
Abstract
Background
The Plasmodium vivax multidrug resistant 1 gene (pvmdr1) codes for a transmembrane protein of the parasite’s digestive vacuole. It is likely that the pvmdr1 gene mutations occur at different sites by convergent evolution. In here, the genetic variation of pvmdr1 at three sites of the Mesoamerican region was studied. Since 1950s, malarious patients of those areas have been treated only with chloroquine and primaquine.
Methods
Blood samples from patients infected with P. vivax were obtained in southern Mexico (SMX), in the Northwest (NIC-NW) and in the northeast (NIC-NE) of Nicaragua. Genomic DNA was obtained and fragments of pvmdr1 were amplified and sequenced. The nucleotide and amino acid changes as well as the haplotype frequency in pvmdr1 were determined per strain and per geographic site. The sequences of pvmdr1 obtained from the studied regions were compared with homologous sequences from the GenBank database to explore the P. vivax genetic structure.
Results
In 141 parasites, eight nucleotide changes (two changes were synonymous and other six were nonsynonymous) were detected in 1536 bp. The PvMDR1 amino acid changes Y976F, F1076FL were predominant in endemic parasites from NIC-NE and outbreak parasites in NIC-NW but absent in SMX. Thirteen haplotypes were resolved, and found to be closely related, but their frequency at each geographic site was different (P = 0.0001). The pvmdr1 codons 925–1083 gene fragment showed higher genetic and haplotype diversity in parasites from NIC-NE than the other areas outside Latin America. The haplotype networks suggested local diversification of pvmdr1 and no significant departure from neutrality. The F ST values were low to moderate regionally, but high between NIC-NE or NIC-NW and other regions inside and outside Latin America.
Conclusions
The pvmdr1 gene might have diversified recently at regional level. In the absence of significant natural, genetic drift might have caused differential pvmdr1 haplotype frequencies at different geographic sites in Mesoamerica. A very recent expansion of divergent pvmdr1 haplotypes in NIC-NE/NIC-NW produced high differentiation between these and parasites from other sites including SMX. These data are useful to set a baseline for epidemiological surveillance.
Similar content being viewed by others


Background
Plasmodium vivax causes most malaria cases in Latin America [1]. About 20 million people are at high risk and, in 2014, there were 390,000 confirmed malaria cases that resulted in 79 deaths [2]. From 2000 to 2015 (the end of the era of millennium development goals elaborated by the WHO), most countries in Mesoamerica (a region extending from central Mexico to Panama) reported a decline of at least 74% in the number of malaria cases [2]. In Nicaragua, still in the control phase, P. vivax and Plasmodium falciparum have been responsible for a fluctuating number of malaria cases during the last decade. From 2008 to 2011, there were less than 1000 cases reported, but the following years registered an increase in this number; in 2014, a total of 1163 cases were confirmed by microscopy. More than 70% of the cases reported in this country are produced in the Northeast or North Caribbean Coast Autonomous Region, while transmission is sporadic (with periodic outbreaks) in the Northwest side of the country. The most affected area of countries Nicaragua and Honduras is the indigenous Miskito region, which lies on both sides of the border (Northeast). Also in 2014, the number of reported malaria cases was 4931 in Honduras and 3380 in Guatemala [2], the latter country between Honduras and Mexico. Since 1999, only P. vivax has been transmitted in Mexico, with 696 cases reported in 2014. This country is in the pre-elimination phase, similar to El Salvador and Belize [2].
In Mesoamerica, two drugs have been used to treat malaria since the 1950s: chloroquine (CQ) and primaquine (PQ) [3]. CQ interferes with haemoglobin digestion by the parasite and causes death of asexual and sexual blood parasites. However, PQ is needed to eliminate dormant stages of P. vivax and young gametocytes of P. falciparum [4]. The first cases of P. vivax resistance to CQ were reported in Oceania and Asia, and later in South America [5]. The multidrug resistance 1 gene (mdr1) codes for a transport protein, which is comprised of two transmembrane domains and two ATP binding sites [6]. In Southeast Asia, a low in vitro sensitivity of P. vivax to CQ was associated with the PvMDR1 amino acid substitution Y976F [7, 8]. The gene fragment containing this and other polymorphisms (e.g. F1076L, also a transmembrane residue), have been found in distinct geographic sites and at different frequencies [9]. In South America, four haplotypes comprising residues (976/1076; Y/F, Y/L, F/F and F/L) were reported as well as one polymorphism (T958M) at a marginal transmembrane position [6, 7, 10,11,12]. In P. vivax from Honduras, the PvMDR1 haplotypes 976/1076; Y/L, F/F and F/L were described, but the wild type (Y/F; Sal I strain) was the most frequent (>70%) [13]. It is remarkable that in clinical studies at different geographic sites no association between PvMDR1 polymorphism and CQ resistance has been found [12, 14, 15]. Besides, a recent study suggested that Pvmdr1 mutations emerged at different geographic sites possibly by convergent evolution [11].
