
Abstract
Background
Although the use of induced blood stage malaria infection has proven to be a valuable tool for testing the efficacy of vaccines and drugs against Plasmodium falciparum, a limiting factor has been the availability of Good Manufacturing Practice (GMP)—compliant defined P. falciparum strains for in vivo use. The aim of this study was to develop a cost-effective method for the large-scale production of P. falciparum cell banks suitable for use in clinical trials.
Methods
Genetically-attenuated parasites (GAP) were produced by targeted deletion of the gene encoding the knob associated histidine rich protein (kahrp) from P. falciparum strain 3D7. A GAP master cell bank (MCB) was manufactured by culturing parasites in an FDA approved single use, closed system sterile plastic bioreactor. All components used to manufacture the MCB were screened to comply with standards appropriate for in vivo use. The cryopreserved MCB was subjected to extensive testing to ensure GMP compliance for a phase 1 investigational product.
Results
Two hundred vials of the GAP MCB were successfully manufactured. At harvest, the GAP MCB had a parasitaemia of 6.3%, with 96% of parasites at ring stage. Testing confirmed that all release criteria were met (sterility, absence of viral contaminants and endotoxins, parasite viability following cryopreservation, identity and anti-malarial drug sensitivity of parasites).
Conclusion
Large-scale in vitro culture of P. falciparum parasites using a wave bioreactor can be achieved under GMP-compliant conditions. This provides a cost-effective methodology for the production of malaria parasites suitable for administration in clinical trials.
Similar content being viewed by others


Background
The use of controlled human malaria infection (CHMI) for testing the efficacy of vaccines and drugs against Plasmodium falciparum is now well established. Induced blood stage malaria (IBSM) infection was pioneered in the 1990s [1,2,3], and was subsequently refined [4,5,6,7]. A limiting factor in the IBSM system has been the restricted number of defined P. falciparum strains available. This is in part due to difficulties obtaining suitable material for IBSM studies. Until recently it has been necessary to rely on access to deliberately infected volunteers or malaria-infected travellers returning from overseas. In both cases, ethical approval is required to collect, store and use this material and there are concerns about contaminating infectious agents.
A process for culturing sufficient volumes of P. falciparum in tissue culture flasks for in vitro production of Good Manufacturing Practice (GMP) grade blood stage malaria cell banks was recently described [8]. This provides a culture-based alternative to sourcing parasites from individuals who have either experimental or natural infection (travellers returning from malaria endemic areas). This allows more flexibility in that new banks can be created as required, and not only when a suitable donor becomes available. It also allows for tight control over all components used in the cell bank manufacture and ensures that cell banks are renewable resources.
However, bulk preparation of P. falciparum cultures is both time consuming and technically demanding; yields can be low and parasite quality can be variable when grown in static culture. Multiply-infected erythrocytes (containing two or more parasites) are also observed more frequently than in suspension cultures [9]. The presence of erythrocytes containing multiple parasites is undesirable for IBSM studies where the parasite dose should be carefully controlled.
Dalton et al. have developed a method for production of large scale suspension cultures of asexual and sexual blood stage P. falciparum using a wave bioreactor (Wave Bioreactor™ 20/50 EHT system) [9, 10]. Growth of P. falciparum in the wave bioreactor system is superior to static flask cultures because parasites retain synchrony over at least 3 cycles of invasion. Additionally, the development of multiply infected erythrocytes is reduced; this was shown to be both consistent and reproducible [9, 10]. Disposable plastic bioreactors also allow for careful control and monitoring of the cellular environment, and have been widely used for the culture of plant, animal and microbial cells, and for the production of proteins, antibodies and other biologics conforming to GMP standards [11].
Here, the manufacture and release of a cell bank that complies with the GMP requirements for a phase 1 investigational product is described. The cell bank was manufactured using a wave bioreactor and consists of erythrocytes infected with genetically modified P. falciparum parasites. This cost-effective methodology using appropriate GMP compliant conditions allows for the production of sufficient quantities of material for IBSM studies and whole blood-stage vaccine trials, with sufficient material available for quality control and ongoing stability testing. The manufacture of the master cell bank was conducted in accordance with the United States Food and Drug Administration (FDA) Guidance for Industry: CGMP for Phase 1 Investigational Drugs.
Methods
Production of Plasmodium falciparum 3D7-KAHRP KO GAP pre-seed bank
Genetically attenuated parasites (GAP) were produced by targeted deletion of the gene encoding the knob-associated histidine-rich protein (kahrp) from P. falciparum strain 3D7, in a similar manner as previously described for this and other genes [12, 13]. Briefly, deletion of kahrp was achieved by a positive–negative selection strategy [14] whereby the entire kahrp gene was replaced through double cross-over homologous recombination with a cassette encoding human dihydrofolate reductase (dhfr), a selectable marker that renders transfectants resistant to WR99210 (Jacobus Pharmaceuticals, NJ).
