1 antibody atezolizumab showed an unexpectedly high overall response rate, prolonged patient survival, and became a potential first-line treatment option for HCC [116]. Similar treatment success has been seen in metastatic renal cell carcinoma (RCC) [117].
After extravasation from tumor vessels, CTLs are likely to infiltrate into the tumor parenchyma in most malignancies. However, they can also be retained in the tumor margins because some solid tumors, such as PDAC, can establish another physical barrier, the robust stroma. Disruption of the massy stroma might promote CTL penetration and facilitate the antitumor immune response. Focal adhesion kinase (FAK) was identified as a significant contributor to the fibrotic TME and correlated negatively with CD8+ CTL infiltration in human and murine PDACs. By reducing the fibrotic stroma and subsequently enhancing CTL entry, FAK inhibition not only slowed tumor progression but also rendered the previously unresponsive rodent PDAC responsive to PD-1 antagonists [118]. However, whether the additional FAK inhibitor improves tumor sensitivity to ICBs in human PDAC remains elusive. Although FAK inhibitors have been proven as safe and promising [119], to date, few clinical studies have investigated the antitumor effect of combined FAK inhibitors and ICBs in human cancers.
Chemotherapeutic and targeted agents restrain immunosuppressive cells
The depletion of immunosuppressive cells is involved in the antitumor effect of several agents. Gemcitabine, a nucleoside analog that is commonly used to treat PDAC, depletes circulating, or intra-tumoral MDSCs in multiple cancers. Such depletion favors the restoration of CTL infiltration and cytotoxic activity in both rodent and human cancers [85].
Cyclophosphamide is a nitrogen mustard derivative that is activated intracellularly by phosphoramides or phosphatase, becoming cytotoxic. Tregs were considered to be susceptible to the toxic effects of cyclophosphamide, likely because of their low levels of intracellular antidotes, like glutathione, and their lack of ATP-binding cassette transports, which help to exclude the active metabolite of cyclophosphamide [120, 121]. A low dose of cyclophosphamide has been noted to not only decrease the number but also inhibited the function, of Tregs in rodent tumors [122]. A recent study found that cyclophosphamide preferentially targeted CCR2+ Tregs in a highly active and proliferating state, i.e., the effector Tregs [48]. In humans, metronomic (a repetitive low dose administration) cyclophosphamide treatment effectively reduced both peripheral naïve and activated Tregs, thereby favoring effector T cell subsets in patients with mesothelioma [123]. A clinical trial has also demonstrated that in patients with end-stage metastatic CRC, repetitive low doses of cyclophosphamide induced Treg-deletion and boosted antitumor immunity, which eventually contributed to prolonged progressive-free survival [49]. Similar to cyclophosphamide, camptothecin, a topoisomerase I inhibitor, can also restrain the generation and function of Tregs. It inactivated the transcriptional activity of the NR4A (nuclear receptor subfamily 4 group A) family of nuclear orphan receptors, which inhibited the expression of Forkhead box P3 (FOXP3) and eventually reduced Tregs generation. By removing the suppression by Tregs, irinotecan, a prodrug of camptothecin, promoted the priming and proliferation of CD8+ T cells in the draining lymph nodes and suppressed the growth of murine lung and colon cancer in a CD8+ T cell-dependent manner [50]. Similarly, the chemotherapeutic regimen containing irinotecan, FOLFIRI, was reported to decrease the suppressive activity of peripheral Tregs in patients with CRC [56].
Multi-targeted TKIs, such as sunitinib, inhibit the downstream signaling of receptors, including VEGFR, platelet-derived growth factor receptor (PDGFR), stem cell factor receptor (c-Kit), and colony-stimulating factor -1 (CSF-1) receptor, preventing neoplastic proliferation and tumor angiogenesis [124]. However, these tumor-promoting pathways also play a crucial role in the generation of MDSCs and Tregs. For example, c-Kit receptor signaling and the VEGFA-VEGFR2 pathway are required to generate MDSCs and Tregs [125, 126]. The administration of sunitinib significantly diminished the levels of circulating and intra-tumoral MDSCs, thereby expanding the number of activated tumor-specific CD8+ T cells in murine tumors [127]. Such a relief of MDSC-mediated immunosuppression was also observed in patients with RCC or other metastatic diseases [80, 128]. Similar to MDSCs, Tregs are vulnerable to sunitinib. Recent observations of rodent HCC showed that sunitinib significantly reduced the frequency and function of tumor-infiltrating Tregs, which recovered the cytotoxicity of tumor-specific CD8+ T cells [81]. A combination of sunitinib and anti-PD1 antibodies powerfully activated the antitumor immune response and suppressed tumor growth [82].
The plasticity of macrophages provides an alternative approach to recover antitumor immunity. This comprises repolarizing M2-TAMs toward the pro-inflammatory state (M1 phenotype) in which they act as APCs that facilitate the antitumor immune response. Such a functional transformation has been observed in human PDAC after GEM-based neoadjuvant chemotherapy [55]. Similarly, paclitaxel, one of the most effective cytotoxic agents, which is considered as the standard of care for breast cancer and ovarian cancer, can also repolarize M2-TAMs. Whereas previous studies have shown that TAMs were recruited by cancer cells after paclitaxel treatment and blocked the CD8+ T cell-dependent chemotherapy response [129], paclitaxel was newly identified as an agonist of TLR4 on TAMs and directly polarized this anti-inflammation population into a pro-inflammatory phenotype [59, 60]. The upregulated antigen-presenting ability in this phenotype reversion released CTL-dependent tumor regression [59]. Along this line, patients with breast cancer treated with paclitaxel showed a peripheral pro-inflammatory profile [130]. In addition, an enrichment of genes linked to the inflammatory macrophage phenotype in the TME was reported in patients with ovarian cancer after paclitaxel treatment [59]. Furthermore, TAM repolarization by paclitaxel provides a rationale for combination therapy with ICBs in the treatment of TNBC, in which a high infiltration of immunosuppressive TAMs is associated with a lower response to ICBs [131]. As expected, the combination of atezolizumab and nab-paclitaxel prolonged progression-free survival of patients with metastatic TNBC [132].
