
Abstract
Context
Multiple observational studies have reported an inverse relationship between 25-hydroxyvitamin D concentrations (25(OH)D) and type 2 diabetes (T2D). However, the results of short- and long-term interventional trials concerning the relationship between 25(OH)D and T2D risk have been inconsistent.
Objectives and methods
To evaluate the causal role of reduced blood 25(OH)D in T2D, here we have performed a bidirectional Mendelian randomization study using 59,890 individuals (5,862 T2D cases and 54,028 controls) from European and Asian Indian ancestries. We used six known SNPs, including three T2D SNPs and three vitamin D pathway SNPs, as a genetic instrument to evaluate the causality and direction of the association between T2D and circulating 25(OH)D concentration.
Results
Results of the combined meta-analysis of eight participating studies showed that a composite score of three T2D SNPs would significantly increase T2D risk by an odds ratio (OR) of 1.24, p = 1.82 × 10–32; Z score 11.86, which, however, had no significant association with 25(OH)D status (Beta -0.02nmol/L ± SE 0.01nmol/L; p = 0.83; Z score -0.21). Likewise, the genetically instrumented composite score of 25(OH)D lowering alleles significantly decreased 25(OH)D concentrations (-2.1nmol/L ± SE 0.1nmol/L, p = 7.92 × 10–78; Z score -18.68) but was not associated with increased risk for T2D (OR 1.00, p = 0.12; Z score 1.54). However, using 25(OH)D synthesis SNP (DHCR7; rs12785878) as an individual genetic instrument, a per allele reduction of 25(OH)D concentration (-4.2nmol/L ± SE 0.3nmol/L) was predicted to increase T2D risk by 5%, p = 0.004; Z score 2.84. This effect, however, was not seen in other 25(OH)D SNPs (GC rs2282679, CYP2R1 rs12794714) when used as an individual instrument.
Conclusion
Our new data on this bidirectional Mendelian randomization study suggests that genetically instrumented T2D risk does not cause changes in 25(OH)D levels. However, genetically regulated 25(OH)D deficiency due to vitamin D synthesis gene (DHCR7) may influence the risk of T2D.
Similar content being viewed by others


Introduction
Type 2 diabetes mellitus (T2D) has become a global health epidemic of twenty-first century. The International Diabetes Federation (IDF) data showed that the number of people affected with T2D would rise from 382 million in 2011 to 592 million by 2035 [1]. T2D is a complicated disease impacted by the complex interplay of genetic, epigenetic, and environmental factors [2,3,4]. In addition, several lifestyle factors such as sedentary lifestyle, westernized diet, smoking, and genetic predisposition enhance T2D risk in some ethnic groups [5, 6].
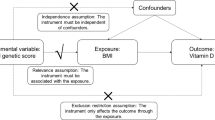
Vitamin D deficiency increases in conjunction with T2D, Type 1 diabetes mellitus (T1D), obesity, and cardiovascular disease [7]. The inverse relationship between circulating 25(OH)D concentrations and T2D is confirmed in several observational and prospective studies from the US, Australia, Europe, and Asia [8,9,10,11]. However, a causal link has not been established between low blood 25(OH)D levels and T2D risk. Recently, a large clinical trial of vitamin D3 supplementation on 2423 participants for 2.5 years concluded that a dose of 4000 IU of 25(OH)D3 did not protect people from developing T2D [12]. Findings from observational studies may be subject to residual confounding because it is difficult to measure individual variation specifically related to sun exposure and cultural variations in such studies. It may also be challenging to rule out the reverse causality accounting for any association. On the other hand, human genetic information is used by the methodology of Mendelian randomization, which takes account of genetic instruments to provide an unconfounded estimate of the association. The Mendelian randomization strategy is based on the principle that the individual genotypes are randomly assigned and can be used as a genetic instrument with the assumption that their involvement affects the outcome only by modifying the biomarkers (i.e., circulating 25(OH)D) and can also be used to test the direction of causation [13, 14].
The blood level of 25(OH)D is the best and commonly used method to determine vitamin D status. According to the US Endocrinology Society, vitamin D deficiency is defined as a 25(OH)D level of ≤ 20 ng/mL; insufficiency as 21 to 29 ng/mL; and sufficiency as > 30 ng/mL or more [15]. Low 25(OH)D status (deficiency or insufficiency) is highly prevalent among elderly, postmenopausal women, and certain ethnic population groups and is influenced by both genetic and environmental factors [16,17,18,19]. Mainly, populations due to specific ethnic backgrounds (South Asian, East Asian, and African) show a high prevalence of blood 25(OH)D deficiency [10, 20, 21]. Genetically regulated 25(OH)D concentration in a longitudinal follow-up study of 95,766 Europeans of Danish ancestry showed that reduced 25(OH)D levels were associated with all-cause mortality, cancer, and mortality from other causes [11].
Common genetic variants in ~ six loci have been identified to affect blood 25(OH)D concentrations in genome-wide association studies (GWAS) performed in European whites [22], Hispanics [23], and Asian Indians [24]. Also, multiple GWAS on T2D have identified the association of common variants in over 100 genes in ethnically diverse cohorts [4]. However, whether genetically influenced reduced 25(OH)D concentrations can be causal or contributes to enhancing T2D risk, or conversely, whether gene variants that are now widely accepted to be robustly associated with increased risk for T2D could be involved in altering blood 25(OH)D levels is unclear and inconsistent.