The loxP-DHFR-loxP-pCC1 plasmid was created by insertion of a loxP-hDHFR-loxP cassette into the pCC1 plasmid backbone [15], as previously described [16]. The KAHRP-KO plasmid was created by cloning kahrp 5′ and 3′ sequences into this plasmid. Plasmid DNA was transfected into P. falciparum 3D7 by electroporation. Following WR positive selection, parasites were grown without WR to deselect those containing the WR-containing plasmid, thereafter WR was reapplied to select those with integration of the dhfr cassette into the genome. These were then placed under negative selection with 5–fluorocytosine (5-FC; Sigma) to select for homologous recombination. 5-FC resistant parasites were genotyped by PCR.
KAHRP-KO parasites were cloned by limiting dilution in 96-well flat bottom plates and the resulting clonal line, 3D7-KAHRP-KO, was characterized by whole genome sequencing (Accession Number PRJEB12838).
The P. falciparum 3D7 parasites used for transfection with the kahrp knockout vector were created from the 3D7 parental strain described previously [17]. This strain was used for the original IBSM studies [1] and has been extensively used for subsequent studies.
Production of 3D7-KAHRP KO GAP seed bank
The GAP seed bank was produced by thawing the contents of one vial of the GAP pre-seed bank following a standard protocol [18] and expanding the parasites in culture (RPMI 1640 media supplemented with 10% heat-treated pooled human serum and 4% human erythrocytes) using standard methods [19]. At each passage, fresh complete media and erythrocytes were added and the culture was expanded to achieve the required volume for cell banking. When the culture contained a minimum of 80% ring-stage parasites at 3–5% parasitaemia, the seed bank was cryopreserved using Glycerolyte 57 (Fenwal Inc.) in a 1:2 ratio, aliquoted into 1 mL cryovials, and stored in vapour phase liquid nitrogen. The final GAP seed bank was transferred to a Therapeutic Goods Administration (TGA) licensed GMP facility (Q-Gen; QIMR Berghofer Medical Research Institute [QIMRB]) for further expansion and production of the GAP master cell bank.
Production of 3D7 KAHRP-KO GAP master cell bank
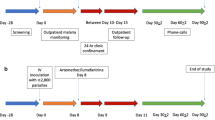
The 3D7 KAHRP-KO GAP master cell bank (GAP MCB) was manufactured in a ISO Class 5 Biological Safety Cabinet within an ISO Class 7 clean room using tissue culture flasks and a bioreactor (GE Wave 25™ System). The GAP MCB dossier was reviewed by the United States Food and Drug Administration (FDA) under the investigational new drug (IND) program. A summary of the process is shown in Fig. 1 and described in detail below.

Process overview. The steps involved in production of the 3D7 KAHRP-KO GAP master cell bank
Parasite thaw and culture in flasks
A vial of the 3D7 KAHRP-KO GAP seed bank was thawed at 37 °C with the stepwise addition of 12 and 1.6% sodium chloride following a standard protocol [18]. The resulting red blood cell (RBC) pellet was washed in 0.9% sodium chloride and resuspended in 8 mL of complete RPMI-Hepes 1640 media (Life Technologies) containing 100 µL of 50% haematocrit (HCT) washed RBCs, and the cell suspension was transferred to a T25 cm2 flask.
Parasites were cultured in flasks under standard malaria culture conditions [19] in an atmosphere of 5% carbon dioxide, 5% oxygen and 90% nitrogen at 37 °C and media was changed every 24 h. Parasitaemia was monitored by examination of Giemsa-stained thin blood films. Additional media and fresh RBCs were added as required to maintain the culture at a parasitaemia of between 0.5 and 4%, and with a 2–5% HCT. The parasite culture was expanded to a volume of approximately 100 mL and the parasites were synchronized by a single treatment with 5% sorbitol [20] prior to transfer to a 2 L cellbag of the Wave 25™ Bioreactor.
Parasite culture using wave bioreactor
The Ready to Process WAVE 25™ system (GE Healthcare) was used for this study and configuration of the equipment is shown in Fig. 2. The system consists of a pre-sterile single use cultivation container (cellbag) made of FDA approved plastic. Each cellbag has built-in inlet and outlet air filters, and ports that allow the addition of culture medium and extraction of samples (Fig. 2a). The cellbag is placed on a temperature-controlled rocking platform (Fig. 2b); gas transfer and mixing of the cell culture is facilitated by movement of the rocker unit which induces a wave motion in the culture. Temperature, rocking speed, rocking angle and atmospheric conditions (Fig. 2c) are monitored and controlled by the cellbag control unit (CBCU). The system is controlled from a PC running UNICORN software version 6.3.2 or later.