Combination therapies
Numerous clinical trials have been carried out to investigate the combination therapy of immune-modulatory agents and ICBs. An overview of the key studies with their reported results is presented in Table 2.
Currently, the majority of combination therapies comprise adding concurrent ICBs to existing chemotherapy or targeted regimens. The chemo-immunotherapy usually consists of several cycles of induction concurrent therapy and subsequent maintenance ICB monotherapy, while targeted-immunotherapy is a continuous concurrent regimen. In most combinations, compounds are given in a full dose. The synergistic antitumor effects of these combination therapies have been demonstrated in various cancers, as mentioned before. Several regimens have been suggested as new first-line treatments [133, 152, 155].
Although encouraging progress has been made, the therapeutic efficacy of the current combination therapy remains unsatisfactory. Besides, increased toxicity is another critical issue that should not be overlooked. The incidence of high-grade treatment-related adverse effects (TRAEs) resulting from combination therapy is usually higher than 50%. Unacceptable hepatic toxicity has been seen in the combination of ipilimumab and the BRAF inhibitor vemurafenib during the treatment of melanoma and has resulted in the interruption of this trial [159]. A similar failure happened in the combination of nivolumab and crizotinib in NSCLC [149]. Furthermore, even though in most cases, TRAEs can be managed through the reduction or interruption of drugs, this impairs therapeutic efficacy [146].
Few trials have investigated how the sequence of administration affects the benefit. However, preclinical studies have demonstrated the relationship. Pretreatment with the MEK inhibitor selumetinib significantly augmented the antitumor efficacy of subsequent anti-CTLA4 monotherapy, while the concurrent regimen did not [71]. Similarly, cyclophosphamide administered 1 day before could enhance the antitumor effect of anti-CTLA4 antibody, whereas, treatment with the reversed-sequence regimen led to the apoptosis of proliferating tumor-specific CD8+ T cells and then attenuated tumor control [160]. These observations indicated that induction of chemotherapy or targeted therapy might optimize the TME, thereby supporting the efficacy of subsequent ICBs, at least in ipilimumab-based combination therapies. A previous clinical trial demonstrated that a phased regimen (inductive chemotherapy alone, before concurrent ipilimumab) but not a concurrent regimen (initiating concurrent chemo-ipilimumab) improved the PFS of patients with extensive-disease SCLC [145]. Another regimen comprising phased ipilimumab started at the third cycles of carboplatin and etoposide also showed benefit [143].
The dose of compounds is another important factor for combination therapy. Continuous full dose dual MAPK inhibitors might be inappropriate for combination therapy with ICBs because the increment in intra-tumoral T cells mediated by the inhibitors occurs in the early phase after treatment initiation and become less frequent beyond 2 weeks [161]. Besides, the toxicity of concomitant full-dose of triplet BRAF, MEK, and anti-PD1/PD-L1 inhibitors significantly limited their efficacy [110, 111]. A phase II study has investigated different doses of BRAF and MEK inhibitors in combination therapy with pembrolizumab in BRAFV600-mutant melanoma. Lower rates of high-grade TRAEs and higher objective response rates (ORR) were found in patients treated with short-term intermittent dual MAPK inhibitions, rather than pembrolizumab plus continuous targeted treatment [162]. However, because the high ORR did not mean a better survival outcome in all cases, whether the intermittent regimen would be the more efficient and safe approach should be tested in a larger cohort and with longer follow-up. However, these findings did suggest exploring the optimal dose in future combination trials.
Perspective and conclusion
ICB-based cancer immunotherapy removes the checkpoint constraints on adaptive antitumor immunity, thereby releasing the cytotoxicity of tumor-specific CD8+ T cells. It can induce tumor shrinkage, durable disease control, and prolonged survival, but only in a minority of patients, likely because the CD8+ T cell-mediated antitumor immune response is impaired in most cancers. Antitumor immunity is mainly driven or suppressed by cellular factors in the TME; therefore, treatment strategies that can specifically modify the TME toward an inflamed phenotype are expected to be combined with ICBs to augment their therapeutic efficacy.
The therapeutic effects of chemotherapy and targeted therapy are traditionally considered to rely on tumor cell-intrinsic sensitivity or cancer-specific alterations. However, the empirical selection of clinically efficient therapeutic regimens might also imply enhanced anticancer immunosurveillance. For example, anthracyclines-based regimens are commonly used to treat breast cancers which are usually considered as nonimmunogenic with rare CD8+ T cell infiltration. Gemcitabine is the frontline treatment option for PDAC, which is characterized by abundant MDSCs and TAMs. These “coincidences” suggest that the TME or cancer-immunity cycles might insensibly affect the selections of standard chemotherapy. Furthermore, it indicates that chemotherapy and targeted therapy are promising candidates to sensitize tumors to ICB therapy because they can target the impaired steps of the cancer-immunity cycle in certain tumors. Indeed, most of the current combination regimens comprise the concurrent administration of ICBs and the existing chemotherapeutic regimen and are currently the best choice to improve patient survival.
Optimization of the drug combination, dose, and sequence is still needed to achieve maximum therapeutic efficacy. Although the existing clinically efficient chemotherapeutic regimens might have specifically enhanced anticancer immunosurveillance for each cancer, whether these empirical regimens are optimal for combination therapy with ICBs remains unknown. A comprehensive understanding of the composition and immune situation of the TME and a careful examination of the immunological characteristics of the currently used chemotherapeutics would be conducive to selecting the most ideal drugs for combination therapy of cancer. In addition to drug combinations, the dose and sequence of administration of chemotherapeutics and ICBs can also affect the therapeutic effect. The long-term and full-dose administration of chemotherapeutics might be unnecessary in the combination regimen because it not only results in more serious toxicity but also damages, rather than enhances, anti-tumor immunity. These findings suggest that there is a need to investigate the optimal doses of chemotherapeutics in combination therapy. Besides, chemotherapy induction before ICB administration seems to be beneficial. In such induction phases, chemotherapeutics promote the generation and infiltration of CTLs or delete the immunosuppressive cells, thereby optimizing the TME for subsequent ICB therapy. Serial histological examination during chemotherapy can reveal the dynamic changes of the cancer immune context, thus favoring the selection of the best combination treatment time and sequence.
