
Abstract
Epithelial-mesenchymal transition (EMT) and cancer stem-like cells (CSC) are becoming highly relevant targets in anticancer drug discovery. A large body of evidence suggests that epithelial-mesenchymal transitioned tumor cells (EMT tumor cells) and CSCs have similar functions. There is also an overlap regarding the stimuli that can induce the generation of EMT tumor cells and CSCs. Moreover, direct evidence has been brought that EMT can give rise to CSCs. It is unclear however, whether EMT tumor cells should be considered CSCs or if they have to undergo further changes. In this article we summarize available evidence suggesting that, indeed, additional programs must be engaged and we propose that macroautophagy (hereafter, autophagy) represents a key trait distinguishing CSCs from EMT tumor cells. Thus, CSCs have often been reported to be in an autophagic state and blockade of autophagy inhibits CSCs. On the other hand, there is ample evidence showing that EMT and autophagy are distinct events. CSCs, however, represent, by themselves, a heterogeneous population. Thus, CSCs have been distinguished in predominantly non-cycling and cycling CSCs, the latter representing CSCs that self-renew and replenish the pool of differentiated tumor cells. We now suggest that the non-cycling CSC subpopulation is in an autophagic state. We propose also two models to explain the relationship between EMT tumor cells and these two major CSC subpopulations: a branching model in which EMT tumor cells can give rise to cycling or non-cycling CSCs, respectively, and a hierarchical model in which EMT tumor cells are first induced to become autophagic CSCs and, subsequently, cycling CSCs. Finally, we address the therapeutic consequences of these insights.
Similar content being viewed by others


Background: Epithelial-mesenchymal transition and cancer stem-like cells
Epithelial-mesenchymal transition (EMT), i.e., the conversion of cells with an epithelial phenotype into cells with a mesenchymal phenotype [1, 2] involves changes that lead to loss of cell–cell adhesion and cell polarity. EMT is critical for embryonic development. In adults it occurs during wound healing, tissue regeneration, organ fibrosis, and tumor progression. Epithelial-mesenchymal transitioned tumor cells (EMT tumor cells) have been reported to possess increased motility and invasiveness, tumor-propagating potential, and resistance to apoptosis and antitumor drugs [3, 4].
CSCs are a subpopulation of tumor cells that have high tumor-propagating potential [5], enhanced metastasis-forming potential [6] and are resistant to antitumor drugs [7]. There is a large overlap in the characteristics of EMT tumor cells and CSCs also as regards the stimuli that can induce the generation of EMT tumor cells and CSCs. Thus, both are the result of two main events. The first is the genetic and epigenetic instability of tumor cells [8–12]. The second event is represented by stimuli from the tumor microenvironment (TME) that promote a cross-talk between different cell types within the TME and that is largely effected by paracrine factors that are released in response to the stimuli and interact with their corresponding receptors on tumor cells [13–15]. Ligand-receptor pairs like hepatocyte growth factor/c-MET, transforming growth factor (TGF)-β/TGF-β receptor, interleukin-6 (IL-6)/IL-6 receptor, platelet-derived growth factor (PDGF)/PDGF receptor (PDGFR), epidermal growth factor (EGF)/EGF receptor, fibroblast growth factor (FGF)/FGF receptor (FGFR), Gas6/AXL, WNT/Frizzled, Hedgehog/Smoothened and Notch ligands/Notch are examples of paracrine factors and receptors that have been shown to promote the induction of both EMT and CSCs [7, 15–20]. Eventually, direct evidence has been brought showing that EMT can give rise to CSCs. Thus, induction of EMT in immortalized human mammary epithelial cells through the ectopic expression of EMT-promoting transcription factors resulted in the acquisition of mesenchymal traits and expression of stem-cell markers [21]. These cells had an increased ability to form mammospheres, a property associated with epithelial stem cells [21]. Further demonstrating the close linkage between EMT and CSCs, it was shown that down-regulation of the receptor tyrosine kinase AXL reversed EMT in human epithelial cells and breast CSCs attenuating self-renewal and restoring chemosensitivity of breast CSCs [16].
Given the multiplicity of genetic and environmental stimuli that are at the origin of EMT and CSCs, it is not surprising that a large number of overlapping intracellular signaling pathways have been reported to be involved in the induction of both. Intracellular signaling hubs like focal adhesion kinase and SRC, pathways like phosphoinositide 3-kinase–AKT–mechanistic target of rapamycin, RAF-RAS-mitogen-activated protein kinase, transcription factors like small mother against decapentaplegic (SMAD), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), signal transducer and activator of transcription (STAT) 3, and reactive oxygen species have been shown being involved in the induction of EMT and CSCs [7, 15, 22–26].
