
Abstract
Background
Activation of hypnozoites of vivax malaria causes multiple clinical relapses, which contribute to the Plasmodium vivax burden and continuing transmission. Artemisinin-based combination therapy (ACT) is effective against blood-stage P. vivax but requires co-administration with primaquine to achieve radical cure. The therapeutic efficacy of primaquine depends on the generation of a therapeutically active metabolite via cytochrome P450 2D6 (CYP2D6). Impaired CYP2D6 metabolism has been associated with primaquine treatment failure. This study investigated the association between impaired CYP2D6 genotypes, drug-exposure to the long-acting ACT component (schizonticidal drugs) and tolerance and efficacy.
Methods
Adult patients with acute vivax malaria were enrolled in a recently completed trial and treated with artesunate–mefloquine, chloroquine or artemether–lumefantrine. All received concomitant primaquine (0.5 mg/kg/day for 7–9 days). The association between efficacy and safety and drug exposure was explored using area-under-the-curve (AUC) and half-life (t1/2) estimates obtained by non-compartmental analysis of the long half-life drugs. Parasite recurrences by day 63 were categorized as related relapses or re-infections/unrelated hypnozoite activation by genotyping three microsatellite loci and two polymorphic loci of merozoite surface antigen-1. The CYP2D6 genotype was identified with Taqman assays by real-time PCR to 9 polymorphisms (8 SNPs and one deletion). Impaired CYP2D6 activity was inferred using the Activity Score System.
Results
Most recurrences in the ASMQ (67%), CQ (80%) and AL (85%) groups were considered related relapses. Eight of nine (88.9%) of the patients with impaired CYP2D6 activity relapsed with related parasite compared to 18/25 (72%) with normal activity (RR = 1.23, 0.88; 1.72, p = 0.40). There were no associations between the measured PK parameters and recurrence. Patients with longer chloroquine half-lives had more pruritus (RR = 1.09, 1.03; 1.14, p = 0.001). Higher CQ AUCs were associated with reduced falls in haemoglobin by day 14 (Coef − 0.02, − 0.005; − 0.03, p = 0.01). All regimens were well tolerated.
Conclusion
Genotyping of P. vivax showed that activation of related (homologous) hypnozoites was the most frequent cause of recurrence. The high proportion of the impaired CYP2D6 activity among patients with recurrent infections suggests that slow primaquine metabolism might influence related relapse rates in Brazil among patients receiving primaquine for radical cure, although confirmatory studies are needed. There was no association between drug exposure of the long-acting ACT component (schizonticidal drugs) and risk of related relapse. ACT was well tolerated. These results provide further re-assurance about the safety and efficacy of ACT when combined with short course primaquine to treat uncomplicated malaria vivax in Brazil.
Trial registration RBR-79s56s (http://www.ensaiosclinicos.gov.br/rg/RBR-79s56s/)
Similar content being viewed by others

Background
The biological features of vivax malaria provide major challenges for pre-elimination and elimination programmes in areas co-endemic for Plasmodium falciparum and Plasmodium vivax [1]. These features include the early appearance of gametocytes, a high proportion of asymptomatic or chronic carriers [2] and the parasite latent form hypnozoites that produce relapses. Relapses make a major contribution to the global P. vivax burden [3]. It was estimated that relapses constituted 76–90% and 79% of total infections in Papua New Guinea and Thailand, respectively [4]. To eliminate this reservoir of latent infections, simple effective radical treatment of vivax using 8-aminoquinolines, such as primaquine or the recently approved analogue tafenoquine, is required.
In most countries with endemic P. vivax, the preferred first-line radical treatment for P. vivax remains chloroquine, combined with 7- to 14-day primaquine regimens, and this has barely changed since the 1950s [5]. Chloroquine is no longer recommended for the case management of falciparum malaria due to the spread of parasite resistance [6], resulting in the use of different treatment regimens for P. vivax and P. falciparum in areas where these species are co-endemic. This is programmatically complicated. The use of a single regimen to treat all species of malaria would simplify malaria treatment guidelines [7]. Artemisinin-based combination therapy (ACT) is emerging as the best option in this context, particularly in settings where there are concerns about chloroquine-resistant P. vivax [8]. ACT is effective against the blood stage of P. vivax [9], but must be co-administered with primaquine to eliminate P. vivax hypnozoites [8, 10]. Short-course primaquine regimens are preferable as they have been proven to have an efficacy not inferior to the standard 14 days’ regimens [11,12,13,14]. However, there is a relative lack of data on the safety, pharmacodynamics and pharmacokinetics of ACT when provided in combination with daily primaquine regimens for the radical cure of P. vivax [15]. Safety is a major concern when deploying new treatments, but there are also concerns that primaquine/ACT drug interactions may reduce the overall regimen efficacy by inhibiting CYP2D6 or reducing synergistic effect of the current regimen. There are uncertainties about the best partner ACT, as drugs with a longer half-life may prevent early relapses.