Similar to chemotherapeutics, radiotherapy has profound immunomodulatory effects. Through inducing DNA damage, radiotherapy can expand the spectrum of neoantigens and upregulate the antigen-presenting machinery in tumor cells [163]. Besides, radiation has been proved as an ICD-inducer and can promote the recruitment of DCs and CTLs through DAMPs and chemokines [164]. However, the radiotherapy-mediated immuno-stimulations are usually blunted by the cancer cell-intrinsic DNA damage response (DDR) and immunosuppressive cells, including MDSCs and TAMs. DDR inhibitors have been shown to further potentialize the radiation-induced inflammation in TME [165]. Furthermore, as chemotherapeutics like paclitaxel can deplete or convert the suppressor cells, the combinations of radiotherapy with chemo-/targeted therapy might further enhance the antitumor immune response and ICB therapy. However, few studies have investigated whether the triplet of chemo-radio-immunotherapy actually works.
In summary, the immunomodulatory effect of chemotherapeutics provides a strong cancer biology rationale for their combination with ICBs. Such combinations will not only directly inhibit malignant cells but also augment the immune recognition and elimination of tumor cells. Furthermore, it establishes long-term antitumor memory and thus might represent a curative treatment. The synergistic antitumor efficacy of combination therapy has been demonstrated in various cancers; however, the maximum benefit has not yet been achieved. Future studies that evaluate the therapeutic efficacy of regimens with different drug combinations, doses, and sequences will help to develop the optimal combination therapy for each cancer.
Availability of data and materials
Not applicable.
Abbreviations
- ALK:
-
Anaplastic lymphoma kinase
- ANGPT2:
-
Angiopoietin-2
- APC:
-
Antigen-presenting cells
- ATP:
-
Adenosine-triphosphate
- AUC:
-
Area under the curve
- BID:
-
Twice daily
- BRAF:
-
B-Raf proto-oncogene, serine/threonine kinase
- BRCA:
-
Breast cancer susceptibility gene
- Breg:
-
Regulatory B cell
- CCR2:
-
Chemokine (C-C motif) receptor 2
- cGAS-STING:
-
Cyclic GMP–AMP synthase/stimulators of interferon gene
- CRC:
-
Colorectal cancer
- CSF-1:
-
Colony-stimulating factor -1
- CTL:
-
Cytotoxic T lymphocytes
- CTLA-4:
-
Cytotoxic T lymphocyte-associated protein 4
- CXCL9/10/11:
-
C-X-C motif chemokine ligand 9/10/11
- CXCR3:
-
C-X-C motif chemokine receptor 3
- DAMP:
-
Damage-associated molecular pattern
- DC:
-
Dendritic cells
- DDR:
-
DNA damage response
- EGFR:
-
Epidermal growth factor receptor
- ERK:
-
Extracellular regulated kinase
- FAK:
-
Focal adhesion kinase
- FOLFIRI:
-
Eolinic acid, 5-fluorouracil
- FOXP3:
-
Forkhead Box P3
- γ/δ T cell:
-
Gamma/delta T cell
- GSDM:
-
Gasdermin
- HCC:
-
Hepatocellular carcinoma
- HMGB1:
-
High-mobility group box1
- HNSCC:
-
Head and neck squamous cell carcinoma
- ICAM-1:
-
Intercellular adhesion molecule–1
- ICB:
-
Immune checkpoint blockade
- ICD:
-
Immunogenic cell death
- IFN-γ:
-
Interferon-gamma
- MDSC:
-
Myeloid-derived suppressor cell
- NK cells:
-
Natural killer cells
- NR4A:
-
Nuclear receptor subfamily 4 group A
- NSCLC:
-
Non-small cell lung cancer
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- PARPi:
-
Poly (ADP-ribose) polymerase inhibitor
- PD-1:
-
Programmed cell death 1
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PDGFR:
-
Platelet-derived growth factor receptor
- PD-L1:
-
Programmed death-ligand 1
- PFS:
-
Progression-free survival
- PGE2:
-
Prostaglandin E2
- QD:
-
Once daily
- QW/Q2W/Q3W:
-
Every 1/2/3 weeks
- RCC:
-
Renal cell carcinoma
- ROS1:
-
ROS proto-oncogene 1, receptor tyrosine kinase
- SCLC:
-
Small cell lung cancer
- TAM:
-
Tumor-associated macrophage
- TAN:
-
Tumor-associated neutrophils
- TCR:
-
T-cell receptor
- TGF-β:
-
Transforming growth factor-beta
- TKI:
-
Tyrosine kinase inhibitor
- TLR:
-
Toll-like receptor
- TLS:
-
Tertiary lymphoid structures
- TMB:
-
Tumor mutation burden
- TME:
-
Tumor microenvironment
- TNBC:
-
Triple-negative breast cancer
- TRAE:
-
Treatment-related adverse effect
- Treg:
-
Regulatory T cell
- VCAM-1:
-
Vascular cell adhesion molecule–1
- VEGF-A:
-
Vascular endothelial growth factor A
References
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375:1856–67.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34.
Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K, Chung HC, Chen JS, Muro K, Kang WK, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:2461–71.
Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv324.
Anandappa AJ, Wu CJ, Ott PA. Directing Traffic: How to Effectively Drive T Cells into Tumors. Cancer Discov. 2020;10:185–97.
Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med. 2018;378:2078–92.
Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, Hermes B, Cay Senler F, Csoszi T, Fulop A, et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med. 2018;379:2040–51.
Tallon de Lara P, Cecconi V, Hiltbrunner S, Yagita H, Friess M, Bode B, Opitz I, Vrugt B, Weder W, Stolzmann P, et al. Gemcitabine Synergizes with Immune Checkpoint Inhibitors and Overcomes Resistance in a Preclinical Model and Mesothelioma Patients. Clin Cancer Res. 2018;24:6345–54.
Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, Venugopal B, Kollmannsberger C, Negrier S, Uemura M, et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. 2019;380:1103–15.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10.
Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol. 2016;27:1492–504.
Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111.
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61.
Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med. 2017;377:2500–1.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13.
Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, Diaz LA Jr. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. 2019;16:361–75.
Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, Rosenberg SA. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33:828–33.
Yuan J, Zhou J, Dong Z, Tandon S, Kuk D, Panageas KS, Wong P, Wu X, Naidoo J, Page DB, et al. Pretreatment serum VEGF is associated with clinical response and overall survival in advanced melanoma patients treated with ipilimumab. Cancer Immunol Res. 2014;2:127–32.
Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39:61–73.
Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20:607–15.
Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16:356–71.
Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016;37:208–20.
DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19:369–82.
Teijeira Á, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, de Andrea C, Ochoa MC, Otano I, Etxeberria I, et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity. 2020;52:856–871.e858.
Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, Condeelis J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022–9.
Su S, Liu Q, Chen J, Chen J, Chen F, He C, Huang D, Wu W, Lin L, Huang W, et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell. 2014;25:605–20.
Huang D, Song SJ, Wu ZZ, Wu W, Cui XY, Chen JN, Zeng MS, Su SC. Epstein-Barr Virus-Induced VEGF and GM-CSF Drive Nasopharyngeal Carcinoma Metastasis via Recruitment and Activation of Macrophages. Cancer Res. 2017;77:3591–604.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416.
Sangaletti S, Di Carlo E, Gariboldi S, Miotti S, Cappetti B, Parenza M, Rumio C, Brekken RA, Chiodoni C, Colombo MP. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 2008;68:9050–9.
Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–52.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51.
Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, Kato Y, Li J, Pollard JW. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. 2015;212:1043–59.
Li X, Liu R, Su X, Pan Y, Han X, Shao C, Shi Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer. 2019;18:177.
Galluzzi L, Chan TA, Kroemer G, Wolchok JD, López-Soto A. The hallmarks of successful anticancer immunotherapy. Sci Transl Med. 2018;10.
Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol. 2019;16:601–20.
Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, Reis ESC. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell. 2018;172:1022–1037.e1014.
Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, Nelson AE, Loo K, Kumar R, Rosenblum MD, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med. 2018;24:1178–91.
Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, Majani Y, Gileadi T, Wallace R, Gustafsson K, et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol Ther. 2018;26:354–65.
Petitprez F, de Reyniès A, Keung EZ, Chen TW, Sun CM, Calderaro J, Jeng YM, Hsiao LP, Lacroix L, Bougoüin A, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577:556–60.
Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577:549–55.
Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561–5.
Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remedios C, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20:1301–9.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61.
Shi L, Lin H, Li G, Sun Y, Shen J, Xu J, Lin C, Yeh S, Cai X, Chang C. Cisplatin enhances NK cells immunotherapy efficacy to suppress HCC progression via altering the androgen receptor (AR)-ULBP2 signals. Cancer Lett. 2016;373:45–56.
Jiménez-Sánchez A, Cybulska P, Mager KL, Koplev S, Cast O, Couturier DL, Memon D, Selenica P, Nikolovski I, Mazaheri Y, et al. Unraveling tumor-immune heterogeneity in advanced ovarian cancer uncovers immunogenic effect of chemotherapy. Nat Genet. 2020;52:582–93.
Loyher PL, Rochefort J, Baudesson de Chanville C, Hamon P, Lescaille G, Bertolus C, Guillot-Delost M, Krummel MF, Lemoine FM, Combadiere C, Boissonnas A. CCR2 Influences T Regulatory Cell Migration to Tumors and Serves as a Biomarker of Cyclophosphamide Sensitivity. Cancer Res. 2016;76:6483–94.
Scurr M, Pembroke T, Bloom A, Roberts D, Thomson A, Smart K, Bridgeman H, Adams R, Brewster A, Jones R, et al. Low-Dose Cyclophosphamide Induces Antitumor T-Cell Responses, which Associate with Survival in Metastatic Colorectal Cancer. Clin Cancer Res. 2017;23:6771–80.
Hibino S, Chikuma S, Kondo T, Ito M, Nakatsukasa H, Omata-Mise S, Yoshimura A. Inhibition of Nr4a Receptors Enhances Antitumor Immunity by Breaking Treg-Mediated Immune Tolerance. Cancer Res. 2018;78:3027–40.
Gao Q, Wang S, Chen X, Cheng S, Zhang Z, Li F, Huang L, Yang Y, Zhou B, Yue D, et al. Cancer-cell-secreted CXCL11 promoted CD8(+) T cells infiltration through docetaxel-induced-release of HMGB1 in NSCLC. J Immunother Cancer. 2019;7:42.
Lu Y, Zhao Q, Liao JY, Song E, Xia Q, Pan J, Li Y, Li J, Zhou B, Ye Y, et al. Complement Signals Determine Opposite Effects of B Cells in Chemotherapy-Induced Immunity. Cell. 2020;180:1081–1097.e1024.
Kanterman J, Sade-Feldman M, Biton M, Ish-Shalom E, Lasry A, Goldshtein A, Hubert A, Baniyash M. Adverse immunoregulatory effects of 5FU and CPT11 chemotherapy on myeloid-derived suppressor cells and colorectal cancer outcomes. Cancer Res. 2014;74:6022–35.
Principe DR, Narbutis M, Kumar S, Park A, Viswakarma N, Dorman MJ, Kamath SD, Grippo PJ, Fishel ML, Hwang RF, et al. Long-Term Gemcitabine Treatment Reshapes the Pancreatic Tumor Microenvironment and Sensitizes Murine Carcinoma to Combination Immunotherapy. Cancer Res. 2020.
Di Caro G, Cortese N, Castino GF, Grizzi F, Gavazzi F, Ridolfi C, Capretti G, Mineri R, Todoric J, Zerbi A, et al. Dual prognostic significance of tumour-associated macrophages in human pancreatic adenocarcinoma treated or untreated with chemotherapy. Gut. 2016;65:1710–20.