Here, we have used genetic instrumental variable methods to obtain estimates of the causal association between circulating 25(OH)D concentrations and T2D, including the direction of causality, by performing two-directional reciprocal Mendelian randomization study [25]. The selection of index SNPs from six genetic loci as an instrumental variable was based on the bonafide gene-regions identified in GWAS and metanalysis studies for affecting T2D and blood 25(OH)D concentrations and their availability in all participant studies [22, 24, 26,27,28]. These included two clusters of 3 index SNPs, each associated with 25(OH)D concentrations (GC rs2282679, DHCR7 rs12785878, CYP2R1 rs12794714), and T2D (IGF2BP2 rs1470579, TCF7L2 rs7903146, and KCNQ1 rs2237896), respectively. Even though multiple GWAS studies have pointed out the role of variants in genes involved in the synthesis, transport, and metabolism for influencing circulating 25(OH)D concentrations, the results of earlier published Mendelian randomization studies to determine the causal link between vitamin D insufficiency and T2D risk have been inconsistent and controversial. With few exceptions, these studies neither clearly support nor exclude the causal association of 25(OH)D levels with T2D [11, 29,30,31]. Moreover, these studies have examined mainly individuals from European populations, except one recent study included East Asians with Europeans [31]. Thus, in this study, we, for the first time, have included data from South Asian Indians with Europeans. People from the Indian sub-continent have 3 to sixfold higher T2D prevalence than Europeans and are a highly understudied population for vitamin D genetic studies. There is a paucity of data showing the relationship between vitamin D status and T2D in South Asian Indians [32].
Research design and methods
Ethical statement
All participating studies were reviewed and approved by the respective Universities or Institutional Review Boards, including the primary institute of the University of Oklahoma Health Sciences Center’s Institutional Review Board, as well as the Human Subject Protection Committees at the participating hospitals and institutes in India. All participants provided written informed consent for investigations.
Study design and phenotypic measurements
Our study with data from up to 59,890 participants from eight cohorts included: the Asian Indian Diabetic Heart Study/Sikh Diabetes Study (AIDHS/SDS); the Indian Migration Study (IMS); Twins UK; The British 1958 birth cohort (1958 BC); the Copenhagen City Heart Study (CCHS), the Copenhagen City General Population Study (CGPS), and the Copenhagen Ischemic Heart Disease Study (CIHDS); The UK Household Longitudinal Study (UKHLS); and The Prevention of Renal and Vascular End-Stage Disease (PREVEND) study. Details of study design and experimental work flow is presented in the Flow Chart (online Supplementary Section). Phenotypic measurements including T2D, 25(OH)D and other recruitment details are presented separately for each cohort (online Supplementary Section). Measures of blood 25(OH)D levels were not available on UKHLS (European) and MS India (Asian Indian) cohorts. UV index values were calculated for the city nearest the blood draw location and the month of recruitment using data from the National Weather Service Climate Prediction Center (http://www.cpc.ncep.noaa.gov) and by averaging of the previous three months UV index as described previously [33]. The clinical characteristics of each study cohort stratified by T2D are summarized in Table 1.
SNP Genotyping and Quality Control (QC) and statistical analysis
The selection of SNPs as genetic instruments was based on their strong global association with respective exposure phenotypes of T2D and 25(OH)D reported in multiethnic GWAS and meta-analysis studies [22, 26,27,28]. Additional criteria for selecting instrumental variables were based on their availability in most of the participating studies. We have provided genotyping, sample, and SNP QC details separately by each cohort in the online Supplementary Section. Genome-wide genotype data for each cohort were checked for population structure using principal components, and outliers were removed before the analysis. To ensure that the Mendelian randomization assumptions were not violated, all SNPs selected as genetic instruments were strongly associated with the outcome (i.e. gene variants associated with T2D for increasing T2D outcome; and gene variants associated with vitamin D for affecting plasma 25(OH)D outcome). None of the SNPs used as Mendelian instruments for T2D and 25(OH)D were in linkage disequilibrium (r2 = 0.001). None of the instrumental variables were associated with the outcome via exposure to other factors (BMI, age, and gender).
For 25(OH)D analysis, linear regression and additive genetic model were used with the natural log-transformed 25(OH)D level adjusted with age, gender, BMI, and study-specific covariates (e.g., T2D, UV index). Similarly, the three GWAS SNPs known to affect T2D risk were analyzed for their association with T2D using logistic regression and an additive genetic model. The T2D binary trait was the dependent variable with covariates age, gender, BMI. Each cohort analysis was limited to a single ancestral group (e.g. European ancestry only, South Asian only). If selected SNPs were unavailable, the best proxy SNP (SNP on the local haplotype having strong linkage disequilibrium (LD) with the reference SNP) was used using SNAP [34].
Upon completing the association analysis, the final list of SNPs was determined for gene score construction for each trait. The SNP, risk allele, number of risk alleles, and effect size (of each allele regarding the effect allele for log-transformed 25(OH)D levels) were required to construct a weighted gene score using the PLINK program [35]. Samples with missing 25(OH)D levels were excluded from the analysis (see Supplementary Section). A weighted gene score construction for T2D required the SNP, risk allele, number of risk alleles, and odds ratio (of each allele concerning the effect allele for T2D) for gene construction through PLINK as described previously [22]. Samples missing T2D status data were excluded, and SNPs that were directly genotyped are included to construct both gene scores. The additional parameter “–score-no-mean-imputation” was used in PLINK to construct both gene scores. The “–score-no-mean-imputation” was used so that the missing allele was not accounted as the most common allele based on the sample allele frequencies while constructing gene scores. The constructed gene scores were each normalized to a quantitative variable at an appropriate scale.