Wave bioreactor configuration. a Each cellbag has built-in inlet and outlet air filters, and ports that allow the addition of culture medium and extraction of samples. b The Ready to Process WAVE 25™ system (GE Healthcare) was used for this study. c Temperature, rocking speed, rocking angle and atmospheric conditions are monitored and controlled by the cellbag control unit (CBCU)
The synchronous parasite culture was transferred into a 2 L cellbag and maintained under conditions controlled electronically by the bioreactor system (Fig. 2c). Parasitaemia was monitored by examination of Giemsa-stained thin blood films. The culture medium was changed daily, with washed RBCs added as required to maintain a parasitaemia of 0.5–1.2% and a 2–5% HCT. When the culture volume reached 500 mL it was transferred to a 10 L cellbag and the parasite culture was expanded to a final volume of approximately 2 L in line with the manufacturers recommended culture volumes for 2 and 10 L cellbags. To facilitate media change and transfer between cellbags the cell culture was removed from the in use cellbag under sterile conditions, centrifuged in 250 mL conical bottles (Corning) and the supernatant removed. The required volume of fresh media was pumped into the destination cell bag under the control of the CBC unit and the required volume of inoculum added under gravity flow.
The culture was monitored to ensure synchronicity and was maintained such that the % of ring stage parasites (as a % of the total parasites) present in the culture did not fall below 80%. When the required volume was reached, the culture was maintained for one further invasion cycle before parasites were harvested for banking at a parasitaemia target of > 3% and a minimum of 80% ring stage parasites. Progression through the final multiplication cycle was monitored by examination of Giemsa-stained thin blood films every 4–6 h. The cell culture supernatant was tested for microbial contamination prior to culture in the Wave 25™ Bioreactor system and prior to harvesting. At harvest the entire cell culture was removed under sterile conditions and centrifuged in 250 mL conical bottles to pellet the RBCs prior to freezing.
Culture media
RPMI-Hepes 1640 media (Life Technologies) was supplemented with 10% heat inactivated (HI) pooled human serum and hypoxanthine (HT) supplement (0.1 mM sodium hypoxanthine and 0.016 mM thymidine). Complete media was used immediately or dispensed in aliquots and stored at a temperature of between 2 and 8 °C for a maximum of 7 days.
The serum used was supplied, screened and tested by Key Biologics (Memphis USA), an FDA registered blood collection and processing establishment. Whole blood units were collected from donors who were screened and found acceptable for donation of transfusable blood components based on all applicable FDA requirements. Infectious disease testing as required for qualification of blood components for transfusion to humans was carried out and was required to be non-reactive or negative. Serum from individual donors was tested (using FDA approved test kits) for human immunodeficiency virus (HIV)-I and HIV-II, human T-lymphotropic virus (HTLV) types I and II, hepatitis B virus (HBV), hepatitis C virus (HCV), Trypanasoma cruzi and syphilis (by serology), and underwent nucleic acid testing for HBV, HCV, HIV, and West Nile virus (WNV). Serum was then pooled from the multiple donors, frozen, stored and shipped under controlled conditions to QIMRB. On arrival the serum was thawed, heat inactivated at 56 °C, filtered and stored at − 20 °C in 50 mL aliquots until required. The pooled serum was additionally tested for Epstein Barr virus, Parvovirus B19 and herpes virus type 6 by PCR at Pathology Queensland. Endotoxin testing and sterility testing was performed at Q-Gen.
Leukodepleted packed RBCs (Group O Rh (D) negative) used for in vitro cultivation of P. falciparum parasites were obtained from the Australian Red Cross Blood Service (Blood Service). All supplied RBCs were screened according to Blood Service protocols to eliminate individuals with risk of exposure to transmissible spongiform encephalopathy and blood borne agents. The blood was collected and leukodepleted at the Blood Service and the donor’s blood sample was tested and certified non-reactive for HIV, HBV, HCV, HTLV and syphilis using diagnostic kits approved by the TGA. A blood sample was additionally tested by Pathology Queensland for infectious diseases that are not included in mandatory testing for blood and blood products including anti-Flavivirus IgM antibodies, Trypanasoma cruzi IgG antibodies, Barmah Forest virus IgM and IgG antibodies, and Ross River virus IgM and IgG antibodies. Samples were also screened (by PCR testing) for Epstein Barr virus (EBV), Cytomegalovirus (CMV), Parvovirus B19, Human Herpes virus type 6 and type 7(HHV-6 and HHV-7), and WNV. On receipt at QIMRB the cells were transferred from the Blood Service pack to a sterile container and a 2 mL aliquot was removed for sterility testing. Aliquots of RBCs were washed three times in RPMI and resuspended in complete RPMI to give a HCT of 50%. Blood was washed as required in aliquots not exceeding 20 mL and stored at 2–8 °C for up to 7 days after washing.