Roselli M, Formica V, Cereda V, Jochems C, Richards J, Grenga I, Orlandi A, Ferroni P, Guadagni F, Schlom J. The association of clinical outcome and peripheral T-cell subsets in metastatic colorectal cancer patients receiving first-line FOLFIRI plus bevacizumab therapy. Oncoimmunology. 2016;5:e1188243.
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–8.
Garg AD, Elsen S, Krysko DV, Vandenabeele P, de Witte P, Agostinis P. Resistance to anticancer vaccination effect is controlled by a cancer cell-autonomous phenotype that disrupts immunogenic phagocytic removal. Oncotarget. 2015;6:26841–60.
Wanderley CW, Colon DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA, Pereira JA, Silva CM, Silva CR, Silva RL, et al. Paclitaxel Reduces Tumor Growth by Reprogramming Tumor-Associated Macrophages to an M1 Profile in a TLR4-Dependent Manner. Cancer Res. 2018;78:5891–900.
Cullis J, Siolas D, Avanzi A, Barui S, Maitra A, Bar-Sagi D. Macropinocytosis of Nab-paclitaxel Drives Macrophage Activation in Pancreatic Cancer. Cancer Immunol Res. 2017;5:182–90.
Schaer DA, Geeganage S, Amaladas N, Lu ZH, Rasmussen ER, Sonyi A, Chin D, Capen A, Li Y, Meyer CM, et al. The Folate Pathway Inhibitor Pemetrexed Pleiotropically Enhances Effects of Cancer Immunotherapy. Clin Cancer Res. 2019;25:7175–88.
Wang Z, Chen J, Hu J, Zhang H, Xu F, He W, Wang X, Li M, Lu W, Zeng G, et al. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. J Clin Invest. 2019;130:4850–62.
Zhang QF, Li J, Jiang K, Wang R, Ge JL, Yang H, Liu SJ, Jia LT, Wang L, Chen BL. CDK4/6 inhibition promotes immune infiltration in ovarian cancer and synergizes with PD-1 blockade in a B cell-dependent manner. Theranostics. 2020;10:10619–33.
Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, Hernandez G, Mier J, He X, Hodi FS, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624.
Erkes DA, Cai W, Sanchez IM, Purwin TJ, Rogers C, Field CO, Berger AC, Hartsough EJ, Rodeck U, Alnemri ES, Aplin AE. Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov. 2020;10:254–69.
Luna JI, Grossenbacher SK, Sturgill IR, Ames E, Judge SJ, Bouzid LA, Darrow MA, Murphy WJ, Canter RJ. Bortezomib Augments Natural Killer Cell Targeting of Stem-Like Tumor Cells. Cancers (Basel). 2019;11.
Pozzi C, Cuomo A, Spadoni I, Magni E, Silvola A, Conte A, Sigismund S, Ravenda PS, Bonaldi T, Zampino MG, et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat Med. 2016;22:624–31.
Glodde N, Bald T, van den Boorn-Konijnenberg D, Nakamura K, O'Donnell JS, Szczepanski S, Brandes M, Eickhoff S, Das I, Shridhar N, et al. Reactive Neutrophil Responses Dependent on the Receptor Tyrosine Kinase c-MET Limit Cancer Immunotherapy. Immunity. 2017;47:789–802.e789.
Liu P, Zhao L, Pol J, Levesque S, Petrazzuolo A, Pfirschke C, Engblom C, Rickelt S, Yamazaki T, Iribarren K, et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat Commun. 2019;10:1486.
Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, Gould SE, Maecker H, Irving BA, Kim JM, et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity. 2016;44:609–21.
Poon E, Mullins S, Watkins A, Williams GS, Koopmann JO, Di Genova G, Cumberbatch M, Veldman-Jones M, Grosskurth SE, Sah V, et al. The MEK inhibitor selumetinib complements CTLA-4 blockade by reprogramming the tumor immune microenvironment. J Immunother Cancer. 2017;5:63.
Pantelidou C, Sonzogni O, De Oliveria TM, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019;9:722–37.
Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, Cristea S, Nguyen T, Diao L, Li L, et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019;9:646–61.
Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, Yap TA, Mills GB, Peng G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019;79:311–9.
Zhang X, Shen L, Liu Q, Hou L, Huang L. Inhibiting PI3 kinase-γ in both myeloid and plasma cells remodels the suppressive tumor microenvironment in desmoplastic tumors. J Control Release. 2019;309:173–80.
Lowe DB, Bose A, Taylor JL, Tawbi H, Lin Y, Kirkwood JM, Storkus WJ. Dasatinib promotes the expansion of a therapeutically superior T-cell repertoire in response to dendritic cell vaccination against melanoma. Oncoimmunology. 2014;3:e27589.
Hekim C, Ilander M, Yan J, Michaud E, Smykla R, Vähä-Koskela M, Savola P, Tähtinen S, Saikko L, Hemminki A, et al. Dasatinib Changes Immune Cell Profiles Concomitant with Reduced Tumor Growth in Several Murine Solid Tumor Models. Cancer Immunol Res. 2017;5:157–69.
Patnaik A, Swanson KD, Csizmadia E, Solanki A, Landon-Brace N, Gehring MP, Helenius K, Olson BM, Pyzer AR, Wang LC, et al. Cabozantinib Eradicates Advanced Murine Prostate Cancer by Activating Antitumor Innate Immunity. Cancer Discov. 2017;7:750–65.
Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, Wood L, Elson P, Garcia J, Dreicer R, Bukowski R. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14:6674–82.
Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148–57.
Liu D, Li G, Avella DM, Kimchi ET, Kaifi JT, Rubinstein MP, Camp ER, Rockey DC, Schell TD, Staveley-O'Carroll KF. Sunitinib represses regulatory T cells to overcome immunotolerance in a murine model of hepatocellular cancer. Oncoimmunology. 2017;7:e1372079.