An association analysis was performed on the constructed gene scores for each cohort. A linear regression and an additive genetic model were used with blood 25(OH)D levels as the dependent variable adjusted by gender, age, BMI, and T2D. Similarly, a logistic regression and an additive genetic model were used with T2D as the dependent variable adjusted by the gender, age, BMI. To combine the association analysis results of each trait by SNP for each cohort, a fixed effect inverse variance meta-analysis implemented in METAL [36] was performed. This required the minor allele frequency, effect size, standard error, and p-value for each SNP across the different ethnic cohorts. The combined results were stratified by their respective trait association; the three SNPs associated with 25(OH)D levels were assessed for their association with T2D and 25(OH)D. Similarly, the three significant SNPs for T2D were evaluated for their association with T2D and blood 25(OH)D. Each cohort provided their statistical data summaries for the SNPs, and meta-analysis was performed on these SNPs by trait using METAL and Forest Plot Viewer (http://ntp.niehs.nih.gov/ntp/ohat/forestplot/), and PRISM (https://www.graphpad.com/scientific-software/prism/) was used to generate the forest plots. We did not observe heterogeneity of association of the genetic instruments of T2D and 25(OH)D with their respective phenotypes between studies; thus, fixed-effect models were used in metanalysis to drive odds ratios and confidence intervals. Finally, to assess the robustness of our conclusions, we applied conservative Bonferroni’s correction and used a corrected significant threshold of 0.0063 (0.05/ 8 test variables including six instrument SNPs, T2D, and 25(OH)D concentration)
Results
Demographics and clinical characteristics of all study cohorts are summarized in Table 1. As expected, there were significant differences in the distribution of BMI and 25(OH)D among cases and controls in most participating cohorts. The overall distribution of blood 25(OH)D was consistent with previously published reports on these studies, and mean 25(OH)D levels were much lower across South Asian, compared to European cohorts. Data on 25(OH)D concentration was available for only a few individuals in the South Asian cohorts, except for the Punjabi Sikh studies (AIDHS/SDS).
Association of T2D susceptibility genes with T2D and blood 25(OH)D concentrations
The association of genetic variants with an increased risk for T2D was analyzed using three known and well-established T2D associated variants from previous studies. Meta-analysis of association of IGF2BP2 (rs1470579) with T2D (adjusted for age, gender, and BMI) showed an OR (95% CI) of 1.12 (1.08, 1.16), P= 7.2x10-7, and Z score = 4.96 (Supplementary Table 1). Similarly, the adjusted per- allele effect for TCF7L2 (rs7903146) was associated with an increased risk for T2D (OR 1.25 (95% CI (1.23, 1.28), P= 1.3 x10-32, and Z score = 11.31 (Supplementary Table 1). Similarly, the adjusted per-allele effect of KCNQ1 (rs2237896) was 1.49 (1.43, 1.55), P= 5.07 x10-7, and Z score = 5.02 (Supplementary Table 1). We then analyzed the same T2D variants for their association with blood 25(OH)D concentrations in the same population cohorts. None of the variants associated with increased or decreased risk for T2D showed any association for affecting blood 25(OH) D concentrations. The per-allele association of IGF2BP2 (rs1470579) for affecting blood 25(OH)D concentration was Beta ± SE (0.0058, 0.005) P= 0.20, Z score = 1.29 (Supplementary Table 2). The per allele effect for TCF7L2 (rs7903146) for affecting blood 25(OH)D concentration was marginally significant Beta ± SE (-0.011, 0.005), P= 0.02, Z score = -2.30, and no association was observed in KCNQ1 (rs2237896) with 25(OH)D concentration Beta ± SE (-0.005, 0.01]), P= 0.26, Z score = -1.18 after adjusting for age, gender, and T2D (Supplementary Table 2).
Genes involved in 25(OH)D synthesis, metabolism, and transport and their effects on 25(OH)D concentrations and T2D
Interestingly, not only was there a significant association, the direction of the minor (effect) allele of GC (rs2282679) and CYP2R1 (rs12794714) and DHCR7 (rs12785878) with blood 25(OH)D were largely consistent across chorts (Supplementary Table 3). The minor (effect) allele frequency was significantly higher in South Asian (0.73 Sikhs) populations compared to Caucasian cohortso (which ranged between 0.22-0.32) (Supplementary Table 3). In the combined meta-analysis of 30,058 samples, the per allele effect of the GC variant (rs2282679) for its association with 25(OH)D levels was (Beta -0.091 ± SE 0.005); P= 2.87x10-61; and Z score = -16.52 nmol/L after adjusting for age, gender and BMI. For CYP2R1, age, gender, and BMI adjusted association was (Beta -0.039 ± SE 0.0032); P=7.56x10-34; and Z score -11.66 nmol/L, while the association of the DHCR7 variant (rs12785878) resulted in Beta -0.042 ± SE 0.0032, P= 9.0x10-32; and Z score -11.73 nmol/L for a total of 49,433 samples (Supplementary Table 3).