Characterization of parasites and patients’ drug metabolism is needed to ascertain the pharmacodynamics, pharmacokinetics and therapeutic success of these regimens. The main malaria clinical trials outcomes are parasitological clearance and recurrence rate [16]. Vivax recurrence includes: (i) recrudescence of parasites that have been previously cleared and microscopically undetectable; (ii) re-infection from another mosquito bite; and, (iii) relapses, i.e., activation of hypnozoites, genetically related (homologous) or unrelated (heterologous). The primaquine metabolite that is active against human hypnozoite is unknown [17], but the metabolism of primaquine to its active metabolite is dependent on the cytochrome P450 enzyme CYP2D6 [17,18,19]. Low CYP2D6 activity results in slow metabolism of primaquine to the active metabolite. CYP2D6 activity may be a proxy of primaquine’s active metabolite exposure and a risk factor for relapse among primaquine recipients [20,21,22]. Similarly, the cytochrome P450 enzymes CYP2C8 was investigated as it is known to participate in the metabolism of chloroquine [23].
A previous randomized clinical trial in Brazil of the treatment of uncomplicated vivax malaria compared the safety and efficacy of the fixed-dose ACT artemether–lumefantrine and artesunate–mefloquine against the standard treatment with chloroquine, all three in combination with short-course primaquine (0.5 mg/kg/day for 7–9 days) [24]. This current study investigated the pharmacokinetics of the long-acting ACT component (schizonticidal drugs) with concomitant primaquine upon the safety and efficacy of these three treatment regimens and the influence of genetic variability of parasite and host, including the frequency of mutations in CYP2D6 gene over relapse rate.
Methods
Overview study design
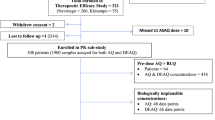
The patients included in the current analysis were enrolled in a larger clinical trial previously published and designed in accordance with WHO guidelines [16]. The trial was designed to evaluate the safety and efficacy of the schizonticidal drugs with concomitant use of primaquine for vivax cure. The details of the trial design and methods have been reported elsewhere [24]; in brief, patients were eligible if they had acute uncomplicated malaria due to P. vivax mono-infection confirmed by microscopy, with fever or a history of fever in the previous 48 h, were aged 18 to 70 years old, weighed between 50 and 90 kg, and had parasite densities > 250/μL and haemoglobin levels > 7.0 g/dL. G6DP deficiency was not an exclusion criterion. They were randomly allocated to three treatment groups: (a) artesunate–mefloquine (100 + 200 mg QD for 3 days) (ASMQ); (b) chloroquine (CQ) (600 mg on day 1, and 450 mg on days 2 and 3); and, (c) artemether–lumefantrine (20 + 120 mg BID for 3 days) (AL). All three arms received the same concomitant primaquine regimen (7–9 days: 0.5 mg/kg/day).
Patients were assessed on the day of enrolment and days 1, 2, 3, 7, 14, 21, 28, 42, and 63. The main endpoints were either treatment failure or adequate clinical and parasitological response. Blood samples were collected for parasite counts at every scheduled visit, on any day of treatment failure and for drug levels on days 0, 3, 7, 14, 21, 28, 42, and 63. Samples (100 μL) were transferred to Whatman (USA) ET 31 CHR E 3MM filter papers for later pharmacokinetic analysis and parasite genotyping [25].
Parasitological densities were estimated using Giemsa-stained blood slides at a magnification of 1000× using WHO-recommended methods [16]. Adverse events (AE) were assessed at each follow-up visit, and patients were encouraged to return to the clinic if they were ill in between scheduled visits. All AEs, including laboratory abnormalities, were categorized by body system.
Pharmacokinetics/pharmacodynamics
Whole blood concentrations of mefloquine (MQ), chloroquine (CQ) and lumefantrine (LMF) were measured using a validated HPLC-MS/MS method in accordance with Brazilian [26] and international regulatory requirements for bio-analytical methods [27]. The pharmacokinetics assays were conducted at the Equivalence and Pharmacokinetics Service (SEFAR)/Oswaldo Cruz Foundation, which is accredited by the Brazilian regulatory agency, Agência Nacional de Vigilância Sanitária (ANVISA). Non-compartmental analysis was performed for CQ, LMF and MQ using the Pmetrics® [28] package to estimate two main parameters; the overall area under the curve (AUC)(3–63 days) and the terminal elimination half-life (t1/2). The terminal elimination half-life was only calculated for subjects with five or more available samples. These two parameters were used as proxy indicators for overall drug exposure for correlation analyses with drug efficacy and safety profiles.