Li G, Liu D, Cooper TK, Kimchi ET, Qi X, Avella DM, Li N, Yang QX, Kester M, Rountree CB, et al. Successful chemoimmunotherapy against hepatocellular cancer in a novel murine model. J Hepatol. 2017;66:75–85.
Tada Y, Togashi Y, Kotani D, Kuwata T, Sato E, Kawazoe A, Doi T, Wada H, Nishikawa H, Shitara K. Targeting VEGFR2 with Ramucirumab strongly impacts effector/ activated regulatory T cells and CD8(+) T cells in the tumor microenvironment. J Immunother Cancer. 2018;6:106.
Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, Feyen K, Tawney J, Hanahan D, Michael IP, Bergers G. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med. 2017;9.
Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell. 2015;28:690–714.
Rufo N, Garg AD, Agostinis P. The Unfolded Protein Response in Immunogenic Cell Death and Cancer Immunotherapy. Trends Cancer. 2017;3:643–58.
Heinhuis KM, Ros W, Kok M, Steeghs N, Beijnen JH, Schellens JHM. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann Oncol. 2019;30:219–35.
Lu J, Liu X, Liao YP, Wang X, Ahmed A, Jiang W, Ji Y, Meng H, Nel AE. Breast Cancer Chemo-immunotherapy through Liposomal Delivery of an Immunogenic Cell Death Stimulus Plus Interference in the IDO-1 Pathway. ACS Nano. 2018;12:11041–61.
Mei L, Liu Y, Rao J, Tang X, Li M, Zhang Z, He Q. Enhanced Tumor Retention Effect by Click Chemistry for Improved Cancer Immunochemotherapy. ACS Appl Mater Interfaces. 2018;10:17582–93.
Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, Nederlof I, Kluin RJC, Warren S, Ong S, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med. 2019;25:920–8.
Song W, Shen L, Wang Y, Liu Q, Goodwin TJ, Li J, Dorosheva O, Liu T, Liu R, Huang L. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat Commun. 2018;9:2237.
Bansal S, Bajaj P, Pandey S, Tandon V. Topoisomerases: Resistance versus Sensitivity, How Far We Can Go? Med Res Rev. 2017;37:404–38.
Corrales L, McWhirter SM, Dubensky TW Jr, Gajewski TF. The host STING pathway at the interface of cancer and immunity. J Clin Invest. 2016;126:2404–11.
Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014;35:88–93.
Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, McCloskey KD, Buckley NE, Savage KI, Salto-Tellez M, et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J Natl Cancer Inst. 2017;109.
George A, Kaye S, Banerjee S. Delivering widespread BRCA testing and PARP inhibition to patients with ovarian cancer. Nat Rev Clin Oncol. 2017;14:284–96.
Gonzalez-Martin A, Pothuri B, Vergote I, DePont CR, Graybill W, Mirza MR, McCormick C, Lorusso D, Hoskins P, Freyer G, et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med. 2019;381:2391–402.
Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med. 2016;375:2154–64.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, Hsu JL, Yu WH, Du Y, Lee HH, et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res. 2017;23:3711–20.
Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, Van Le L, Sachdev JC, Chapman-Davis E, Colon-Otero G, et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019;5:1141–9.
Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, Wahner-Hendrickson AE, Forero A, Anders C, Wulf GM, et al. Open-Label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019;5:1132–40.
Färkkilä A, Gulhan DC, Casado J, Jacobson CA, Nguyen H, Kochupurakkal B, Maliga Z, Yapp C, Chen YA, Schapiro D, et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat Commun. 2020;11:1459.
Thomas A, Vilimas R, Trindade C, Erwin-Cohen R, Roper N, Xi L, Krishnasamy V, Levy E, Mammen A, Nichols S, et al. Durvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a Phase II Study. J Thorac Oncol. 2019;14:1447–57.
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8.
Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103.
Ma Y, Pitt JM, Li Q, Yang H. The renaissance of anti-neoplastic immunity from tumor cell demise. Immunol Rev. 2017;280:194–206.
Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, Junqueira C, Meza-Sosa KF, Mok TMY, Ansara J, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579:415–20.
Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, Pinheiro EM, Koya RC, Graeber TG, Comin-Anduix B, Ribas A. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med. 2015;7:279ra241.
Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH Jr, Carlino MS, Fisher R, Long GV, Hodi FS, Tsoi J, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med. 2019;25:936–40.
Ascierto PA, Ferrucci PF, Fisher R, Del Vecchio M, Atkinson V, Schmidt H, Schachter J, Queirolo P, Long GV, Di Giacomo AM, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med. 2019;25:941–6.
Lu H, Zhang S, Wu J, Chen M, Cai MC, Fu Y, Li W, Wang J, Zhao X, Yu Z, et al. Molecular Targeted Therapies Elicit Concurrent Apoptotic and GSDME-Dependent Pyroptotic Tumor Cell Death. Clin Cancer Res. 2018;24:6066–77.
Shigeta K, Datta M, Hato T, Kitahara S, Chen IX, Matsui A, Kikuchi H, Mamessier E, Aoki S, Ramjiawan RR, et al. Dual Programmed Death Receptor-1 and Vascular Endothelial Growth Factor Receptor-2 Blockade Promotes Vascular Normalization and Enhances Antitumor Immune Responses in Hepatocellular Carcinoma. Hepatology. 2020;71:1247–61.
Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, Kienast Y, Mueller HJ, Ooi CH, Laoui D, De Palma M. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med. 2017;9.
Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell. 2017;169:1342–1356.e1316.
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, Kudo M, Breder V, Merle P, Kaseb AO, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382:1894–905.
Rini BI, Powles T, Atkins MB, Escudier B, McDermott DF, Suarez C, Bracarda S, Stadler WM, Donskov F, Lee JL, et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open-label, phase 3, randomised controlled trial. Lancet. 2019;393:2404–15.
Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851–60.
Infante JR, Camidge DR, Mileshkin LR, Chen EX, Hicks RJ, Rischin D, Fingert H, Pierce KJ, Xu H, Roberts WG, et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J Clin Oncol. 2012;30:1527–33.