Seeking to extend the genetic characterization of current P. vivax in Mesoamerica, parasites were obtained from three different geographic sites. Then, the occurrence of natural selection and the genetic relationships of P. vivax studying mdr1 polymorphisms and haplotype frequency were investigated.
Methods
Blood samples and geographic origin
All infected blood samples were obtained from symptomatic patients seeking malaria diagnosis in Mexico and Nicaragua (in each case, after informed consent was given). From 2008 to 2010 in Mexico, 93 samples were obtained in the laboratory facility at the Regional Research Center for Public Health (CRISP-INSP) from patients living in the Tapachula municipality and its surroundings in southern Chiapas (SMX), the southernmost point of Mexico that borders with Guatemala [16]. From 2011 to 2012 in Nicaragua, 107 samples were selected from the sentinel laboratory network established by the Health Ministry at head municipalities, 83 from the Northeast (NIC-NE; RACCN, North Caribbean Coast Autonomous Region) and 24 from the Northwest (NIC-NW; North Pacific Coast). Another 14 samples were included that had been obtained in NIC-NW during 2006–2007 [17]. Distances between these sites are the following: ≈611 km from SMX to NIC-NW, ≈853 km from SMX to NIC-NE, and ≈331 km from NIC-NE to NIC-NW. The NIC-NE in comprised by the Miskito and the mining regions.
The diagnosis of P. vivax was carried out by microscopic examination of stained thick blood smears, coming from capillary blood samples (before treatment) used to impregnate filter paper (Whatman #2). Then the participants were administered the CQ-PQ combination treatment, which is in accordance with health standards in Mexico [18, 19] and in Nicaragua [20]. Genomic DNA was extracted from P. vivax-infected bloods impregnated on filter paper, using a commercially available QIAmp DNA blood Minikit (Qiagen, USA), and following the manufacturer’s instructions.
PCR amplification and sequencing
A gene fragment of about 600 bp and containing codon positions 976 and 1076 was amplified using the following oligonucleotides: F3.2 5′˗ACC AGG ATA GTC ATG CCC˗3′ (nt 2747–2764) and R3 5′˗TCT CCC TTT AGG GAC ATC AAC˗3′ (nt 3384–3368). The PCR reaction was prepared as follows: 10 μL of 5× PCR buffer, 5 μL of MgCl2 (25 mM), 2.5 μL of the dNTPs (1.25 mM), 2.5 μL of each primer (10 μM), 0.5 μL of Go Taq DNA polymerase, 5 u/μL and 2–4 μL of sample DNA for a final volume of 50 μL. The PCR reaction conditions were as follows: 5 min at 94 °C followed by 35 cycles: 1 min at 94 °C, 1 min at 60 °C, and 1 min at 72 °C; afterwards, there was a final extension of 72 °C for 10 min.
To investigate the presence of other nucleotide changes reported previously [6], two additional nucleotide fragments were PCR amplified. To that, primers F1 5′˗GAG GGA GAT GTC ATC ATC AAC GA˗3′ (nt 1315–1337) and R1 5′˗CTT CTG TCC ACC TGA CAA CTT AG˗3′(nt 1733–1755), and F2 5′˗CAA GGA CAG CAA TGA GAA GAA˗3′ (nt 2013–2033) and R2 5′˗CGC GAT GAC TAA GAT GTA GAG G˗3′ (nt 2601–2622) were used. The PCR reaction was prepared as follows: 10 μL of 5× PCR buffer, 4 μL of MgCl2 (25 mM), 2.5 μL of dNTPs (1.25 mM), 1.9 μL of each primer (10 μM), 0.5 μL of Go Taq DNA polymerase 5 u/μL (Invitrogen Corporation, Carlsbad, CA), and 2–4 μL of extracted DNA for a final volume of 50 μL. The PCR reaction conditions were as follows: 3 min at 94 °C followed by 35 cycles: 40 s at 94 °C, 40 s at 54 °C, and 1 min at 72 °C; afterwards, there was a final extension of 72 °C for 5 min. All PCR reactions were run in a MyCycler (BioRad, Hercules, CA, USA).