The correlation between drug exposure and treatment failure was evaluated using the Mann–Whitney test. The effect of pharmacokinetic parameters on the frequency of AEs likely or probably related to the test drug was expressed as the relative risks (RR) obtained from random effects generalized estimation equation (GEE) log-binomial regression models. Linear regression was used to test the effects of PK parameters on the fall in haemoglobin (Hb) concentrations by day 14 relative to enrolment values [29]. The effects of these pharmacokinetic parameters on treatment failure (both early and later failures) were evaluated as odds ratios (OR) from binomial logit link regression models, using generalized linear models (GLM). Time to failure was tested using standard Cox regression. The proportion of clonal variability at recurrence was compared between treatment arms using Fisher’s exact test. Two-sided p-values of < 0.05 were considered statistically significant.
DNA extraction and genotyping of parasites microsatellites and polymorphic blocks of MSP-1 and patients’ CYP2D6 and CYP2C8
DNA was extracted from dried blood using QIAamp DNA blood mini kit (Qiagen, Hilden, Germany) following the instructions of the manufacturer. Three microsatellite loci (MS2, MS6, MS7) and two polymorphic loci (blocks 2 and 10) of Merozoite surface antigen 1 (MSP-1) were amplified using specific primers and conditions as previously described [30, 31]. The exact length and relative abundance (fluorescence levels) of each PCR product were determined in the DNA automatic sequencer (ABI 3730, Applied Biosystems, Thermo Fischer Scientific, Waltham, MA, USA) with fluorescein-labelled forward primers and an internal size standard (GeneScan 500 LIZ, Applied Biosystems). The predominant allele for each locus was identified as the highest peak of fluorescence in the electropherogram using GeneMapper 4.1 software (Applied Biosystems). The multiplicity of parasite variants was estimated measuring extra peaks in the electropherogram with fluorescence above the cut-off (150 arbitrary fluorescence units) and at least one-third the high of the main peak. Parasite recurrences within 63 days were categorized as ‘related’, including totally identical (homologues) if all five polymorphic loci (MS2; MS6; MS7; MSP1B2; MSP1B10) were identical and ‘similar’ if 80% of their alleles were identical; and otherwise as unrelated (heterologous) (Additional file 1). Number of alleles and heterozigosity expected were calculated in Arlequim software v. 3.5.2.2 (http://cmpg.unibe.ch/software/arlequin35/).
The cytochrome P450 enzymes CYP2D6 [17, 19] and CYP2C8 [23] are known to participate in the metabolism of the primaquine and chloroquine, respectively. Two SNPs (G416A[rs11572080] and A805T [rs11572103]) were genotyped in the CYP2C8 gene. In the CYP2D6 gene eight SNPs were genotyped; (G-1584C [rs1080985], C100T [rs1065852], C1023T [rs28371706], G1846A [rs3892097], C2850T [rs16947], G2988A [rs28371725], G3183A [rs59421388] and G4180C [rs1135840]) and one deletion (2615-2617delAAG [rs5030656]) were genotyped. The copy number was also determined. All SNPs genotyping were performed by real-time PCR using specific hydrolysis probe [22] in ViiA 7 Real-time PCR system (Applied Biosystems). Haplotypes and CYP2D6 star alleles were inferred using the Phase software (version 2.1). CYP2D6 gene copy number was determined with Hs00010001_cn assay (Applied Biosystem) in Real-time PCR [20]. Each allele got one value that was used to calculate the CYP2D6 activity score (AS). Patient were categorized based on their AS score into normal metabolizer fast (gNM-F) (AS = 1.5 or 2.0), normal metabolizer slow (gNM-S) (AS = 1), intermediate metabolizer (gIM) (AS = 0.5), poor metabolizer (gPM) (AS = 0), and ultra metabolizer (gUM) (more than 2 copies of the normal allele) (AS ≥ 2.0). Impaired CYP2D6 activity was defined as AS scores less than 1.5 (Additional file 2).
Results
Population
The study is based on a sub-set of samples from the original trial. Only 2/3 of samples (1400 samples of 175 patients) were evaluated in pharmacokinetics/pharmacodynamics analysis due to logistical issues, however the baseline characteristics of this sub-set were similar across the three arms (Table 1). Characterization of parasites and drug metabolism was conducted in all 35 patients with parasite recurrence within 63 days.