Given the great similarity between the functions, inducing stimuli and intracellular signaling pathways of EMT tumor cells and CSCs, and the observation that EMT can give rise to CSCs, one is led to ask whether EMT tumor cells are identical to CSCs. Answering this question has considerable translational relevance, because EMT and CSCs have become important targets in drug discovery and several anti-EMT and anti-CSC compounds are now in active clinical development [15, 27].
Main text
Differences between EMT tumor cells and CSCs
While EMT tumor cells and CSCs are induced by similar stimuli and apparently discharge similar tasks, there is now considerable evidence suggesting that the two cell types are distinct and may represent different stages of a tumor cell dedifferentiation process. Thus, salinomycin, a natural, fused polypyran ionophore, is a widely used anti-coccidiosis agent. It was found to have anti-CSC activity in a chemical screen designed to discover compounds toxic for breast CSCs [28]. In head and neck squamous cell carcinoma stem cells, salinomycin significantly inhibited sphere forming-capability, repressed the expression of CSC markers and reduced invasiveness of CSCs [29]. On the other hand, salinomycin induced the expression of EMT markers and decreased expression of E-cadherin, a hallmark trait of epithelial tumor cells. Thus, salinomycin appears to inhibit CSCs while, at the same time, promoting EMT. Unfortunately, several different mechanisms of action have been ascribed to salinomycin and, therefore, no conclusion can be drawn as to the signaling pathway(s) or factor(s) dictating this switch.
Other work has shown that the homeobox transcription factor paired related homeobox 1 (Prrx1) can be such a switch factor. Prrx1 induced EMT in cooperation with the other transcription factor Twist1, conferring migratory and invasive properties [30]. Loss of Prrx1, on the other hand, was required for cancer cells to metastasize and acquire CSC properties and markers. Importantly, this article showed that, contrarily to common knowledge, functions of EMT tumor cells and CSCs are not coincident. Rather, EMT tumor cells were shown to have migratory and invasive properties, while CSCs metastasized.
It has also been reported that tumor cell subpopulations expressing a strong epithelial gene program are enriched in highly metastatic CSCs, while subpopulations with stable mesenchymal traits (i.e. EMT tumor cells) are impoverished in CSCs, confirming that EMT tumor cells and CSCs are functionally and phenotypically separate entities [31]. The same study showed that both tumor cell subpopulations cooperate so that nonmetastatic cells promote the escape of metastatic cells for metastatic colonization. These results, as well as the previous ones, while suggesting that EMT tumor cells and CSCs are indeed separate entities, do not exclude the possibility that they are developmentally related.
A further turn of complexity to this picture was introduced by the observation that CSCs themselves can undergo an EMT [32]. In fact, in cell lines derived from oral and skin carcinomas, EMT occurred within the CD44high CSC fraction resulting in two CSC phenotypes, one predominantly epithelial with high expression of epithelial specific antigen (ESA), and another with EMT tumor cell characteristics and low expression of ESA. CSCs could switch between these two phenotypes with EMT tumor cells being relatively quiescent [33].
A dichotomy between EMT tumor cells and CSCs has also been confirmed in samples of different subtypes of breast cancers from tumor patients [34]. A method for scoring transcriptomic EMT signatures in different types of cancer showed that tumors of predominantly mesenchymal phenotype do not always show resistance to chemotherapy and suggested that it is the CSC phenotype, rather than the EMT phenotype that engenders drug resistance [35].
Overall, there is now considerable evidence that EMT tumor cells and CSCs are neither phenotypically nor functionally identical. Some of the reports that have been discussed suggest even that EMT tumor cells and CSCs are two (de)differentiation pathways that can cross each other, but are, nevertheless, distinct. This is in apparent contrast with the commonly held view that EMT can lead to the generation of CSCs and that EMT tumor cells and CSCs exert largely overlapping tasks.
Autophagy as a key trait that distinguishes CSCs from EMT tumor cells
In addition to the differences that have been discussed above, we propose that also macroautophagy (herein, autophagy) [36] is a key trait that distinguishes CSCs from EMT tumor cells. Autophagy is an adaptive catabolic process of cells that stop dividing and enter quiescence, and occurs in response to different forms of environmental stress, including nutrient deprivation, growth factor depletion, and hypoxia [36]. Autophagy involves the delivery of cytoplasmic cargoes sequestered inside double-membrane vesicles to lysosomes. Autophagosomes are then formed and this is where the captured material is degraded. This process of self-digestion provides nutrients to maintain vital cellular functions during fasting and other forms of stress. Autophagy has a suppressor role in initial steps of tumorigenesis, but has a prosurvival effect in established tumors by allowing tumor cells to cope with environmental and therapy-induced stress [36].