Cryopreservation of the GAP MCB
Percentage parasitaemia and parasite life cycle stage were assessed by microscopic examination of thin blood films and when cultures achieved > 3% parasitaemia with at least 80% ring-stage parasites the cell bank was cryopreserved using Glycerolyte 57 in a 1:2 ratio (i.e. two volumes of Glycerolyte 57 were added to each volume of pRBC) as previously described [1].
The cell bank was dispensed in 1 mL aliquots in cryovials, cryopreserved using a controlled rate freezer as previously described [8] and stored in vapour phase liquid nitrogen at –196 °C. A total of 200 vials were produced; two vials were allocated as retention samples. The remainder of the cell bank vials were allocated for release testing. The release testing of the MCB complied with the requirements of the FDA’s Guidance for Industry- Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications, the United States Pharmacopeia (USP) < 85 > for Bacterial Endotoxin Testing, USP < 63 > mycoplasma/spiroplasma testing, and the British Pharmacopoeia Appendix XVI A: Test for Sterility. The parameters assessed, acceptance criteria, test methods and place of testing are listed in Table 1.
Parasite viability following cryopreservation
Parasite viability post-freezing was determined by measurement of cloning efficiency via a limiting dilution assay [8, 21] with positivity determined by production of P. falciparum histidine-rich protein II (HRP-II).
In order to determine post-freeze viability, a vial of the GAP MCB was thawed following a standard protocol [18] and a sample was removed for RBC counting using a haemocytometer. The number of parasitized/infected RBCs (pRBCs) was determined via microscopic examination of a Giemsa-stained thin blood smear.
Dilutions of pRBCs were prepared in complete media and dispensed in 100 μL aliquots in 96-well flat bottom plates at 2% HCT at theoretical concentrations of 40, 20, 10, 5, 2.5, 1.25, 0.625 and 0.3125 parasites/well. Thirty-two replicates of each concentration were plated out and non-parasitized RBCs were included as a negative control. Plates were incubated in an atmosphere of 5% O2, 5% CO2 and 90% N2 at 37 °C for 7 days and media was replenished on Day 4. On Day 7, supernatant was removed from each well and processed immediately or frozen for future testing. The supernatant was tested for P. falciparum HRPII using the SD Malaria Antigen Pf ELISA kit, according to the manufacturer’s instructions.
Briefly, supernatants from each well were transferred to the corresponding well of an uncoated microplate and incubated for 15 min following the addition of 150 µL enzyme conjugate (anti-mouse P. falciparum HRP II Ig conjugated to horseradish peroxidase in lysis buffer). A 100 µL sample was then transferred from each well of the uncoated microplate to the corresponding well of the pre-coated assay microplate and incubated. Plates were washed and 100 µL of tetramethylbenzidine substrate added to each well. After a 10 min incubation, the reaction was stopped by the addition of 100 µL of 1 mL/L (1 N) hydrochloric acid. The absorbance was read at 450 nm with reference wavelength at 620 nm. The mean absorbance values (optical density [OD]) for the positive and negative controls were calculated in order to ensure the validity of the assay.
The cut off value for parasite positivity was calculated using the absorbance values of the 32 two wells containing the RBC negative control. The cut off value was the mean plus 3 standard deviations and this value was applied to the test samples to calculate the number of positive and negative wells. Limiting dilution analysis was performed to determine the frequency of viable parasites as previously described [22].
In vitro anti-malarial drug sensitivity testing of the GAP MCB
Anti-malarial drug sensitivity testing was carried out on the GAP MCB parasites using the standard [3H]-hypoxanthine uptake inhibition assay [23] to ensure that the drug resistance phenotype of the parasites had not changed during culture. The in vitro sensitivity of the GAP MCB parasites to nine anti-malarial drugs was assessed (chloroquine, dihydroartemisinin, piperaquine, lumefantrine, amodiaquine, atovaquone, pyronaridine, mefloquine, quinine). The parental 3D7 line (chloroquine sensitive) and W2 (chloroquine resistant) were included as controls. The assays were performed at the Army Malaria Institute, Queensland, Australia.