Characterization of parasites and drug metabolism in the population with treatment failure
The frequency of related (homologous and similar) and unrelated (heterologous) parasites among 35 patients with parasite recurrence within 63 days by study arm is shown in Table 2. Overall, 67, 80 and 85% of the recurrent malaria in the ASMQ, CQ and AL groups, respectively, were considered related relapses. Among the recurrences the pooled percentage of related relapses across the three arms was 77.1%.
Genetic analysis of the parasite populations comparing all recurrences also demonstrated that 77.1% (27/35) of patients presented with a single clonal infection at the initial infection and at the recurrence. The number of parasite variants between initial infection and recurrence remained the same in 23 (65.7%) of the 35 patients with recurrent infections and increased in 10 (28.5%) and decreased in 2 (5.7%). There was no difference in multiplicity of parasite clones at recurrence between treatment arms (p = 0.51).
Cyp2d6
Eight of 9 (88.9%) patients classified to have reduced enzymatic activity for primaquine metabolism based on their CYP2D6 genotypes relapsed with related parasites (RR = 1.23 95% CI (0.88–1.72) p = 0.40) (Table 3). Eighteen out of 25 (72%) normal metabolisers had related relapses.
Cyp2c8
The frequency of two polymorphisms in CYP2C8 gene in the population who failed is presented in Table 4. Out of the 10 patients in the CQ arm with parasite recurrence, one had the SNP G416A genotype indicative of reduced enzyme activity of CYP2C8. The AUC and half-life for chloroquine of this patient were 91.9 µg/mL h and 11.32 days, respectively, compared with 102.3 µg/mL h and 19.3 days in the 9 CYP2C8 non-mutated genotypes in the CQ arm. These results for the patient with the mutated genotype fall within the 95% CI for the overall population (Table 5).
Pharmacokinetics/pharmacodynamics
AUC0–63 day values were calculated for each patient in each arm; the terminal elimination half-life (t1/2) could only be calculated for those receiving CQ or MQ. Figure 1 shows the terminal PK profile generated for MQ, CQ and LMF.

Pharmacokinetic profile of Mefloquine (MQ), Lumefantrine (LMF) and Chloroquine (CQ). Profiles were generated from 58 to 60 subjects for each drug. Data shows median with 95 confidence interval (CI). a Mefloquine concentration over the time, b lumefantrine concentration over the time and c chloroquine concentration over the time
Drug exposure (defined by AUC3–63 day or elimination half-life) under the therapeutic threshold could potentially explain the relapses and recrudescence, but not re-infections. The relationship of drug exposure (defined by AUC3–63 day or elimination half-life) to recurrence was investigated by comparing the PK parameters in the cured population with those with confirmed related relapse (homologous + similar). There were no statistically significant differences in AUC3–63 day or elimination half-life for MQ, CQ or LMF between patients without recurrent infections by day 63 and those with relapses in univariate (Table 5). Weight and gender were also not associated with parasite recurrence by day 63 (Additional file 3), parasite clearance by day 3 (Additional file 4), or time to failure (Additional file 5).
Higher drug exposures could be associated with a higher frequency of AEs. The influence of AUC, half-life and weight on the frequency of AEs likely and probably related to the treatment per body systems (n ≥ 30) are presented in Additional file 6. The relative risk of having pruritus increased as the half-life of CQ increased (RR 1.09, 95% CI 1.03–1.14, p = 0.001). Conversely, the influence of drug exposure on the Hb drop (defined as Hb at day 14 − Hb at baseline/Hb at baseline) shown that the higher the AUC of CQ the lower the reduction in the Hb decline (Coef − 0.02, 95% CI − 0.03; − 0.00, p = 0.01) (Additional file 7). Please number in sequence i.e. first 6 then 7).
Discussion
This study showed by genotyping polymorphic loci of P. vivax that relapse due to related parasites was the most frequent cause of recurrence by day 63 in all three treatment arms of the previous trial [32], where 13–16% of infections recurred by day 63. The current study also showed that the treatment failure rate in patients with reduced inferred CYP2D6 activity (26%) was higher than in the general population (≅ 11%) [33]. Moreover, 8 out of 9 recurrences among patients with low CYP2D6 activity were related relapses compared to 18 out of 25 with inferred normal CYP2D6 activity. Low CYP2D6 activity and presumptive low exposure to the active metabolite of primaquine or disruption of the PQ-enzyme interactions might influence related relapse rates in Brazil, although further studies are need to elucidate this effect. Likewise, the impaired activity of CYP2C8 may result in a lower exposure to desethylchloroquine, the chloroquine active metabolite. The PK parameters and clinical outcomes of the single patient with impaired CYP2C8 in CQ arm did not differ from the overall population.