CSCs have often been reported to be in an autophagic state [37–39] and blockade of autophagy reduces their activity [40] and sensitizes them to antitumor drugs [41]. Moreover, one of the most commonly used markers for CSCs of several tumor types, CD133, promotes the autophagocytic activity of hepatoma CSCs [42], suggesting a functional link between CSCs and autophagy.
On the other hand, there is ample evidence that EMT and autophagy are distinct and even mutually exclusive events. Thus, autophagy induction impaired migration and invasion by inhibiting EMT in glioblastoma cells through down-regulation of the EMT-promoting transcription factors Snail and Slug [43]. Vice versa, silencing autophagy-inducing proteins restored the mesenchymal phenotype [43]. The Aurora kinase A inhibitor alisertib induced cell cycle arrest and autophagy and suppressed EMT in human pancreatic cancer cells [44]. Vice versa, Aurora kinase A suppressed autophagy and autophagic cell death by activating mechanistic in mTOR signaling in breast cancer cells [45]. Induction of EMT and metastasis upon inhibition of autophagy was observed also in gastric cancer cells [46].
In several instances a molecular cross-talk between the two pathways has been demonstrated to dictate the choice between EMT and autophagy. Thus, suppression of autophagy was shown to promote tumor growth and metastasis through stabilization of the EMT-promoting transcription factor Twist 1 by the selective autophagy substrate p62 that accumulated due to autophagy inhibition [47]. Similarly, autophagy inhibition by EMT in a p62-dependent manner has been reported in normal hepatocytes [48]. In metastatic breast cancer cells, the death-effector domain-containing DNA-binding protein (DEDD) interacted with class III phosphatidylinositol 3-kinase (PI3KC3) to activate autophagy and attenuated EMT [49]. DEDD physically interacted with PI3KC3 and this led to its stabilization and activation, and autophagic degradation of the EMT-promoting transcription factors Snail and Twist.
Altogether, these results suggest that EMT tumor cells and autophagic CSCs are distinct states of dedifferentiation that ensue in response to similar stimuli. Autophagic tumor cells, however, can induce other tumor cells to undergo EMT upon release of EMT-inducing paracrine factors [50, 51]. We suggest this to be a positive feed-back mechanism whereby autophagic CSCs release danger signals (i.e. paracrine factors) that induce an increasing number of tumor cells to enter a state, EMT, allowing them to cope with stress conditions in the TME. This view is consistent with the knowledge that EMT is the result of a cross-talk between tumor cells and tumor-associated cells and is mediated by the release of paracrine factors [14].
While CSCs can be in an autophagic state that appears to be mutually exclusive with that of EMT tumor cells, one main function of CSCs is to self-renew and to differentiate into mature tumor cells. This process implies active proliferation of CSCs. When cells undergo autophagy, however, they stop dividing and become quiescent, a condition opposite to the previous one.
Proliferating and autophagic CSCs
There is now increasing evidence that CSCs are themselves heterogeneous [52–54]. Phenotypic [55] and functional [56] heterogeneity of CSCs has been described. These characteristics can change over time [57] and differences between CSC subpopulations are not strictly qualitative but, rather, quantitative [58]. These observations suggest that different CSC subpopulations can switch from one to another in a dynamic manner [58].
Of particular relevance in the present context is the observation that CSCs can be distinguished in predominantly non-cycling CSCs and cycling CSCs [54, 58–60]. This is reminiscent of the proliferating and dormant subpopulations of somatic stem cells, with the dormant stem cell pool representing the most primitive stem cells [55]. Cycling CSCs are associated with cytokine production and cytokine receptor expression and this may be causally related with their replicative potential [61].
We propose that autophagic CSCs correspond to the non-cycling CSC subpopulation. To this regard, in squamous cell carcinoma, two CSC subpopulations have been identified on the basis of their capacity to promote tumor growth or invasion and metastasis, respectively. FGFR 1 inhibition reduced tumor growth without blocking metastasis, whereas PDGFR α inhibition reduced invasion and metastasis, but not tumor growth [62]. PDGFR signaling had been previously shown to induce formation of CSCs from non-CSCs [63]. Interestingly, PDGFR signaling has been reported to be an essential promoter of hypoxia-induced autophagy in tumor cells by prolonging the half-life of hypoxia-inducible factor-1α [64]. This lends support to our view that non-cycling CSCs are in an autophagic state.