Next, we tested the association of the same variants with the risk of T2D. As summarized in Supplementary Table 4, neither variants for GC and CYP2R1 were associated with the risk of increasing T2D in any of the individual cohorts in the combined meta-analysis. The per-allele effect for increasing T2D risk yielded an OR (95% CI’s) of 1.03 (0.97,1.09), P=0.207, and Z score = 0.16 for the GC variant (rs2282679), and 1.01 (0.97,1.05), P=0.453, Z score = 1.09 for the CYP2R1 (rs12794714). However, the per-allele effect of the DHCR7 (rs12785878) variant showed a significantly increased risk for T2D OR 1.05 95% CI (1.0, 1.11), P=0.004, and Z score =2.84 in the meta-analysis (Supplementary Table 4). We also adjusted the effects of UV index on modulating association of 25(OH)D SNPs on 25(OH)D concentrations (Supplementary Tables 5), and also for their association with T2D (Supplementary Tables 6). The overall outcome of association remained unchanged as the data on the UV index was only available in AIDHS/SDS.
Gene score association analysis
The gene score construction for SNP alleles associated with an increased risk for T2D, IGF2BP2 (rs1470579), TCF7L2 (rs7903146), and KCNQ1 (rs2237896) showed a significant association with increased risk for T2D in all studies OR (95% CI) 1.24 (1.22, 1.26), P= 1.82x10-32, Z score 11.86 in 51,816 subjects (Table 2; Supplementary Figure 1A). However, the same allelic score showed no association for affecting blood 25(OH)D concentration (Beta ± SE [-0.0002 ± 0.0001], P= 0.829, Z score = -0.212). These results appear to suggest that diabetes genetic loci increase T2D risk independently without modulating 25(OH)D concentrations (Table 3 and Supplementary Figure 1B).
Next, we combined the allelic effects of genes affecting blood 25(OH)D concentrations and gene variants associated with T2D risk by constructing a gene score. Table 4 shows a significant association between the gene score of 25(OH)D lowering alleles in GC (rs2282679), CYP2R1 (rs12794714), and DHCR7 (rs12785878) in a total of 41,136 individuals which yielded Beta ± SE (-0.021 ± 0.001), i= 7.92x10-78, and Z score = -18.68 (Supplementary Figure 2A). However, the same gene score of the 25(OH)D lowering allele was not associated with the increased risk for T2D OR, 1.002 (1.001-1.005), p= 0.12, and Z score = 1.54 (Table 5 and Supplementary Figure 2B).
Discussion
Here we performed a Mendelian randomization study to investigate whether genetically reduced vitamin D levels could be causally related to increasing T2D risk using data from up to 59,890 participants (5,862 cases and 54,028 controls). Essentially, our findings suggest that even though the candidate genetic variants (individually) or via composite gene score are associated with increased susceptibility to T2D, these are not involved in affecting blood 25(OH)D concentration. This means that the genetically instrumented T2D risk could be independent of the pathway (s) linked with vitamin D synthesis, transport, or early stages of metabolism. Additionally, our data showed that the genetically instrumented reduction of 25(OH)D levels does not increase T2D susceptibility in a composite gene score. More specifically, gene variants in the vitamin D transporter (GC) and metabolism (hydroxylation) (CYP2R1) regulate 25(OH)D concentrations independent of influencing T2D risk. On the other hand, in the (DHCR7; the gene involved in 25(OH)D synthesis), the genetically instrumented decrease of -4.2 nmol/L ± 0.3 nmol/L SE of 25(OH)D concentration (Supplementary Table 3) was predicted to increase T2D risk by 5% [95% CI 0%,11%], p = 0.004, with a Z score of 2.84 (Supplementary Table 4). These results agree with an earlier published study on 96,423 Danes (one of the participating cohorts from Copenhagen) in which the DHCR7 allelic effect was associated with a modestly increased risk of T2D (OR 1.51 [95% CI 0.98, 2.33]; p trend = 0.04), whereas no association was observed for CYP2R1 (OR 1.02 [95% CI 0.75, 1.37]; p = 0.86) in that study [11].
During exposure to sunlight, a precursor molecule, 7-dehydrocholesterol (DHC), is converted to cholesterol by the action of ultraviolet light (UV) on the skin through a thermal isomerization. Hydroxylation of pre-vitamin D3 occurs in the liver from the actions of CYPs (CYP2R1). The CYP27A1 drives the conversion of 25(OH)D to 1,25(OH)D in the kidney (Supplementary Fig. 3). While our present study was in progress, a study using the Mendelian randomization approach showed that a combined gene score of 25(OH)D genes involved in synthesis (DHCR7 rs12785878) and metabolism (CYP2R1 rs10741657) was associated with an increased risk for T2D in European and Chinese adults [31]. However, our results could not confirm the role of CYP2R1 variants in the risk for T2D. This discrepancy could be due to the use of a different CYP2R1 SNP (rs12794714) in the present study or due to the overlapping role of multiple cytochrome P450 (CYP) enzymes on the hydrolysis of 25(OH)D in hepatocytes [37].
As multiple CYPs in addition to CYP2R1 and CYP27A1 could evidently be involved in vitamin D hydroxylation in the liver [38]. Therefore, the role of other CYPs resulting in compensation for 25 hydroxylase activity cannot be ruled out [37]. Thus, in light of these findings, our negative findings on genetically reduced vitamin D by CYP2R1 are not surprising.