Other causes of low exposure to primaquine are also potential risk factors for relapses, such as low adherence or impaired bioavailability. Similarly, others factors which affect the success of anti-infective therapeutics may influence the responses to the radical treatment, such as differences in the biology of the parasite, the immune status of the patients and the density of latent hypnozoites [34].
This was the first time that some of the ACT combinations, namely ASMQ, were evaluated for the cure of P. vivax in clinical trial conditions, and thus in combination with daily primaquine (0.5 mg/kg/day for 7–9 days) regimens. The correlation between drug exposure to the long-acting components of the ACT and the risk of recurrence and AEs was also assessed. Potential interactions between primaquine and the ACT, and also the safety of these regimens, have not been extensively assessed [15]. Drug interactions such as lumefantrine inhibiting CYP2D6 [35] may reduce the overall regimen efficacy; on the other hand, drugs with a longer half-life may prevent early relapses. This study could not demonstrate a significant associations between the PK parameters of the long half-life drugs and the risk of recurrence, or the risk of relapse due to either homologous and heterologous relapses or time to recurrence (Additional files 3, 4, 5), although the numbers of relapses were small. Patients with longer chloroquine elimination half-life estimates were more likely to report pruritus. Transient, mild to moderate pruritus is a well-known adverse effect of chloroquine [36] and a threat to treatment adherence. A smaller drop in haemoglobin by day 14 was associated with higher CQ exposure (AUC), which may reflect better therapeutic efficacy achieved with higher concentrations of CQ [37].
This study has several limitations. The blood sampling schedule was designed to evaluate the blood levels of the long half-life drugs; the pharmacokinetic data allowed the prediction of the drug exposures up to 63 days post-treatment with up to 8 sample points available for each patient. It did not allow a proper modelling of primaquine levels and limited the calculation of the elimination half-life of lumefantrine. The absence of desethylchloroquine blood levels measurement is another study limitation. A trial designed with 6 months follow-up would have been able to evaluate relapses with more accuracy, as the median time to vivax recurrence in Brazil is 71 days [13]. The characterization of parasites and drug metabolism were conducted only in 35 patients with treatment failure limiting the comparisons. Genotyping vivax parasites to infer relapse frequencies also presents limitations. Genotyping in vivax does not allow differentiation between new infections (reinfections) or activation of unrelated (heterologous) hypnozoites. In this study, only recurrences with homologous and similar parasites were considered relapses. However, relapses are often heterologous activation of hypnozoites [31, 38]. The recrudescence of sub-microscopic parasite population [39] is also a biologically plausible explanation for homologous parasites in two samples. Future use of more sensitive parasite detection strategies, such as ultrasensitive PCR of all consecutive samples of the patients who failed could elucidate these results.
Conclusion
The genotyping of polymorphic loci of P. vivax showed that relapse due to genetically related parasites was the most frequent cause of recurrence in all three treatment arms. The high proportion of CYP2D6 genetic polymorphisms among patients with recurrent infections suggests that impaired primaquine metabolism might influence the related relapse rates in Brazil among patients receiving primaquine for radical cure, further studies are needed to confirm this finding. The three ACT regimens were very effective, and there was no association between drug exposure levels of the long-acting components of the ACT and risk of relapse. The ACT was well tolerated overall. These results provided further reassurance about the safety of the combined use of ACT and short-course primaquine (0.5 mg/kg/day for 7–9 days) to treat uncomplicated malaria vivax in Brazil.
Availability of data and materials
The evaluation of pharmacokinetics’ parameters, gender and weight as predictors of failures per treatment drug (LGM, binomial logit link); evaluation of pharmacokinetics’ parameters and weight as predictors of D3 failures per treatment drug (LGM, binomial logit link); and evaluation of pharmacokinetics’ parameters, gender and weight as predictors of time to failures per treatment drug (Cox regression, binomial logit link) are provided in the additional file.