In squamous cell carcinoma CSCs the choice between cycling and non-cycling state is induced by TGF-β, which bestows the properties of the non-cycling subpopulation [65]. While non-responding CSCs proliferated faster and accelerated tumor growth, TGF-β-responding progenies invaded and showed increased protection against anti-cancer drugs.
Regarding the functions of cycling and non-cycling CSCs, we have already referred to reports suggesting that the cycling subpopulation accelerates tumor growth, while the non-cycling, possibly autophagic one, promotes invasion, migration and metastasis [60, 62, 65]. Several other reports are in accordance with these results [7, 33, 66], some suggesting that the cycling subpopulation has a predominantly epithelial phenotype in contrast to the predominantly mesenchymal phenotype of the non-cycling subpopulation [33, 60]. Moreover, mesenchymal-like breast CSCs were characterized as CD24−CD44+, primarily quiescent and located at the tumor-invasive front, whereas epithelial-like CSCs expressed aldehyde dehydrogenase, proliferated and were located more centrally within tumors [60].
Regarding the relationship between cycling and non-cycling CSCs, we propose that CSCs develop into one or the other phenotype depending on stimuli from the TME. This view is supported by the observation that CSCs with predominantly epithelial phenotype relied mainly on oxygen metabolism, whereas predominantly mesenchymal CSCs showed decreased mitochondrial mass and membrane potential, consumed less oxygen per cell and produced markedly reduced levels of reactive oxygen species, suggesting that this subpopulation relied mainly on glycolysis for energy production [32]. Importantly, the shift towards a mesenchymal phenotype was induced by stimuli from the TME like hypoxia or tumor necrosis factor. These results are of interest because CSC heterogeneity may well explain the contradictory results on CSC metabolism, with several reports showing oxidative phosphorylation as the predominant energy source, while several others show that CSCs rely on glycolysis for energy production (see, for example, Refs. [67, 68]).
From EMT tumor cells to CSCs: What roads are taken?
As we have already discussed, it has been shown that EMT can give rise to CSCs [6, 21]. EMT tumor cells and CSCs have also been reported to have similar functions, although some results discussed above contrast this view [30, 31]. Moreover, most recent evidence has put into question that EMT tumor cells can metastasize [69, 70]. Yet, if we accept the view that there are two major subpopulations of CSCs, a cycling and a non-cycling one, then we have to address the issue as to which is the relationship between EMT tumor cells and these CSC subpopulations, and between the CSC subpopulations themselves. We have proposed that cycling and non-cycling CSCs are the result of different classes of cues from the TME. In support, it has been demonstrated that CSCs may display a background of genetic instability that is similar to that of differentiated tumor cells suggesting that, at least in some cases, environmental cues may play the predominant, if not the sole role in giving rise to CSCs [71]. This does not exclude, however, that in other instances stimuli from the TME may interact with a background of genetic instability. On the other hand, we have not addressed the issue as to whether the two subpopulations represent two consecutive steps of a single developmental pathway or if they are the result of two different developmental pathways. Moreover, before illustrating possible models that can accommodate acquired knowledge, it is important to mention that both EMT tumor cells as well as CSCs can derive from and can revert back to differentiated tumor cells [72, 73].
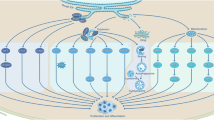
Given that knowledge, we propose two models that can explain the relationship between EMT and the two main CSC subpopulations, non-cycling, autophagic CSCs and cycling CSCs. We refer to the first model as the branching model (Fig. 1a). In this model, EMT tumor cells give rise to the cycling CSC compartment in response to paracrine factors within a niche of the TME that is conducive to the development of this CSC subpopulation. Alternatively, EMT tumor cells give rise to the non-cycling, autophagic CSC compartment in response to environmental cues that are represented by stress conditions such as nutrient shortage, mechanical stress, hypoxia etc. It is possible that this is a default pathway that occurs in the absence of paracrine factors. EMT tumor cells that are not diverted into one of the two pathways may remain in their state or may even revert back to differentiated tumor cells.