Presumably, the differences in linkage disequilibrium patterns among different populations, population-specific risk factors, and pleiotropy may have masked the potential cumulative effects of gene scores. It appears that perhaps due to similar limitations, the study of Lu et al. [31] also could not capture the impact of genetically regulated 25(OH)D concentration on T2D using a genetically instrumented gene score of four SNPs GC (rs2282679), CYP2R1 (rs10741657), Cyp27A1 (rs6013897) and DHCR7 (rs12785878) with T2D (p = 0.07) in meta-analysis despite using a sample size of up to 476,099 of Chinese and European adults.
We undertook an additional meta-analysis combining the results of our analyses with those of Lu et al. [31]. These findings for the DHCR7 SNP showed that the genetically instrumented reduction in 25(OH)D of Beta ± SE (-4.3 nmol/L ± 0.3 nmol/L), p = 1.26 × 10–98, Z score -21.08 (Supplementary Table 7B) would significantly increase T2D risk from 5% (current study) to 7%; P = 5 × 10–4 Z score = 3.51 in a total of 493,057 individuals (Supplementary Table 8B). However, we did not observe this effect in the meta-analysis of the GC variant with the results of Lu et al. [31] (Supplementary Tables 7A and 8A). These data further confirm the possible causal effects associated with synthesis (DHCR) SNPs for increasing the risk of T2D.
The strengths of our study include well-characterized multiethnic cohorts to detect allelic association of vitamin D genes with T2D risk. We reduced the confounding effects of age, gender, and obesity across all studies. Principal components were used in each individual cohort to control for the potential of confounding by population stratification. Sample size–weighted Z score method was used to reduce inter-study variation in the diverse ancestries and differences in 25(OH)D assays between cohorts as described earlier [22]. Limitations of this study include insufficient data on 25(OH)D measures and T2D phenotypes in some cohorts; over-representation of European cohorts; lack of data on 25(OH)D measures in most South Asian studies that were available with GWAS; and data access was limited to summary statistics for each study cohorts. It is possible that the differences in serum 25(OH)D levels and T2D prevalence between South Asians and Europeans may influence the outcome of the association of biomarker with the genetic instrument. However, this would less likely be the case because; 1) the genetic instruments used for T2D and 25(OH)D were from bonafide candidate genes; 2) the application of sample-size weighted Z-score method would reduce inter-study variation, and 3) our metanalysis results do not show much heterogeneity in the SNP-phenotype association across all cohorts.
Additionally, seasonal differences in sun exposure (UV index) for effecting 25(OH)D concentration in most studies were not available except for our Punjabi Sikh cohort (AIDHS/SDS). Nonetheless, the geographic location of most participating European cohorts was nearby (Northern Europe), which might not have confounded the results due to latitude (to derive distance from the equator) variation (which all ranged from 540 to 550 N) (See Supplementary Table 10). Nevertheless, adjusting for the effects of the UV index in our Punjabi sample (AIDHS/SDS) did not change the overall impact of genetically instrumented 25(OH)D on T2D susceptibility in AIDHS/SDS and overall across studies.
It is possible to speculate that the lack of association with 25(OH)D metabolism and transport genes, but the association with synthesis gene (DHCR7) could suggest that any association with T2D may be through a UV-dependent, vitamin D-independent effect as described [39]. The power of capturing the causal association would be improved by the expansion and the refinement of the appropriateness of the genetic instrument to determine causality. Moreover, because of some inherent limitations of Mendelian randomization design, including pleiotropy, buffering effects of environmental and genetic forces (redundancy due to more than one gene), and the effects of developmental compensation, large sample size may be required to logically confirm the causal association of 25(OH)D status with T2D.
Conclusions
For the first time, we have performed a bidirectional multiethnic Mendelian randomization study to determine the causal relationship between T2D and vitamin D concentrations, including data from the population of South Asians with Europeans. Even though our research has not found strong evidence of causation, it also does not rule out the possible contribution of genetic influence of vitamin D synthesis SNPs in increasing T2D risk. As the long-standing question of the role of vitamin D in influencing the risk of T2D remains unaddressed, our data stresses the need for the population-specific design for future observational and randomized clinical trials on this important and controversial topic of immense clinical importance.
In summary, our bidirectional Mendelian randomization study having data from South Asian Indians with Europeans suggests that genetically instrumented T2D risk may not be causing changes in 25(OH)D levels. However, we cannot entirely exclude the likelihood of DHCR7 genetic variants to influence the risk of T2D.
Availability of data and materials
On request from corresponding author.
References
Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract. 2014;103(2):137–49.
Barnett AH, Eff C, Leslie RD, Pyke DA. Diabetes in identical twins. A study of 200 pairs. Diabetologia. 1981;20(2):87–93.
Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP. The continuing epidemics of obesity and diabetes in the United States. JAMA. 2001;286(10):1195–200.
Sanghera DK, Blackett PR. Type 2 Diabetes Genetics: Beyond GWAS. J Diabetes Metab. 2012;3(198).
McKeigue PM, Shah B, Marmot MG. Relation of central obesity and insulin resistance with high diabetes prevalence and cardiovascular risk in South Asians. Lancet. 1991;337(8738):382–6.
Chambers JC, Eda S, Bassett P, Karim Y, Thompson SG, Gallimore JR, et al. C-reactive protein, insulin resistance, central obesity, and coronary heart disease risk in Indian Asians from the United Kingdom compared with European whites. Circulation. 2001;104(2):145–50.