Briefly, aliquots of parasites from the GAP MCB were grown in vitro and synchronized to give 95% ring stage parasites. The parasite culture was dispensed into a 96-well plate containing twofold dilutions of each anti-malarial drug for testing. [3H]-hypoxanthine was added and the amount incorporated into parasites was measured after a 48-h incubation. The IC50 values for each drug were determined by nonlinear regression analysis. This calculation was performed using GraphPad Prism software (GraphPad Prism version 6.00). Drug threshold values were expressed as mean IC50 (95% confidence interval) in nmol/L. The results were compared to published threshold values for drug resistance [24].
Genotyping of the GAP MCB
The genotype of the GAP MCB was determined by PCR using primers to confirm KAHRP gene deletion and hDHFR integration. In vitro culture samples were taken at the beginning of the process (Day 0) and at harvest (Day 16) and placed in Qiagen lysis buffer. Genomic DNA was extracted from paired samples using a Qiagen DNAmp DNA Blood Mini Kit and gDNA from the parent strain 3D7 was used as a control. Standard PCR was set up using 4 pairs of primers (Table 2) in PCR mix as follows:
- Mix 1. :
-
Primers mo517 and mo518. No band expected when used with gDNA from P. falciparum 3D7 KAHRP-KO.
- Mix 2. :
-
Primers mo517 and Aw560 Presence of a 1463 bp band expected when used with gDNA from P. falciparum 3D7 KAHRP-KO.
- Mix 3. :
-
Primers mo519 and mo520 No band expected when used with gDNA from P. falciparum 3D7 KAHRP-KO.
- Mix 4. :
-
Primers Aw171 and mo520 Presence of 1128 bp expected when used with gDNA from P. falciparum 3D7 KAHRP-KO.
Amplification of the PCR product was performed using Taq DNA polymerase under the following cycling conditions: One initial cycle at 45 °C for 15 min, denaturing at 94 °C for 30 s, annealing at 52 °C for 40 s, extension at 68 °C for 105 s (30 cycles), one final extension cycle at 68 °C for 7 min.
Quality check of GAP MCB by qRT-PCR
To further validate the successful genetic attenuation of P. falciparum 3D7 and verify hDHFR integration, a real time PCR assay was set up using primers designed at various regions of the P. falciparum 3D7 GAP (Fig. 3). Primer sequences are listed in Table 2.

qRT-PCR primers mapped on KAHRP gene. To validate the successful genetic attenuation of P. falciparum 3D7 and verify hDHFR integration, a real time PCR assay was set up using primers designed at various regions of the P. falciparum 3D7 GAP
To determine limit of detection of possible contamination with wildtype P. falciparum 3D7, two single copy genes (pfmdr-1 and pfldh) were amplified and used as reference for copy number estimation. A standard curve containing five ten-fold serial dilutions of wildtype P. falciparum 3D7 was prepared, starting from 2.0 ng/μL to amplify KAHRP excised region and MDR1 and LDH genes.
Triplicate PCR reactions were set up using the Roche Fast Start Essential DNA Green Master Mix (Cat. No. 06 402 712 001), 10 μM of each primer (Table 2) and 1 μL of each gDNA sample in a 12 μL reaction volume. Quantitative PCR was performed using a Roche Light Cycler 96 with the following cycling conditions: 94 °C for 3 min; 45 cycles of 94 °C for 30 s, 55 °C for 30 s and 68 °C for 30 s, and one final extension cycle of 68 °C for 5 min.
Other testing performed on the Gap MCB
A summary of testing performed on the GAP MCB is shown in Table 1. Sterility testing complying with the requirements of the British Pharmacopoeia (BP) Appendix XVI A: Test for Sterility was carried out using direct inoculation as well as testing the supernatant following membrane filtration. Endotoxin inhibition testing of the GAP MCB and Glycerolyte 57 was performed to comply with the requirements of USP < 85 > Bacterial Endotoxin Test. Testing for Adventitious Agents complied with the requirements outlined in the FDA’s Guidance for Industry: Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications. Assays for HIV 1 and 2, HTLV I&II, EBV, CMV, HCV, HBV, human parvovirus (B19), West Nile virus (WNV), adeno-associated virus (AAV), human herpes virus type 7, human herpes virus type 6 and retroviruses were performed in addition to testing for spiroplasma and mycoplasma. Sections were examined by quantitative transmission electron microscopy for the detection of viruses, fungi, yeasts, and bacteria (200 cell profiles). In vitro assays for the detection of viral contaminants and inapparent viruses was also performed.
Results
A cell bank consisting of erythrocytes infected with genetically-modified P. falciparum parasites was successfully manufactured using a wave bioreactor. Two hundred vials of the GAP MCB were produced that met the requirements for use in Phase I clinical trials.