Two Models for EMT Tumor Cells Giving Rise to CSCs. a. Branching Model. In this model EMT tumor cells give rise to the cycling CSC compartment in response to paracrine factors within a niche in the TME that is conducive to the development of this CSC subpopulation. Alternatively, EMT tumor cells give rise to the non-cycling, autophagic CSC compartment in response to stress conditions such as nutrient shortage, mechanical stress, hypoxia etc. It is possible that this is a default pathway occurring in the absence of paracrine factors. b. Hierarchical Model. Here, EMT tumor cells are induced to become autophagic CSCs in response to stress conditions in the TME. Once these stimuli are relieved and replaced or complemented by paracrine factors, autophagic CSCs become cycling CSCs that self-renew and replenish the pool of differentiated tumor cells. CSC, cancer stem-like cell; EMT, epithelial-mesenchymal transition; TME, tumor microenvironment
The second, hierarchical, model (Fig. 1b) is more traditional and has already been proposed for both somatic stem cells as well as CSCs [55, 73]. Here, EMT tumor cells are induced to become autophagic CSCs in response to cues from the TME. Once these stimuli are relieved and/or are replaced by paracrine factors, autophagic CSCs become cycling (progenitor) CSCs that self-renew and replenish the pool of differentiated tumor cells. Both models foresee the possibility of bidirectional conversion, i.e. from EMT tumor cells to CSCs and vice versa [74]. The latter model is more consistent with a traditional view of stem cell and CSC development and differentiation, while the former is better apt to explain the consequences of regional differences of the TME in giving rise to one or the other CSC subpopulation [60].
Conclusions
In this article we reviewed the differences between EMT tumor cells and CSCs and proposed two models to explain the cross-talk between EMT tumor cells and two main CSC subpopulations, one non-cycling and autophagic, the other cycling. It is likely, however, that the differences between differentiated tumor cells, EMT tumor cells, and CSC subpopulations are not strict. Thus, it has been demonstrated that EMT tumor cells can exist in different transitions states, from cells with a predominantly epithelial phenotype to cells with a predominantly mesenchymal phenotype [75]. Similarly, it is conceivable that EMT tumor cells acquire characteristics of autophagic CSCs (i.e. activation of autophagy-related gene products) while not entirely losing those of EMT tumor cells, and reacquire predominantly epithelial characteristics when they develop into cycling CSCs. The phenotypic heterogeneity of CSCs supports this possibility [55]. The existence of phenotypic transition states would also imply functional transition states as regards the capacity to invade, migrate and metastasize, resist to apoptosis and antitumor drugs, or to self-renew and differentiate into mature tumor cells.
One key aspect that remains to be addressed concerns the therapeutic consequences deriving from these insights. Given the heterogeneity of the tumor cell population that encompasses differentiated tumor cells, EMT tumor cells, and CSC subpopulations, it appears unlikely that tumor eradication can be achieved without targeting each of these different tumor cell populations. We ignore, however, whether cytotoxic drugs are equally active on differentiated tumor cells and on cycling CSCs. Moreover, we don’t know whether anti-CSC compounds that are in active clinical development [27], including monoclonal antibodies against putative CSC markers [76–78], are equally active on cycling and non-cycling CSCs. Likewise, we don’t know whether compounds that are potentially active on both EMT tumor cells and CSCs are equally active on both populations and on CSC subpopulations. Progress in these directions is warranted. Nevertheless, available knowledge has allowed for the preclinical testing of combination therapies that target some of these tumor cell populations. For instance, while curcumin has antitumor effects but, at the same time, promotes the development of autophagic CSCs, these cells could be depleted by targeting a CSC marker [79]. The combination of a chemotherapeutic drug (temozolomide) targeting cycling tumor cells with a drug targeting quiescent tumor cells has yielded promising results in a genetically engineered mouse model of glioblastoma [80]. Combination of a glycolytic inhibitor targeting glioblastoma CSCs and the cytotoxic drug carmustine significantly impaired the sphere-forming ability of glioblastoma CSCs in vitro and tumor formation in vivo, leading to increase in the overall survival of mice bearing orthotopic inoculation of glioblastoma CSCs [81]. Other similar approaches of combination therapies targeting different tumor cell populations, including CSCs, have been reported [53]. Eventually, as regards autophagic CSCs, it appears logical to test compounds that inhibit autophagy or induce autophagic cell death in combination with drugs that target the cycling tumor cell compartment (mature tumor cells and cycling CSCs). Compounds that inhibit autophagy and that are being tested as anti-CSC compounds in clinical studies are chloroquine or hydroxychloroquine [27, 82, 83], while other compounds of this class are in earlier stages of development [84].
Further preclinical and, eventually, clinical testing of these or forthcoming combination therapies will tell us if our increasing knowledge of EMT and CSC biology can translate into improved therapeutic efficacy.