Dimeloe S, Frick C, Fischer M, Gubser PM, Razik L, Bantug GR, Ravon M, Langenkamp A, Hess C. Human regulatory T cells lack the cyclophosphamide-extruding transporter ABCB1 and are more susceptible to cyclophosphamide-induced apoptosis. Eur J Immunol. 2014;44:3614–20.
Zhao J, Cao Y, Lei Z, Yang Z, Zhang B, Huang B. Selective depletion of CD4+CD25+Foxp3+ regulatory T cells by low-dose cyclophosphamide is explained by reduced intracellular ATP levels. Cancer Res. 2010;70:4850–8.
Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34:336–44.
Noordam L, Kaijen MEH, Bezemer K, Cornelissen R, Maat L, Hoogsteden HC, Aerts J, Hendriks RW, Hegmans J, Vroman H. Low-dose cyclophosphamide depletes circulating naïve and activated regulatory T cells in malignant pleural mesothelioma patients synergistically treated with dendritic cell-based immunotherapy. Oncoimmunology. 2018;7:e1474318.
Roskoski R Jr. Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun. 2007;356:323–8.
Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY, Chen SH. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–22.
Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, Dubreuil O, Carpentier AF, Tartour E, Taieb JJCR. VEGFA-VEGFR Pathway Blockade Inhibits Tumor-Induced Regulatory T-cell Proliferation in Colorectal Cancer. Cancer Res. 2013;73:539–49.
Draghiciu O, Nijman HW, Hoogeboom BN, Meijerhof T, Daemen T. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology. 2015;4:e989764.
Chen HM, Ma G, Gildener-Leapman N, Eisenstein S, Coakley BA, Ozao J, Mandeli J, Divino C, Schwartz M, Sung M, et al. Myeloid-Derived Suppressor Cells as an Immune Parameter in Patients with Concurrent Sunitinib and Stereotactic Body Radiotherapy. Clin Cancer Res. 2015;21:4073–85.
DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67.
Pusztai L, Mendoza TR, Reuben JM, Martinez MM, Willey JS, Lara J, Syed A, Fritsche HA, Bruera E, Booser D, et al. Changes in plasma levels of inflammatory cytokines in response to paclitaxel chemotherapy. Cytokine. 2004;25:94–102.
Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, Sheng K, Niu Y, Goldstein A, Zhao N, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol. 2019;21:1113–26.
Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im SA, Shaw Wright G, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med. 2018;379:2108–21.
Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, Huemer F, Losonczy G, Johnson ML, Nishio M, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med. 2018;379:2220–9.
Jotte R, Cappuzzo F, Vynnychenko I, Stroyakovskiy D, Rodríguez-Abreu D, Hussein M, Soo R, Conter HJ, Kozuki T, Huang KC, et al. Atezolizumab in Combination With Carboplatin and Nab-Paclitaxel in Advanced Squamous Non-Small-Cell Lung Cancer (IMpower131): Results From a Randomized Phase III Trial. J Thorac Oncol. 2020.
West H, McCleod M, Hussein M, Morabito A, Rittmeyer A, Conter HJ, Kopp HG, Daniel D, McCune S, Mekhail T, et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20:924–37.
Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, Rodríguez-Abreu D, Moro-Sibilot D, Thomas CA, Barlesi F, et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med. 2018;378:2288–301.
Adams S, Diamond JR, Hamilton E, Pohlmann PR, Tolaney SM, Chang CW, Zhang W, Iizuka K, Foster PG, Molinero L, et al. Atezolizumab Plus nab-Paclitaxel in the Treatment of Metastatic Triple-Negative Breast Cancer With 2-Year Survival Follow-up: A Phase 1b Clinical Trial. JAMA Oncol. 2019;5:334–42.
Sullivan RJ, Hamid O, Gonzalez R, Infante JR, Patel MR, Hodi FS, Lewis KD, Tawbi HA, Hernandez G, Wongchenko MJ, et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat Med. 2019;25:929–35.
McArthur GA. Evaluation of atezolizumab (A), cobimetinib (C), and vemurafenib (V) in previously untreated patients with BRAFV600 mutation-positive advanced melanoma: Primary results from the phase 3 IMspire150 trial: AACR 2020 Session VCTPL01 - Opening Clinical Plenary CT012.
Nie J, Wang C, Liu Y, Yang Q, Mei Q, Dong L, Li X, Liu J, Ku W, Zhang Y, et al. Addition of Low-Dose Decitabine to Anti-PD-1 Antibody Camrelizumab in Relapsed/Refractory Classical Hodgkin Lymphoma. J Clin Oncol. 2019;37:1479–89.
Fang W, Yang Y, Ma Y, Hong S, Lin L, He X, Xiong J, Li P, Zhao H, Huang Y, et al. Camrelizumab (SHR-1210) alone or in combination with gemcitabine plus cisplatin for nasopharyngeal carcinoma: results from two single-arm, phase 1 trials. Lancet Oncol. 2018;19:1338–50.
Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, Statsenko G, Hochmair MJ, Özgüroğlu M, Ji JH, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet. 2019;394:1929–39.
Arriola E, Wheater M, Galea I, Cross N, Maishman T, Hamid D, Stanton L, Cave J, Geldart T, Mulatero C, et al. Outcome and Biomarker Analysis from a Multicenter Phase 2 Study of Ipilimumab in Combination with Carboplatin and Etoposide as First-Line Therapy for Extensive-Stage SCLC. J Thorac Oncol. 2016;11:1511–21.
Reck M, Luft A, Szczesna A, Havel L, Kim SW, Akerley W, Pietanza MC, Wu YL, Zielinski C, Thomas M, et al. Phase III Randomized Trial of Ipilimumab Plus Etoposide and Platinum Versus Placebo Plus Etoposide and Platinum in Extensive-Stage Small-Cell Lung Cancer. J Clin Oncol. 2016;34:3740–8.
Reck M, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, Sebastian M, Lu H, Cuillerot JM, Lynch TJ. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol. 2013;24:75–83.