Growth of P. falciparum 3D7-KAHRP KO GAP in a bioreactor
GAP parasites maintained an asexual blood stage cycle of approximately 39 h during the in vitro culture period in the bioreactor, and showed a normal maturation pattern. The percent parasitaemia and percent ring stage parasites of the culture over time is illustrated in Fig. 4. Parasitaemia was maintained between 0.5 and 2% by the addition of RBCs during the expansion phase (Day 0–10). During the banking phase (Day 10–15) the parasitaemia was maintained below 4%. At harvest the culture had a parasitaemia of 6.3%, with 96% of parasites at ring stage (high percentage of ring stage parasites is illustrated in Fig. 5). Thus, the pre-defined acceptable criteria of > 3% parasitized RBC and ≥ 80% ring stage parasites (Table 1) were met.

Profile of parasitaemia and parasite life cycle stages during growth in biowave reactor system. Parasites maintained an asexual blood stage cycle of approximately 39 h during the in vitro culture period, and showed a normal maturation pattern. The percent parasitaemia and percent ring stage parasites of the culture over time were determined by microscopy

Giemsa-stained smear showing GAP MCB parasites at time of cryopreservation. At harvest the culture contained a high percentage of ring stage parasites
Identity testing of the GAP MCB
PCR assays confirmed kahrp gene deletion and hDHFR integration in parasite samples collected at the initiation of culture (Day 0) and at harvest (Day 16) (Fig. 6) demonstrating that the genotype of the parasites was maintained during the culture and banking process.

Agarose gel showing the genetic identity of the GAP MCB at Days 0 and 16. PCR assays confirmed kahrp gene deletion and hDHFR integration in parasite samples collected at the initiation of culture (Day 0) and at harvest (Day 16)
Quality check of Pf KAHRP-KO by qRT-PCR
Mean Ct values using primer pairs for KAHRP (inside remaining region), hDHFR and recombination region were < 31 (positive) using KAHRP-KO gDNA as template indicating success of integration. Mean Ct values were > 31 (negative) using P. falciparum 3D7 (wildtype) gDNA as template. Mean Ct value using primer pairs for KAHRP gene (in excised region) was < 31 (positive) only for P. falciparum 3D7 (wildtype) gDNA. Mean Ct values were > 31 (negative) or undetectable (no Ct) for KAHRP-KOa and KAHRP-KOb gDNA as templates indicating success of deletion in the genetically attenuated parasite.
Amplification of the KAHRP excised region on serially diluted wildtype P. falciparum 3D7 and comparing Ct values obtained from serially diluted GAP showed undetectable contamination of the GAP starting from 2.0 ng/μL DNA concentration up to as low as 2.0 × 10−5 ng/μL. This is equivalent to a single copy or less using single copy gene references pfmdr-1 and pfldh.
In vitro drug sensitivity of GAP MCB parasites
The in vitro sensitivity of the GAP MCB parasites to a panel of 9 anti-malarial drugs is shown in Table 3. The mean IC50 (95% confidence interval) results for each drug are presented in Table 4 along with the corresponding results for the control P. falciparum strains 3D7 and W2.
GAP master cell bank parasites demonstrated sensitivity to eight of the anti-malarial drugs tested (chloroquine, artemisinin, piperaquine, lumefantrine, amodiaquine, atovaquone, pyronaridine, quinine) and resistance to mefloquine.
Parasite viability of the GAP MCB following cryopreservation
Parasite viability was tested using samples taken approximately 24 h (Time 0) after freezing and at 3 months post-production. The GAP MCB was determined to contain 52% viable parasites at Time 0 and 64% viable parasites at 3 months post-production. These values conform to the pre-defined acceptable criteria of > 25% viable parasites. The variability between the two time points is within normally observed limits for this assay.
Discussion
Our aim was to produce a GMP compliant cell bank consisting of KAHRP deficient P. falciparum parasites using the wave bioreactor system. Previous studies have shown that the wave bioreactor system is suitable for the production of large volumes of high quality P. falciparum cultures [9]. We have taken this method further to demonstrate that the wave bioreactor can be used to culture parasites for use in phase 1 clinical trials.
The use of a wave bioreactor system has a number of significant advantages over traditional culture of P. falciparum in flasks. Foremost is that it allows close control of culture conditions that can be monitored and maintained electronically by the wave bioreactor system; the cellbags used are single use closed systems approved by the FDA. Significant savings in time and labour were also achieved; 100 mL of culture was transferred to a cellbag on Day 11, expanded to 500 mL on Day 12, and a culture volume of 2 L was achieved on Day 15 with harvest/cryopreservation on Day 16. During this rapid expansion phase the labour required to maintain the culture (monitor parasitaemia, change media, add fresh RBCs) was less than would be required if growing in multiple tissue culture flasks. In addition, the use of multiple flasks can result in cultures spending more time in less than optimal growth conditions, and increase the probability of contamination.