Autier P, Boniol M, Pizot C, Mullie P. Vitamin D status and ill health: a systematic review. Lancet Diabetes Endocrinol. 2014;2(1):76–89.
Scragg R, Sowers M, Bell C, Third National H, Nutrition ES. Serum 25-hydroxyvitamin D, diabetes, and ethnicity in the Third National Health and Nutrition Examination Survey. Diabetes Care. 2004;27(12):2813–8.
Gagnon C, Lu ZX, Magliano DJ, Dunstan DW, Shaw JE, Zimmet PZ, et al. Serum 25-hydroxyvitamin D, calcium intake, and risk of type 2 diabetes after 5 years: results from a national, population-based prospective study (the Australian Diabetes, Obesity and Lifestyle study). Diabetes Care. 2011;34(5):1133–8.
Braun TR, Been LF, Blackett PR, Sanghera DK. Vitamin D Deficiency and Cardio-Metabolic Risk in a North Indian Community with Highly Prevalent Type 2 Diabetes. J Diabetes Metab. 2012;3.
Afzal S, Brondum-Jacobsen P, Bojesen SE, Nordestgaard BG. Vitamin D concentration, obesity, and risk of diabetes: a mendelian randomisation study. Lancet Diabetes Endocrinol. 2014;2(4):298–306.
Pittas AG, Dawson-Hughes B, Sheehan P, Ware JH, Knowler WC, Aroda VR, et al. Vitamin D Supplementation and Prevention of Type 2 Diabetes. N Engl J Med. 2019;381(6):520–30.
Sekula P, Del Greco MF, Pattaro C, Kottgen A. Mendelian Randomization as an Approach to Assess Causality Using Observational Data. J Am Soc Nephrol. 2016;27(11):3253–65.
Smith GD, Ebrahim S. “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22.
Bischoff-Ferrari HA, Giovannucci E, Willett WC, Dietrich T, Dawson-Hughes B. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84(1):18–28.
Holick MF, Siris ES, Binkley N, Beard MK, Khan A, Katzer JT, et al. Prevalence of Vitamin D inadequacy among postmenopausal North American women receiving osteoporosis therapy. J Clin Endocrinol Metab. 2005;90(6):3215–24.
van der Wielen RP, Lowik MR, van den Berg H, de Groot LC, Haller J, Moreiras O, et al. Serum vitamin D concentrations among elderly people in Europe. Lancet. 1995;346(8969):207–10.
Marwaha RK, Tandon N, Garg MK, Kanwar R, Narang A, Sastry A, et al. Vitamin D status in healthy Indians aged 50 years and above. J Assoc Physicians India. 2011;59:706–9.
Karohl C, Su S, Kumari M, Tangpricha V, Veledar E, Vaccarino V, et al. Heritability and seasonal variability of vitamin D concentrations in male twins. Am J Clin Nutr. 2010;92(6):1393–8.
Yan L, Zhou B, Wang X, D’Ath S, Laidlaw A, Laskey MA, et al. Older people in China and the United Kingdom differ in the relationships among parathyroid hormone, vitamin D, and bone mineral status. Bone. 2003;33(4):620–7.
Harris SS. Vitamin D and African Americans. J Nutr. 2006;136(4):1126–9.
Wang TJ, Zhang F, Richards JB, Kestenbaum B, van Meurs JB, Berry D, et al. Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet. 2010;376(9736):180–8.
Engelman CD, Meyers KJ, Ziegler JT, Taylor KD, Palmer ND, Haffner SM, et al. Genome-wide association study of vitamin D concentrations in Hispanic Americans: the IRAS family study. J Steroid Biochem Mol Biol. 2010;122(4):186–92.
Sapkota BR, Hopkins R, Bjonnes A, Ralhan S, Wander GS, Mehra NK, et al. Genome-wide association study of 25(OH) Vitamin D concentrations in Punjabi Sikhs: Results of the Asian Indian diabetic heart study. J Steroid Biochem Mol Biol. 2016;158:149–56.
Timpson NJ, Nordestgaard BG, Harbord RM, Zacho J, Frayling TM, Tybjaerg-Hansen A, et al. C-reactive protein levels and body mass index: elucidating direction of causation through reciprocal Mendelian randomization. International journal of obesity. 2011;35(2):300–8.
Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet;doi:101038/ngxxxx. 2008.
Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet.42(7):579–89.
Saxena R, Saleheen D, Been LF, Garavito ML, Braun T, Bjonnes A, et al. Genome-Wide Association Study Identifies a Novel Locus Contributing to Type 2 Diabetes Susceptibility in Sikhs of Punjabi Origin From India. Diabetes. 2013;62(5):1746–55.
Ye Z, Sharp SJ, Burgess S, Scott RA, Imamura F, InterAct C, et al. Association between circulating 25-hydroxyvitamin D and incident type 2 diabetes: a mendelian randomisation study. Lancet Diabetes Endocrinol. 2015;3(1):35–42.
Jorde R, Schirmer H, Wilsgaard T, Joakimsen RM, Mathiesen EB, Njolstad I, et al. Polymorphisms related to the serum 25-hydroxyvitamin D level and risk of myocardial infarction, diabetes, cancer and mortality. The Tromso Study. PLoS One. 2012;7(5):e37295.