Abbreviations
- CSC:
-
Cancer stem-like cell
- DEDD:
-
Domain-containing DNA-binding protein
- EMT:
-
Epithelial-mesenchymal transition
- EMT tumor cells:
-
Epithelial-mesenchymal transitioned tumor cell
- ESA:
-
Epithelial specific antigen
- mTOR:
-
Mammalian target of rapamycin
- PDGFR:
-
Platelet-derived growth factor receptor
- PI3KC3:
-
Class III phosphatidylinositol 3-kinase
- Prrx1:
-
Paired related homeobox 1
- TME:
-
Tumor microenvironment
References
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90.
Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009;119:1417–9.
Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–73.
Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–51.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11.
Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–23.
Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells – what challenges do they pose? Nat Rev Drug Discov. 2014;13:497–512.
Avgustinova A, Benitah SA. The epigenetics of tumour initiation: cancer stem cells and their chromatin. Curr Opin Genet Dev. 2016;36:8–15.
Shukla S, Meeran SM. Epigenetics of cancer stem cells: Pathways and therapeutics. Biochim Biophys Acta. 2014;1840:3494–502.
Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–13.
Wallin JJ, Guan J, Edgar KA, Zhou W, Francis R, Torres AC, et al. Active PI3K pathway causes an invasive phenotype which can be reversed or promoted by blocking the pathway at divergent nodes. PLoS One. 2012;7:e36402.
Lin Y, Dong C, Zhou BP. Epigenetic regulation of EMT: the Snail story. Curr Pharm Des. 2014;20:1698–705.
Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. 2011;121:3804–9.
Marcucci F, Bellone M, Caserta CA, Corti A. Pushing tumor cells towards a malignant phenotype. Stimuli from the microenvironment, intercellular communications and alternative roads. Int J Cancer. 2014;135:1265–76.
Marcucci F, Stassi G, De Maria R. Epithelial-mesenchymal transition: a new target in anticancer drug discovery. Nat Rev Drug Discov. 2016;15:311–25.
Asiedu MK, Beauchamp-Perez FD, Ingle JN, Behrens MD, Radisky DC, Knutson KL. AXL induces epithelial to mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene. 2014;33:1316–24.
Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12:445–64.
Lim YC, Kang HJ, Moon JH. C-Met pathway promotes self-renewal and tumorigenecity of head and neck squamous cell carcinoma stem-like cell. Oral Oncol. 2014;50:633–9.
Sette G, Salvati V, Memeo L, Fecchi K, Colarossi C, Di Matteo P, et al. EGFR inhibition abrogates leiomyosarcoma cell chemoresistance through inactivation of survival pathways and impairment of CSC potential. PLoS One. 2012;7:e46891.
Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci U S A. 2010;107:21737–42.
Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15.
Keysar SB, Le PN, Miller B, Jackson BC, Eagles JR, Nieto C, et al. Regulation of head and neck squamous cancer stem cells by PI3K and SOX2. J Natl Cancer Inst. 2016. doi:10.1093/jnci/djw189.
Lu H, Clauser KR, Tam WL, Fröse J, Ye X, Eaton EN, et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. 2014;16:1105–17.
Thakur R, Trivedi R, Rastogi N, Singh M, Mishra DP. Inhibition of STAT3, FAK and Src mediated signaling reduces cancer stem cell load, tumorigenic potential and metastasis in breast cancer. Sci Rep. 2015;5:10194.
Zhang L, Zhang L, Li H, Ge C, Zhao F, Tian H, et al. CXCL3 contributes to CD133(+) CSCs maintenance and forms a positive feedback regulation loop with CD133 in HCC via Erk1/2 phosphorylation. Sci Rep. 2016;6:27426.
Zhang Z, Duan Q, Zhao H, Liu T, Wu H, Shen Q, et al. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-κB/STAT3 signaling cascade. Cancer Lett. 2016;382:53–63.
Marcucci F, Rumio C, Lefoulon F. Anti-cancer stem-like cell compounds in clinical development – an overview and critical appraisal. Front Oncol. 2016;6:115.
Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–59.
Kuo SZ, Blair KJ, Rahimy E, Kiang A, Abhold E, Fan JB, et al. Salinomycin induces cell death and differentiation in head and neck squamous cell carcinoma stem cells despite activation of epithelial-mesenchymal transition and Akt. BMC Cancer. 2012;12:556.
Ocaña OH, Córcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell. 2012;22:709–24.
Celià-Terrassa T, Meca-Cortés O, Mateo F, De Paz Martínez A, Rubio N, Arnal-Estapé A, et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest. 2010;122:1849–68.