The continual wave motion of the wave bioreactor ensures gentle movement of cells in culture resulting in a high proportion of single infections [9]. This allows for accurate calculation of parasite numbers which is essential for defined dose delivery in clinical trials. Results confirmed that wave bioreactor cultures contained high quality ring stage parasites with a low number of multiply infected red cells (< 0.5%). Additionally, the effective cryopreservation of the MCB was demonstrated by the fact that parasite viability was over 50% after 3 months.
The ability to use research grade starting material, including genetically-modified parasites, to produce a GMP grade product offers significant opportunities. It allows for genetically-modified parasites to be used as vaccine candidates and for the number of P. falciparum strains available for IBSM studies to be expanded. The availability of multiple antigenically and geographically diverse P. falciparum strains for IBSM studies is essential to allow testing of new anti-malarials against multiple strains before field testing begins.
The methodology described here is highly cost effective, with each of the 200 vials produced containing sufficient material for 3600 doses at an approximate cost of USD 0.14/dose. Although the method requires use of a specialist item of equipment (wave bioreactor system), the fact that these units are becoming widely used for a range of applications (including tissue engineering, cellular and gene therapies, the production of therapeutic proteins and human vaccines) means that they are frequently available within research facilities or hospitals. The use of sterile, single use closed system cell bags means that a wave bioreactor system can be shared between groups without risk of cross contamination.
This methodology may represent a cost effective strategy by allowing extensive testing in a controlled environment (phase 1 trials) before deciding to move to field studies (phase 2 trials) which can be very expensive due to logistic and administrative challenges associated with carrying out clinical trials in a distant research site. The limiting factor for this methodology is that it is currently only useful for P. falciparum as other human malaria species are not easily grown in vitro.
Conclusion
The large scale in vitro culture of P. falciparum parasites using a wave bioreactor can be achieved under GMP compliant conditions. This provides a cost-effective methodology for the production of malaria parasites suitable for administration in clinical trials.
Abbreviations
- IBSM:
-
induced blood stage malaria
- GMP:
-
Good Manufacturing Practice
- GAP:
-
genetically-attenuated parasites
- kahrp:
-
knob-associated histidine-rich protein
- MCB:
-
master cell bank
- FDA:
-
Food and Drug Administration
- CHMI:
-
controlled human malaria infection
- dhfr:
-
human dihydrofolate reductase
- 5-FC:
-
5-fluorocytosine
- TGA:
-
Therapeutic Goods Administration
- QIMRB:
-
QIMR Berghofer Medical Research Institute
- GAP MCB:
-
3D7 KAHRP-KO GAP master cell bank
- IND:
-
investigational new drug
- RBC:
-
red blood cell
- HCT:
-
haematocrit
- HI:
-
heat inactivated
- HT:
-
hypoxanthine
- HIV:
-
human immunodeficiency virus
- HTLV:
-
human T-lymphotropic virus
- HBV:
-
hepatitis B virus
- HCV:
-
hepatitis C virus
- WNV:
-
West Nile virus
- Blood Service:
-
Australian Red Cross Blood Service
- EBV:
-
Epstein Barr virus
- CMV:
-
Cytomegalovirus
- OD:
-
optical density
- pRBC:
-
parasitized/infected RBCs
- HRP-II:
-
histidine-rich protein II
References
Cheng Q, Lawrence G, Reed C, Stowers A, Ranford-Cartwright L, Creasey A, et al. Measurement of Plasmodium falciparum growth rates in vivo: a test of malaria vaccines. Am J Trop Med Hyg. 1997;57:495–500.
Lawrence G, Cheng Q, Reed C, Taylor D, Stowers A, Cloonan N, et al. Effect of vaccination with 3 recombinant asexual-stage malaria antigens on initial growth rates of Plasmodium falciparum in non-immune volunteers. Vaccine. 2000;18:1925–31.
Pombo DJ, Lawrence G, Hirunpetcharat C, Rzepczyk C, Bryden M, Cloonan N, et al. Immunity to malaria after administration of ultra-low doses of red cells infected with Plasmodium falciparum. Lancet. 2002;360:610–7.
McCarthy JS, Marjason J, Elliott S, Fahey P, Bang G, Malkin E, et al. A phase 1 trial of MSP2-C1, a blood-stage malaria vaccine containing 2 isoforms of MSP2 formulated with Montanide(R) ISA 720. PLoS ONE. 2011;6:e24413.
McCarthy JS, Ruckle T, Djeriou E, Cantalloube C, Ter-Minassian D, Baker M, et al. A Phase II pilot trial to evaluate safety and efficacy of ferroquine against early Plasmodium falciparum in an induced blood-stage malaria infection study. Malar J. 2016;15:469.