Lu L, Bennett DA, Millwood IY, Parish S, McCarthy MI, Mahajan A, et al. Association of vitamin D with risk of type 2 diabetes: A Mendelian randomisation study in European and Chinese adults. PLoS Med. 2018;15(5):e1002566.
Sanghera DK, Sapkota BR, Aston CE, Blackett PR. Vitamin D Status, Gender Differences, and Cardiometabolic Health Disparities. Ann Nutr Metab. 2017;70(2):79–87.
Hong J, Hatchell KE, Bradfield JP, Bjonnes A, Chesi A, Lai CQ, et al. Transethnic Evaluation Identifies Low-Frequency Loci Associated With 25-Hydroxyvitamin D Concentrations. J Clin Endocrinol Metab. 2018;103(4):1380–92.
Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O’Donnell CJ, de Bakker PI. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24(24):2938–9.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics.26(17):2190–1.
Bikle DD. Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol. 2014;21(3):319–29.
Zhu JG, Ochalek JT, Kaufmann M, Jones G, Deluca HF. CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proc Natl Acad Sci U S A. 2013;110(39):15650–5.
Gorman S, Lucas RM, Allen-Hall A, Fleury N, Feelisch M. Ultraviolet radiation, vitamin D and the development of obesity, metabolic syndrome and type-2 diabetes. Photochem Photobiol Sci. 2017;16(3):362–73.
Acknowledgements
AIDHS/SDS. This work was supported by NIH grants -R01DK082766, R01DK118427, funded by the National Institute of Health (NIDDK) and NOT-HG-11-009 funded by NHGRI, and a VPR Bridge Grant from University of Oklahoma Health Sciences Center. The authors thank all the participants of AIDHS/SDS and are grateful for their contribution to this study.
TWINS UK. TwinsUK is funded by the Wellcome Trust, Medical Research Council, European Union, the National Institute for Health Research (NIHR)-funded BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London.
1958 BC: This work was supported by the National Health and Medical Research Council, Australia (GNT GNT11123603) and the 25(OH)D measures were funded by the BUPA Foundation. The management of the 1958 Birth Cohort is funded by the Economic and Social Research Council (grant number ES/M001660/1). Access to these resources was enabled via the 58READIE Project funded by Wellcome Trust and Medical Research Council (grant numbers WT095219MA and G1001799). DNA collection was funded by MRC grant G0000934 and cell-line creation by Wellcome Trust grant 068545/Z/02. This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of investigators who contributed to generation of the data is available from the Wellcome Trust Case-Control Consortium website. Funding for the project was provided by the Wellcome Trust under the award 076113. This research used resources provided by the Type 1 Diabetes Genetics Consortium, a collaborative clinical study sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute of Allergy and Infectious Diseases, National Human Genome Research Institute, National Institute of Child Health and Human Development, and Juvenile Diabetes Research Foundation International (JDRF) and supported by U01 DK062418.
CCHS/CGPS/CIHDS. The Danish Heart Foundation and Herlev and Gentofte Hospital, Copenhagen University Hospital, Denmark
PREVEND. PREVEND genetics is supported by the Dutch Kidney Foundation (Grant E033), the EU project grant GENECURE (FP-6 LSHM CT 2006 037697), the National Institutes of Health (grant LM010098), The Netherlands organization for health research and development (NWO-Groot grant 175.010.2007.006, NWO VENI grant 916.761.70, ZonMw grant 90.700.441), and the Dutch Inter University Cardiology Institute Netherlands (ICIN).
UKHLS. The UK Household Longitudinal Study is led by the Institute for Social and Economic Research at the University of Essex. The UK Household Longitudinal Study is funded by the Economic and Social Research Council (ES/H029745/1). The survey was conducted by Nat Cen and the genome-wide scan data were analyzed and deposited by the Wellcome Trust Sanger Institute (WT098051). Information on how to access the data can be found on the Understanding Society website https://www.understandingsociety.ac.uk/.
IMS. This work was supported by the Wellcome Trust, UK (Grant no. 070797 and Grant no. 083541) which provided funding support for phenotyping and genotyping.
Funding
Provided in the acknowledgement section for each study.