Abbreviations
- ACT:
-
artemisinin-based combination therapy
- AE:
-
adverse events
- AL:
-
fixed dose combination of 20 + 120 mg artemether and lumefantrine with concomitant use of primaquine
- ANVISA:
-
National Regulatory Agency
- AS:
-
CYP2D6 activity score
- ASMQ:
-
fixed dose combination of 100 + 200 mg artesunate and mefloquine with concomitant use of primaquine
- AUC:
-
area-under-the-curve
- CQ:
-
chloroquine
- CI:
-
confidence interval
- CQ:
-
chloroquine with concomitant use of primaquine
- G6PD:
-
glucose-6-phosphate dehydrogenase
- GEE:
-
generalized estimation equation
- GLM:
-
generalized linear models
- Hb:
-
haemoglobin
- HPLC-MS/MS:
-
high performance liquid chromatography-tandem mass spectrometry
- LMF:
-
lumefantrine
- MQ:
-
mefloquine
- OR:
-
odds ratio
- PCR:
-
polymerase chain reaction
- RR:
-
relative risks
- SD:
-
standard deviations
- SEFAR:
-
Equivalence and Pharmacokinetics Service
- t1/2 :
-
half-life
- WHO:
-
World Health Organization
References
Gething PW, Elyazar IR, Moyes CL, Smith DL, Battle KE, Guerra CA, et al. A long neglected world malaria map: Plasmodium vivax endemicity in 2010. PLoS Negl Trop Dis. 2012;6:e1814.
Chen I, Clarke SE, Gosling R, Hamainza B, Killeen G, Magill A, et al. “Asymptomatic” malaria: a chronic and debilitating infection that should be treated. PLoS Med. 2016;13:e1001942.
Betuela I, Rosanas-Urgell A, Kiniboro B, Stanisic DI, Samol L, De Lazzari E, et al. Relapses contribute significantly to the risk of Plasmodium vivax infection and disease in Papua New Guinean children 1–5 years of age. J Infect Dis. 2012;206:1771–80.
White MT, Karl S, Koepfli C, Longley RJ, Hofmann NE, Wampfler R, et al. Plasmodium vivax and Plasmodium falciparum infection dynamics: re-infections, recrudescences and relapses. Malar J. 2018;17:170.
Mendis K, Sina BJ, Marchesini P, Carter R. The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg. 2001;64(1–2 Suppl):97–106.
Wellems TE, Plowe CV. Chloroquine-resistant malaria. J Infect Dis. 2001;184:770–6.
Price RN, Douglas NM, Anstey NM, von Seidlein L. Plasmodium vivax treatments: what are we looking for? Curr Opin Infect Dis. 2011;24:578–85.
WHO. Guidelines for the treatment of malaria. 3rd ed. Geneva: World Health Organization; 2015.
Sinclair D, Gogtay N, Brand F, Olliaro P. Artemisinin-based combination therapy for treating uncomplicated Plasmodium vivax malaria. Cochrane Database Syst Rev. 2011;7:CD008492.
Alving AS, Hankey DD, Coatney GR, Jones R Jr, Coker WG, Garrison PL, et al. Korean vivax malaria. II. Curative treatment with pamaquine and primaquine. Am J Trop Med Hyg. 1953;2:970–6.
Taylor R. Improving the radical cure of P. vivax—the efficacy results of the IMPROV study. In: Annual meeting of the American Society of Tropical Medicine and Hygiene, New Orleans. 2018.
Chu CS, Phyo AP, Turner C, Win HH, Poe NP, Yotyingaphiram W, et al. Chloroquine versus dihydroartemisinin–piperaquine with standard high-dose primaquine given either for 7 days or 14 days in Plasmodium vivax malaria. Clin Infect Dis. 2019;68(8):1311–9.
Daher A, Silva J, Stevens A, Marchesini P, Fontes CJ, Ter Kuile FO, et al. Evaluation of Plasmodium vivax malaria recurrence in Brazil. Malar J. 2019;18:18.
Abdon NP, Pinto AY, Silva RDS, de Souza JM. Assessment of the response to reduced treatment schemes for vivax malaria. Rev Soc Bras Med Trop. 2001;34:343–8.
WHO. Control and elimination of vivax malaria: a technical brief. Geneva: World Health Organization; 2015.
WHO. Methods for surveillance of antimalarial drug efficacy. Geneva: World Health Organization; 2009.
Baird JK, Louisa M, Noviyanti R, Ekawati L, Elyazar I, Subekti D, et al. Association of impaired cytochrome P450 2D6 activity genotype and phenotype with therapeutic efficacy of primaquine treatment for latent Plasmodium vivax malaria. JAMA Netw Open. 2018;1:e181449.
Pybus BS, Marcsisin SR, Jin X, Deye G, Sousa JC, Li Q, et al. The metabolism of primaquine to its active metabolite is dependent on CYP 2D6. Malar J. 2013;12:212.
Bennett JW, Pybus BS, Yadava A, Tosh D, Sousa JC, McCarthy WF, et al. Primaquine failure and cytochrome P-450 2D6 in Plasmodium vivax malaria. N Engl J Med. 2013;369:1381–2.