All amplified products were resolved in agarose gels at 1%, and stained with 0.2 µg/mL ethidium bromide using an electrophoresis chamber Midicell primo (Thermo EC330, New York, USA). A molecular marker of 100 bp ladder was used (Invitrogen Corporation, Carlsbad, CA, USA). PCR products were then purified using a MiniElute PCR Purification Kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s instructions. The purified products were Sanger sequenced by using forward and reverse primers (at the High Throughput Genomics Unit, Department of Genome Sciences, University of Washington, Seattle, WA, USA).
The quality of pherograms with the forward and reverse nucleotide sequences was verified manually and by using BioEdit v7.1.3 software. DNA sequences with rare polymorphisms were re-amplified and sequenced. All pherograms showed a single genotype pattern. The consensus sequences obtained for each gene fragment were submitted to the NCBI-Gen Bank [Accession Numbers: KX180164–KX180638].
Data analysis
The Salvador I strain (Sal I) mdr1sequence was used as reference: XM_001613678. Sequences forward and reverse were aligned using ClustalW Multiple Alignment of BioEdit v7.0 [21] and revised manually. Nonsynonymous and synonymous changes were identified, and the frequency of each nucleotide change was calculated per geographic site in Mesoamerica (NIC-NE, NIC-NW and SMX), as well the haplotypes were constructed to background mutations at codons 976/1076 with the dnaSP program v5.1 [22]. Statistical analysis was carried out with STATA v12.1.
In order to explore the regional and global parasite relationships based on pvmdr1, haplotype networks were constructed using TCS 1.21 [23]. Mutational steps represent the connections between haplotypes, and empty squares showed the non-sampled or extinct haplotypes. The colour of the circles represents the geographic origins of each haplotype, while the size of the circle represents the frequency of each haplotype.
Indexes of nucleotide diversity were calculated in dnaSP v5.1. To test whether natural selection and gene flow shaped the evolution of pvmdr1 gene in parasite populations of Mesoamerica, the number of synonymous (s) and nonsynonymous (ns) nucleotide changes and the difference in the rate of nonsynonymous versus synonymous changes (dN˗dS) were determined by using the Nei Gojobori proportion method with 1000 bootstrap replicates in MEGA v6.0 software [24]. Tajima’s D test and the minimal number of recombination events were calculated using dnaSP. To estimate the P. vivax genetic relationships from different locations, the F ST values were estimated by using the two parameters of Kimura in dnaSP; values ranged from 0.0 to 1.0. A value of 0.0 indicates that populations are equal and 1.0 indicates they are completely different [25, 26].
DNA sequences for the global genetic analysis. Groups of DNA sequences that comprised codons 925–1083 (including positions 976/1076) were obtained from the NBCI Gene Bank. Brazil (BRZ): n = 7, EU333973-9 [5]; n = 3, AY571981-3 and 79–83, 85–89 [14]; n = 15, KM016495/502, 04, 05 and 07–17 [10]. Papua New Guinea (PNG): n = 6, AY571975-80 [14]. Iran (IR): n = 4, KM216181-4 (Sharifi-Sarasiabi et al. unpublished). Madagascar (MD): n = 80, EU683813-19 [14]. South Korea (SK): GU476519 (Chen et al. unpublished). India (IND): n = 49, KC818349-78, 80, 82, 85, 87, 89, 90, 92, 93, 95, 96, 98, 99/400, 02, 04, 06–09, 11 and 12 [27]. Cameroon: n = 8, KJ534638-45 (Ngasa et al., pers.comm.). Additionally, other DNA sequences were reconstructed, using the aforementioned sequence reference of the Sal I strain as a template (AY618622, with codon 958 atg/M; AY571984 and XM001613678 with codon 958 acg/T), and only if the length and coordinates of the pvmdr1 gene fragment, synonymous and nonsynonymous nucleotide changes, and haplotype frequency were clearly described: IND, n = 25 [28]. Honduras (HON), n = 37 [13]. Nepal (NEP), n = 39 [29]. Ecuador (ECU), n = 17; Sri Lanka (SLK), n = 119; Pakistan (PK), n = 24; Sudan (SUD), n = 4; Sao Tomé (SAT), n = 3 [11].