Govindan R, Szczesna A, Ahn MJ, Schneider CP, Gonzalez Mella PF, Barlesi F, Han B, Ganea DE, Von Pawel J, Vladimirov V, et al. Phase III Trial of Ipilimumab Combined With Paclitaxel and Carboplatin in Advanced Squamous Non-Small-Cell Lung Cancer. J Clin Oncol. 2017;35:3449–57.
Rizvi NA, Hellmann MD, Brahmer JR, Juergens RA, Borghaei H, Gettinger S, Chow LQ, Gerber DE, Laurie SA, Goldman JW, et al. Nivolumab in Combination With Platinum-Based Doublet Chemotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2016;34:2969–79.
Gettinger S, Hellmann MD, Chow LQM, Borghaei H, Antonia S, Brahmer JR, Goldman JW, Gerber DE, Juergens RA, Shepherd FA, et al. Nivolumab Plus Erlotinib in Patients With EGFR-Mutant Advanced NSCLC. J Thorac Oncol. 2018;13:1363–72.
Spigel DR, Reynolds C, Waterhouse D, Garon EB, Chandler J, Babu S, Thurmes P, Spira A, Jotte R, Zhu J, et al. Phase 1/2 Study of the Safety and Tolerability of Nivolumab Plus Crizotinib for the First-Line Treatment of Anaplastic Lymphoma Kinase Translocation - Positive Advanced Non-Small Cell Lung Cancer (CheckMate 370). J Thorac Oncol. 2018;13:682–8.
Boku N, Ryu MH, Kato K, Chung HC, Minashi K, Lee KW, Cho H, Kang WK, Komatsu Y, Tsuda M, et al. Safety and efficacy of nivolumab in combination with S-1/capecitabine plus oxaliplatin in patients with previously untreated, unresectable, advanced, or recurrent gastric/gastroesophageal junction cancer: interim results of a randomized, phase II trial (ATTRACTION-4). Ann Oncol. 2019;30:250–8.
Amin A, Plimack ER, Ernstoff MS, Lewis LD, Bauer TM, McDermott DF, Carducci M, Kollmannsberger C, Rini BI, Heng DYC, et al. Safety and efficacy of nivolumab in combination with sunitinib or pazopanib in advanced or metastatic renal cell carcinoma: the CheckMate 016 study. J Immunother Cancer. 2018;6:109.
Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, Gentzler RD, Martins RG, Stevenson JP, Jalal SI, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016;17:1497–508.
Toulmonde M, Penel N, Adam J, Chevreau C, Blay JY, Le Cesne A, Bompas E, Piperno-Neumann S, Cousin S, Grellety T, et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas: A Phase 2 Clinical Trial. JAMA Oncol. 2018;4:93–7.
Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, Denkert C, Park YH, Hui R, Harbeck N, et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N Engl J Med. 2020;382:810–21.
Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de Castro G Jr, Psyrri A, Basté N, Neupane P, Bratland Å, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019;394:1915–28.
Atkins MB, Plimack ER, Puzanov I, Fishman MN, McDermott DF, Cho DC, Vaishampayan U, George S, Olencki TE, Tarazi JC, et al. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: a non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. Lancet Oncol. 2018;19:405–15.
Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, Pouliot F, Alekseev B, Soulières D, Melichar B, et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. 2019;380:1116–27.
Sheng X, Yan X, Chi Z, Si L, Cui C, Tang B, Li S, Mao L, Lian B, Wang X, et al. Axitinib in Combination With Toripalimab, a Humanized Immunoglobulin G(4) Monoclonal Antibody Against Programmed Cell Death-1, in Patients With Metastatic Mucosal Melanoma: An Open-Label Phase IB Trial. J Clin Oncol. 2019;37:2987–99.
Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–6.
Iida Y, Harashima N, Motoshima T, Komohara Y, Eto M, Harada M. Contrasting effects of cyclophosphamide on anti-CTL-associated protein 4 blockade therapy in two mouse tumor models. Cancer Sci. 2017;108:1974–84.
Deken MA, Gadiot J, Jordanova ES, Lacroix R, van Gool M, Kroon P, Pineda C, Geukes Foppen MH, Scolyer R, Song JY, et al. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. Oncoimmunology. 2016;5:e1238557.
Rozeman EA, Deken MA, Gadiot J, Geukes Foppen MH, Pronk L, Thienen JV, Haanen JB, Blank CU. Phase II study comparing pembrolizumab (PEM) with intermittent/short‐term dual MAPK pathway inhibition plus PEM in patients harboring the BRAFV600 mutation (IMPemBra). Ann Oncol. 2018;29:mdy424.056.
Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, Vanpouille-Box C, Friedman K, Ferrari de Andrade L, Wucherpfennig KW, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med. 2018;24:1845–51.
McLaughlin M, Patin EC, Pedersen M, Wilkins A, Dillon MT, Melcher AA, Harrington KJ. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer. 2020;20:203–17.
Dillon MT, Bergerhoff KF, Pedersen M, Whittock H, Crespo-Rodriguez E, Patin EC, Pearson A, Smith HG, Paget JTE, Patel RR, et al. ATR Inhibition Potentiates the Radiation-induced Inflammatory Tumor Microenvironment. Clin Cancer Res. 2019;25:3392–403.
Acknowledgments
Not applicable.
Author information
Authors and Affiliations
Contributions
Jun-Yan Li, Yu-Pei Chen, Ying-Qin Li, Na Liu, and Jun Ma were involved in the conception of the manuscript. Jun-Yan Li, Yu-Pei Chen, Ying-Qin Li, and Na Liu wrote and reviewed the original manuscript. Jun-Yan Li, Yu-Pei Chen, Ying-Qin Li, Na Liu, and Jun Ma revised the manuscript. Jun-Yan Li, Yu-Pei Chen, Ying-Qin Li, Na Liu, and Jun Ma approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have declared no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, JY., Chen, YP., Li, YQ. et al. Chemotherapeutic and targeted agents can modulate the tumor microenvironment and increase the efficacy of immune checkpoint blockades. Mol Cancer 20, 27 (2021). https://doi.org/10.1186/s12943-021-01317-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-021-01317-7