Silvino ACR, Costa GL, de Araújo FCF, Ascher DB, Pires DEV, Fontes CJF, et al. Variation in human cytochrome P-450 drug-metabolism genes: a gateway to the understanding of Plasmodium vivax relapses. PLoS ONE. 2016;11:e0160172.
Brasil LW, Rodrigues-Soares F, Santoro AB, Almeida AC, Kühn A, Ramasawmy R, et al. CYP2D6 activity and the risk of recurrence of Plasmodium vivax malaria in the Brazilian Amazon: a prospective cohort study. Malar J. 2018;17:57.
Ladeia-Andrade S, Menezes MJ, de Sousa TN, Silvino ACR, de Carvalho JF, Salla LC, et al. Monitoring the efficacy of chloroquine–primaquine therapy for uncomplicated Plasmodium vivax malaria in the main transmission hot spot of Brazil. Antimicrob Agents Chemother. 2019;63:e01965-18.
Gil JP, Gil Berglund E. CYP2C8 and antimalaria drug efficacy. Pharmacogenomics. 2007;8:187–98.
Pereira D, Daher A, Zanini G, Maia I, Fonseca L, Pitta L, et al. Safety, efficacy and pharmacokinetic evaluations of a new coated chloroquine tablet in a single-arm open-label non-comparative trial in Brazil: a step towards a user-friendly malaria vivax treatment. Malar J. 2016;15:477.
WHO. Methods and techniques for assessing exposure to antimalarial drugs in clinical field studies. Geneva: World Health Organization; 2011.
Minestério da Saúde, ANVISA. Guia para validação de métodos analíticos e bioanalíticos, 2003.
Food and Drug Administration. Bioanalytical method validation—guidance for industry. 2001.
Neely MN, van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit. 2012;34:467–76.
Uthman OA, Graves PM, Saunders R, Gelband H, Richardson M, Garner P. Safety of primaquine given to people with G6PD deficiency: systematic review of prospective studies. Malar J. 2017;16:346.
Rezende AM, Tarazona-Santos E, Fontes CJ, Souza JM, Couto AD, Carvalho LH, et al. Microsatellite loci: determining the genetic variability of Plasmodium vivax. Trop Med Int Health. 2010;15:718–26.
de Araujo FC, de Rezende AM, Fontes CJ, Carvalho LH, de Brito CFA. Multiple-clone activation of hypnozoites is the leading cause of relapse in Plasmodium vivax infection. PLoS ONE. 2012;7:e49871.
Daher A, Pereira D, Lacerda MVG, Alexandre MAA, Nascimento CT, Alves de Lima ESJC, et al. Efficacy and safety of artemisinin-based combination therapy and chloroquine with concomitant primaquine to treat Plasmodium vivax malaria in Brazil: an open label randomized clinical trial. Malar J. 2018;17:45.
Gaedigk A, Sangkuhl K, Whirl-Carrillo M, Klein T, Leeder JS. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med. 2017;19:69–76.
White NJ. Determinants of relapse periodicity in Plasmodium vivax malaria. Malar J. 2011;10:297.
Administration. UFaD. Clinical pharmacology and biopharmaceutics review. Application Number: 22-268. 2008.
Liu Q, Tang Z, Surdenikova L, Kim S, Patel KN, Kim A, et al. Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell. 2009;139:1353–65.
Commons RJ, Simpson JA, Thriemer K, Humphreys GS, Abreha T, Alemu SG, et al. The effect of chloroquine dose and primaquine on Plasmodium vivax recurrence: a WorldWide Antimalarial Resistance Network systematic review and individual patient pooled meta-analysis. Lancet Infect Dis. 2018;18:1025–34.
Imwong M, Snounou G, Pukrittayakamee S, Tanomsing N, Kim JR, Nandy A, et al. Relapses of Plasmodium vivax infection usually result from activation of heterologous hypnozoites. J Infect Dis. 2007;195:927–33.
Nguyen TN, von Seidlein L, Nguyen TV, Truong PN, Hung SD, Pham HT, et al. The persistence and oscillations of submicroscopic Plasmodium falciparum and Plasmodium vivax infections over time in Vietnam: an open cohort study. Lancet Infect Dis. 2018;18:565–72.
Acknowledgements
The authors thank all the patients who participated in this study, and CEPEM, FMT-HVD, VPPLR/Fiocruz and SEFAR/Fiocruz, teams. The authors thank the Program for Technological Development in Tools for Health-PDTIS-FIOCRUZ for the use of the DNA sequencing (RPT01E) and Real-Time PCR (RPT09D) facilities. CAB and TNS thank CNPq for the research scholarship support. ACRS thanks the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for scholarship support (Finance Code 001). DFR thanks Fapemig for scholarship support. MVL is a CNPq fellow. The Brazilian National Council of Research by Science Without Border programme (233618/2014-7) granted funds to AD scholarship.