Gammon L, Biddle A, Heywood HK, Johannessen AC, Mackenzie IC. Sub-sets of cancer stem cells differ intrinsically in their patterns of oxygen metabolism. PLoS One. 2013;8:e62493.
Biddle A, Liang X, Gammon L, Fazil B, Harper LJ, Emich H, et al. Cancer stem cells in squamous cell carcinoma switch between two distinct phenotypes that are preferentially migratory or proliferative. Cancer Res. 2011;71:5317–26.
Pomp V, Leo C, Mauracher A, Korol D, Guo W, Varga Z. Differential expression of epithelial-mesenchymal transition and stem cell markers in intrinsic subtypes of breast cancer. Breast Cancer Res Treat. 2015;154:45–55.
Tan TZ, Miow QH, Miki Y, Noda T, Mori S, Huang RY, et al. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med. 2014;6:1279–93.
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42.
Ojha R, Bhattacharyya S, Singh SK. Autophagy in cancer stem cells: A potential link between chemoresistance, recurrence, and metastasis. Biores Open Access. 2015;4:97–108.
Altman JK, Szilard A, Goussetis DJ, Sassano A, Colamonici M, Gounaris E, et al. Autophagy is a survival mechanism of acute myeloid leukemia precursors during dual mTORC2/mTORC1 targeting. Clin Cancer Res. 2014;20:2400–9.
Rausch V, Liu L, Apel A, Rettig T, Gladkich J, Labsch S, et al. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J Pathol. 2012;227:325–35.
Yang MC, Wang HC, Hou YC, Tung HL, Chiu TJ, Shan YS. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol Cancer. 2015;14:179.
Rothe K, Lin H, Lin KB, Leung A, Wang HM, Malekesmaeili M, et al. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood. 2014;123:3622–34.
Chen H, Luo Z, Dong L, Tan Y, Yang J, Feng G, et al. Wang, CD133/prominin-1-mediated autophagy and glucose uptake beneficial for hepatoma cell survival. PLoS One. 2013;8:e56878.
Catalano M, D’Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C, et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol. 2015;9:1612–25.
Wang F, Li H, Yan XG, Zhou ZW, Yi ZG, He ZX, et al. Alisertib induces cell cycle arrest and autophagy and suppresses epithelial-to-mesenchymal transition involving PI3K/Akt/mTOR and sirtuin 1-mediated signaling pathways in human pancreatic cancer cells. Drug Des Devel Ther. 2015;9:575–601.
Xu LZ, Long ZJ, Peng F, Liu Y, Xu J, Wang C, et al. Aurora kinase a suppresses metabolic stress-induced autophagic cell death by activating mTOR signaling in breast cancer cells. Oncotarget. 2014;5:7498–511.
Qin W, Li C, Zheng W, Guo Q, Zhang Y, Kang M, et al. Inhibition of autophagy promotes metastasis and glycolysis by inducing ROS in gastric cancer cells. Oncotarget. 2015;6:39839–54.
Qiang L, Zhao B, Ming M, Wang N, He T-C, Hwang S, et al. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci U S A. 2014;111:9241–6.
Grassi G, Di Caprio G, Santangelo L, Fimia GM, Cozzolino AM, Komatsu M, et al. Autophagy regulates hepatocyte identity and epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions promoting Snail degradation. Cell Death Dis. 2015;6:e1880.
Lv Q, Wang W, Xue J, Hua F, Mu R, Lin H, et al. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial–mesenchymal transition in human breast cancer. Cancer Res. 2012;72:3238–50.
Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014;4:466–79.
Zhu H, Wang D, Zhang L, Xie X, Wu Y, Liu Y, et al. Upregulation of autophagy by hypoxia-inducible factor-1α promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol Rep. 2014;32:935–42.
Poleszczuk J, Hahnfeldt P, Enderling H. Evolution and phenotypic selection of cancer stem cells. PLoS Comput Biol. 2015;11:e1004025.
Yang T, Rycaj K, Liu ZM, Tang DG. Cancer stem cells: constantly evolving and functionally heterogeneous therapeutic targets. Cancer Res. 2014;74:2922–7.
Roth S, Fodde R. Quiescent stem cells in intestinal homeostasis and cancer. Cell Commun Adhes. 2011;18:33–44.
Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012;22:457–72.
Leth-Larsen R, Terp MG, Christensen AG, Elias D, Kühlwein T, Jensen ON, et al. Functional heterogeneity within the CD44 high human breast cancer stem cell-like compartment reveals a gene signature predictive of distant metastasis. Mol Med. 2012;18:1109–21.