McCarthy JS, Sekuloski S, Griffin PM, Elliott S, Douglas N, Peatey C, et al. A pilot randomised trial of induced blood-stage Plasmodium falciparum infections in healthy volunteers for testing efficacy of new antimalarial drugs. PLoS ONE. 2011;6:e21914.
Sanderson F, Andrews L, Douglas AD, Hunt-Cooke A, Bejon P, Hill AV. Blood-stage challenge for malaria vaccine efficacy trials: a pilot study with discussion of safety and potential value. Am J Trop Med Hyg. 2008;78:878–83.
Stanisic DI, Liu XQ, De SL, Batzloff MR, Forbes T, Davis CB, et al. Development of cultured Plasmodium falciparum blood-stage malaria cell banks for early phase in vivo clinical trial assessment of anti-malaria drugs and vaccines. Malar J. 2015;14:143.
Dalton JP, Demanga CG, Reiling SJ, Wunderlich J, Eng JW, Rohrbach P. Large-scale growth of the Plasmodium falciparum malaria parasite in a wave bioreactor. Int J Parasitol. 2012;42:215–20.
Demanga CG, Eng JWL, Gardiner DL, Roth A, Butterworth A, Adams JH, et al. The development of sexual stage malaria gametocytes in a Wave Bioreactor. Parasit Vectors. 2017;10:216.
Eibl R, Werner S, Eibl D. Bag bioreactor based on wave-induced motion: characteristics and applications. Adv Biochem Eng Biotechnol. 2009;115:55–87.
Crabb BS, Cooke BM, Reeder JC, Waller RF, Caruana SR, Davern KM, et al. Targeted gene disruption shows that knobs enable malaria-infected red cells to cytoadhere under physiological shear stress. Cell. 1997;89:287–96.
VanBuskirk KM, O’Neill MT, De La Vega P, Maier AG, Krzych U, Williams J, et al. Preerythrocytic, live-attenuated Plasmodium falciparum vaccine candidates by design. Proc Natl Acad Sci USA. 2009;106:13004–9.
Maier AG, Braks JA, Waters AP, Cowman AF. Negative selection using yeast cytosine deaminase/uracil phosphoribosyl transferase in Plasmodium falciparum for targeted gene deletion by double crossover recombination. Mol Biochem Parasitol. 2006;150:118–21.
Maier AG, Rug M, O’Neill MT, Brown M, Chakravorty S, Szestak T, et al. Exported proteins required for virulence and rigidity of Plasmodium falciparum-infected human erythrocytes. Cell. 2008;134:48–61.
O’Neill MT, Phuong T, Healer J, Richard D, Cowman AF. Gene deletion from Plasmodium falciparum using FLP and Cre recombinases: implications for applied site-specific recombination. Int J Parasitol. 2011;41:117–23.
Walliker D, Quakyi IA, Wellems TE, McCutchan TF, Szarfman A, London WT, et al. Genetic analysis of the human malaria parasite Plasmodium falciparum. Science. 1987;236:1661–6.
EVIMalaR. Methods in Malaria Research. 6th ed. Glasgow: EVIMalaR; 2013.
Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–5.
Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979;65:418–20.
Butterworth AS, Robertson AJ, Ho MF, Gatton ML, McCarthy JS, Trenholme KR. An improved method for undertaking limiting dilution assays for in vitro cloning of Plasmodium falciparum parasites. Malar J. 2011;10:95.
Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–8.
Estimation of Plasmodium falciparum drug susceptibility by the 3H-hypoxanthine uptake inhibition assay. WorldWide Antimalarial Resistance Network (WWARN); 2012.
Basco LK. Field application of in vitro assays sensitivity of human malaria parasites antimalarial drugs. Geneva: World Health Organization; 2007.
Authors’ contributions
RP, RF, SS, HJ, AC, LS, KT and JM contributed to the design of the study. RP, RF, SS, HJ, JH, TP, PS, CP, KE, NC, JM and KT participated in experimental work, collection and analysis of data and/or interpretation of results. RP, KT, JM, JH and CP participated in writing of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank the Australian Red Cross Blood Service for providing human erythrocytes.
Competing interests
The authors declare they have no competing interests.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
The study was funded by NHMRC Development Grant APP105999 and NHMRC Fellowship Support APP1002835 (LS) and NHMRC Fellowship Support APP1041802 (JM).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pawliw, R., Farrow, R., Sekuloski, S. et al. A bioreactor system for the manufacture of a genetically modified Plasmodium falciparum blood stage malaria cell bank for use in a clinical trial. Malar J 17, 283 (2018). https://doi.org/10.1186/s12936-018-2435-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-018-2435-x