Funding
The Brazilian National Council of Research by the Fiocruz Programme of Excellence in Clinical Research (402131/2011-80) granted funds to this study. The Institute of Drug Technology (Farmanguinhos), Oswaldo Cruz Foundation (Fiocruz), Ministry of Health of Brazil provided additional funding. This work was supported in part by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG).
Author information
Authors and Affiliations
Contributions
AD was the study coordinator. DP and MVL were the principal investigators. MAAA, CTN, collected the clinical data. JCA and AD performed the analysis of the data. LBF, DMDS, DPP performed the pharmacokinetics analysis. GA performed the analysis of the pharmacokinetics data. CFAB, ACRS, TNS and DFR performed the PCR analysis. AD, JCA, DGL, FOtK, CFAB participated in interpretation of the data and critical revisions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Ethics Committee at the Tropical Medicine Research Centre of Rondonia (CEPEM Nº 31/11 CEP/CEPEM e 0018.0.046.000-11 CAAE–SISNEP and Plataforma Brasil Nº 74869 CEP/CEPEM e Nº 05462612.7.0000.0011 CAAE) reviewed and approved the clinical study protocol and informed consent from all participants was acquired before the study initiation, as declared in the ethics and regulatory statement. All signed informed consent forms are kept for at least 5 years after the study end.
Consent for publication
This manuscript does not contain any individualized data. The confidentiality of the patients’ records has been observed according to ethical regulations.
Competing interests
The authors declare following competing interests and financial disclosure: André Daher is an employee of the Institute of Drug Technology (Farmanguinhos), Oswaldo Cruz Foundation (Fiocruz), a Brazilian governmental institution of Ministry of Health. Farmanguinhos is one of the study sponsors. He was involved in the study design, decision to publish, and preparation of the manuscript. Farmanguinhos does not sell medicine on the market. The Brazilian Ministry of Health exclusively drives its drug production. These disclosures do not alter our adherence policies on sharing data and materials. There are no restrictions on the sharing of data and/or materials.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Genotyping of Plasmodium vivax polymorphic loci from patients during initial infection and recurrence. Size of PCR products in base pairs (bp). #Number of patient and day of sample collection: d0—diagnosis and treatment, dX—the day of recurrence. *Allele similar to initial infection present in lower intensity. NA non-amplified.
Additional file 2.
Genotyping of CYP2C8 gene and CYP2D6 gene and predicted phenotype of CYP2D6 activity. a CYP2D6 haplotype inferred using Phase software. b Activity score and inferred phenotype of CYP2D6: AS = 1.5 or 2 − normal metabolizer fast (gNM-F); AS ≥ 2 − ultra metabolizer (gUM) (more than 2 copies of the normal allele); AS = 1 − normal metabolizer slow (gNM-S); AS = 0.5 − intermediate metabolizer (gIM); AS = 0 − poor metabolizer (gPM), according to Gaedigk et al. [20]. c Sum of AS attributed to allele 1 and 2 of CYP2D6 gene. NP not performed.
Additional file 3.
Evaluation of pharmacokinetics’ parameters, gender and weight as predictors of failures per treatment drug (Generalized Linear Model, binomial logit link).
Additional file 4.
Evaluation of pharmacokinetics’ parameters and weight as predictors of D3 failures per treatment drug (Generalized Linear Model, binomial logit link). Only available to chloroquine, all males. ASMQ and AL 100% presented clearance at D3.
Additional file 5.
Evaluation of pharmacokinetics’ parameters, gender and weight as predictors of time to failures per treatment drug (Cox regression).
Additional file 6.
Evaluation of pharmacokinetics’ parameters as predictors of frequent (n ≥ 30) adverse event (possible and likely related to treatment) per system and drug using Generalized Estimation Equation log-binomial regression. *CQ AUC and weigh correlation is significant at the 0.01 level (2-tailed). Weight was excluded as a covariate.
Additional file 7.
Evaluation of pharmacokinetics’ parameters and weight as predictors of the drop in haemoglobin* at day 14 using ordinary least squares. *Hb at day 14 − Hb at baseline/Hb at baseline. **CQ AUC and weigh correlation is significant at the 0.01 level (2-tailed). Weight was excluded as a covariate. NA non-applicable.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Daher, A., Aljayyoussi, G., Pereira, D. et al. Pharmacokinetics/pharmacodynamics of chloroquine and artemisinin-based combination therapy with primaquine. Malar J 18, 325 (2019). https://doi.org/10.1186/s12936-019-2950-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-019-2950-4