Author information
Authors and Affiliations
Contributions
Dharambir K. Sanghera conceived, coordinated the consortium investigators, contributed to genotyping, and phenotyping as a cohort PI, and wrote the manuscript. Cynthia A. Bejar. and Shiwali Goyal performed data analysis, preparation of data, and manuscript. Piers R. Blackett contributed to clinical phenotyping and helped in manuscript writing. Shoaib Afzal contributed to genotyping, phenotyping, data analysis, and writing. Massimo Mangino contributed to data analysis and phenotyping. Ang Zhou, Yanchun Bao, Peter J. van der Most contributed to data analysis and genotyping. Niek Verweij and Peter J. van der Most assisted in phenotyping and data curation. Dorairaj Prabhakaran performed the phenotyping. Sarju Ralhan, Gurpreet S. Wander, Narinder K. Mehra, Jai Rup Singh assisted in data curation, and phenotyping and manuscript review. Elina Hyppönen contributed in genotyping, phenotyping and is a cohort PI. Sanjay Kinra contributed in phenotyping and is a cohort PI. Vipin Gupta, Gagandeep K Walia performed genotyping, phenotyping and data analysis. Martin H. de Borst contributed to data analysis, phenotyping, writing and is a cohort PI. Børge G. Nordestgaard contributed phenotyping, genotyping, and writing and is a cohort PI. Elina Hyppönen and Tim D. Spector are cohort PI’s and reviewed the manuscript. All authors read and provided crucial comments on the manuscript. The authors are grateful to Drs. Robert Clarke and Derrele Bennett for agreeing to provide summary stats for their data from Lu et al., Plos Genetics (2018). The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Asian Indian Diabetic Heart Study/Sikh Diabetes Study (AIDHS/SDS) was reviewed and approved by the University of Oklahoma Health Sciences Center Institutional Review Board (IRB#2911), as well as the Human Subject Protection Committee at the participating hospitals and institutes in India. IMS study was approved by ethics committee of the All India Institute of Medical Sciences, New Delhi, India (IRB# number A-60/4/8/2004). Twins UK study obtained ethical approval from the Ethics Committee at Guy's and St Thomas Hospital Trust, London. Internal Ethical Committee reviewed the 1958 BC study. The Copenhagen City Heart Study (CCHS), the Copenhagen General Population Study (CGPS), and the Copenhagen Ischemic Heart Disease Study (CIHDS) studies were approved by the Danish Ethical Committees and Herlev Hospital, Copenhagen University Hospital. The UK Household Longitudinal Study (UKHLS) got ethical approval from the local ethics committee. The PREVEND study was approved by the local medical ethics committee and was conducted in accordance with the guidelines of the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
We declare that no conflict of interest could be perceived as prejudicing the impartiality of the research reported.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
Association results of individual T2D SNPs used as genetic instruments in Mendelian randomization for their association with T2D. Supplementary Table 2. Association of individual T2D SNPs used as genetic instruments in Mendelian randomization analyses for their association with 25(OH)D concentrations. Supplementary Table 3. Association results of individual vitamin D SNPs used as genetic instruments in Mendelian randomization for their association with circulating 25(OH)D concentration. Supplementary Table 4. Association results of individual vitamin D SNPs used as genetic instruments in Mendelian randomization for their association with T2D. Supplementary Table 5. Association of vitamin D SNPs used as genetic instruments for affecting 25(OH)D concentrations using UV index as a covariate. Supplementary Table 6. Association of vitamin D SNPs used as genetic instruments showing their effect on Type 2 diabetes risk using UV index as a covariate. Supplementary Table 7 A-B. Results of joint metanalysis of current study and a published study (Lu et al, 2018) showing significant associations of variants in vitamin D candidate genes (GC and DHCR7) for effecting 25(OH)D concentrations. Supplementary Table 8 A-B. Results of joint metanalysis of current study and a published study (Lu et al, 2018) showing effects of genetically instrumented vitamin D candidate genes (GC and DHCR7) SNPs for their effects on T2D risk. Supplementary Table 9. Information on samples with missing 25(OH)D levels from 8 different cohorts. Supplementary Table 10. Differences in latitude, 25(OH)D concentration and distribution of allele frequencies of vitamin D and T2D SNPs among South Asian and European cohorts.
Additional file 2: Supplementary (Flow Chart).
Experimental plan including the details of participating cohorts and candidate gene SNPs used as genetic instrument for performing a bidirectional Mendelian randomization study. Supplementary Figure 1 (A-B). Association analysis of composite gene score of T2D SNPs (IGF2BP2, TCF7L2 and KCNQ1) used as a genetic instrument for their joint effect on T2D and 25(OH)D concentrations (https://www.graphpad.com/scientific-software/prism/). Supplementary Figure 2 (A-B). Association analysis of composite gene score of vitamin D SNPs (GC, CYP2R1 and DHCR7) used as a genetic instrument for their effect on 25(OH)D concentrations and T2D (https://www.graphpad.com/scientific-software/prism/). Supplementary Figure 3. This figure illustrates pathways of synthesis, absorption, metabolism, and transportation of vitamin D. Genetic factors known to influence circulating levels of vitamin D are shown in the figure. DHCR7 expresses a reductase which uses nicotinamide adenine dinucleotide phosphate-oxidase to catalyzes the production of cholesterol to 7-dehydrocholesterol (7-DHC). The GC gene catalyzes the vitamin D binding protein formation. The enzyme (25-hydroxylase) responsible for the first hydroxylation step is encoded by CYP2R1. The enzyme in the kidneys responsible for the second hydroxylation is catalyzed by the CYP27B1 gene product (not used in this study). 1,25(OH)2D3 is the most effective and commonly measured vitamin D deficiency marker. Genetic instruments used in this study include gene pathways involved in vitamin D synthesis (DHCR7), metabolism (CYP2R1) and transport (GC) and genetic loci linked with increased T2D risk (IGF2BP2, TCF7L2, and KCNQ1). Note that in our results causality appears to be inferred by direct association of 25(OH)D variants (i.e. DHCR7) with 25(OH)D levels and the T2D phenotype. In contrast the T2D genes are associated with phenotype but not with the intermediate phenotype measured as 25(OH)D.
Additional file 3.
Online Supplemental Information: Phenotypic measurements including T2D, 25(OH)D, and other recruitment details are presented separately for each cohort.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bejar, C.A., Goyal, S., Afzal, S. et al. A Bidirectional Mendelian Randomization Study to evaluate the causal role of reduced blood vitamin D levels with type 2 diabetes risk in South Asians and Europeans. Nutr J 20, 71 (2021). https://doi.org/10.1186/s12937-021-00725-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12937-021-00725-1