Oktem G, Bilir A, Uslu R, Inan SV, Demiray SB, Atmaca H, et al. Expression profiling of stem cell signaling alters with spheroid formation in CD133high/CD44high prostate cancer stem cells. Oncol Lett. 2014;7:2103–9.
Mukhopadhyay P, Farrell T, Sharma G, McGuire TR, O’Kane B, Sharp JG. Heterogeneity of functional properties of clone 66 murine breast cancer cells expressing various stem cell phenotypes. PLoS One. 2013;8:e78725.
Zhang B, Li L, Ho Y, Li M, Marcucci G, Tong W, et al. Heterogeneity of leukemia-initiating capacity of chronic myelogenous leukemia stem cells. J Clin Invest. 2016;126:975–91.
Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports. 2013;2:78–91.
Levina V, Marrangoni AM, DeMarco R, Gorelik E, Lokshin AE. Drug-selected human lung cancer stem cells: cytokine network, tumorigenic and metastatic properties. PLoS One. 2008;3:e3077.
da Silva-Diz V, Simón-Extremera P, Bernat-Peguera A, de Sostoa J, Urpí M, Penín RM, et al. Cancer stem-like cells act via distinct signaling pathways in promoting late stages of malignant progression. Cancer Res. 2016;76:1245–59.
Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, et al. Protein kinase C α is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell. 2013;24:347–64.
Wilkinson S, O’Prey J, Fricker M, Ryan KM. Hypoxia-selective macroautophagy and cell survival signaled by autocrine PDGFR activity. Genes Dev. 2009;23:1283–8.
Oshimori N, Oristain D, Fuchs E. TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell. 2015;160:963–76.
Ruscetti M, Quach B, Dadashian EL, Mulholland DJ, Wu H. Tracking and functional characterization of epithelial–mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res. 2015;75:2749–59.
Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–32.
Ciavardelli D, Rossi C, Barcaroli D, Volpe S, Consalvo A, Zucchelli M, et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014;5:e1336.
Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–30.
Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, Choi H, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472–6.
Klevebring D, Rosin G, Ma R, Lindberg J, Czene K, Kere J, et al. Sequencing of breast cancer stem cell populations indicates a dynamic conversion between differentiation states in vivo. Breast Cancer Res. 2014;16:R72.
Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial − mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22:725–36.
Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15:1010–2.
Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–44.
Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19:1438–49.
Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A. 2012;109:11717–22.
Smith DC, Eisenberg PD, Manikhas G, Chugh R, Gubens MA, Stagg RJ, et al. A phase I dose escalation and expansion study of the anticancer stem cell agent demcizumab (anti-DLL4) in patients with previously treated solid tumors. Clin Cancer Res. 2014;20:6295–303.
Storm EE, Durinck S, de Sousa e Melo F, Tremayne J, Kljavin N, Tan C, et al. Targeting PTPRK-RSPO3 colon tumours promotes differentiation and loss of stem-cell function. Nature. 2016;529:97–100.
Kantara C, O’Connell M, Sarkar S, Moya S, Ullrich R, Singh P. Curcumin promotes autophagic survival of a subset of colon cancer stem cells, which are ablated by DCLK1-siRNA. Cancer Res. 2014;74:2487–98.
Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–6.
Yuan S, Wang F, Chen G, Zhang H, Feng L, Wang L, et al. Effective elimination of cancer stem cells by a novel drug combination strategy. Stem Cells. 2013;31:23–34.
Balic A, Sørensen MD, Trabulo SM, Sainz Jr B, Cioffi M, Vieira CR, et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther. 2014;13:1758–71.
Firat E, Weyerbrock A, Gaedicke S, Grosu AL, Niedermann G. Chloroquine or chloroquine-PI3K/Akt pathway inhibitor combinations strongly promote gamma-irradiation-induced cell death in primary stem-like glioma cells. PLoS One. 2012;7:e47357.
Vakifahmetoglu-Norberg H, Xia H-g, Yuan J. Pharmacologic agents targeting autophagy. J Clin Invest. 2015;125:5–13.
Acknowledgements
Not applicable.
Funding
This activity was not supported by any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Availability of data and materials
Not applicable.
Authors’ contributions
FM, PG and CR designed the review content and wrote the review. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Marcucci, F., Ghezzi, P. & Rumio, C. The role of autophagy in the cross-talk between epithelial-mesenchymal transitioned tumor cells and cancer stem-like cells. Mol Cancer 16, 3 (2017). https://doi.org/10.1186/s12943-016-0573-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-016-0573-8