Results
Pvmdr1 gene polymorphism
Upon analysing 1536 bp of pvmdr1, two synonymous and six nonsynonymous nucleotide changes were detected (Table 1). From 163 parasites (66, 23 and 74 from NIC-NE, NIC-NW and SMX, respectively), two changes (one nonsynonymous and one synonymous) were detected in the gene fragment of 411 bp (nucleotides 1330–1740). The nonsynonymous change was observed (only in NIC-NW) at codon 500 D → N). While the synonymous change at codon 529, was highly frequent in SMX and NIC-NE, but not detected in parasites from NIC-NW. Interestingly, from 155 parasites (65, 26 and 64 from NIC-NE, NIC-NW and SMX, respectively) a second fragment of 549 bp (nucleotides 2767–3315) was obtained, and six nucleotide changes were detected. The nonsynonymous changes at codons Y976F and F1076L were highly frequent in NIC-NE and NIC-NW, but not detected in SMX. Similarly, the synonymous change at codon 1021 was highly frequent in NIC-NE and NIC-NW, but rare in SMX. The change at codon A927T exclusive to NIC-NE, it was detected in parasites from different municipalities of the Miskito and the mining regions. The substitution T958M was highly frequent at the three sites, while the F1070L change was detected in only two isolates from NIC-NE (Rosita and Prinzapolka municipalities) and two isolates from SMX (from the year 2010). The third gene fragment of 576 bp (nucleotides 2032–2607), obtained from 148 parasites (63, 17 and 72 from NIC-NE, NIC-NW and SMX, respectively), was identical to the Sal I strain sequence.
Pvmdr1 haplotypes and its geographic frequency
There were 141 parasite isolates (59 from NIC-NE, 59 from SMX and 23 from NIC-NW) with the nucleotide information at variant codons 500, 529–927, 958, 976, 1021, 1070 and 1076 resolved 13 pvmdr1 haplotypes (m1 → m13; ordered by their frequency) (Fig. 1). There were differences in haplotype frequencies between the three sites (P = 0.000): haplotype m1 GG-GTACTT was predominant (93.2%) in SMX; m3 AA-GTTTTC was highly frequent (78.2%) in NIC-NW; m2 GA-GTTTTC and m4 GG-ATATTC were at 62.7 and 22%, respectively, in NIC-NE. Other haplotypes were detected in two or all sites, although at a very low frequency. For instance, haplotype m1 was also detected in one isolate from NIC-NE and in another from NIC-NW, m6 and m10 were observed at SMX and NIC-NE, and m5 was found in NIC-NW and NIC-NE. One haplotype (m8) was exclusive to SMX and resembled the Sal I strain sequence. Another three haplotypes (m9, m11 and m13) were exclusive to NIC-NE and two (m7 and m12) to NIC-NW (Fig. 1). There were four haplotypes having nucleotide changes at codons 976F (a → t) and 1076L (t → c); m2, m3, m7: GG-GTTCTC, m12: GG-GTTTTC; those haplotypes differed among them by one to three mutations. Two other haplotypes showed only 1076F/L; m4 and m11: GA-ATATTC. The haplotypes m2 and m4, highly frequent at NIC-NE were also frequent in all sampled municipalities: Rosita, Bonanza, Waspam and Puerto Cabezas. The higher number of exclusive haplotypes were detected in Waspam (n = 3), the Miskito region that borders Honduras.

Geographic distribution of the P. vivax mdr1 haplotypes in Mesoamerica. Gene polymorphism was detected at codons 500, 529–927, 958, 976, 1021, 1070 and 1076. It includes 141 isolates (59, 59 and 23 from SMX, NIC-NE and NIC-NW, respectively). (A n = 59) SMX and (B n = 59) NIC-NE. In NIC-NW, six haplotypes were detected in 2006–2007 (C1 n = 7), while only one was detected in 2012 (C2 n = 16) (Asterisk synonymous changes). The circle indicates pvmdr1 haplotypes (n = 13; by colours); Sal I; pvmdr1, XM001613678. One-letter code indicates amino acid: D aspartic acid; T threonine; A alanine; Y tyrosine; F phenylalanine; N asparagine; M methionine; L leucine. The amino acid changes at codons 976 and 1096 are shown in red. n number of samples
Genetic relationship of pvmdr1 haplotypes from SMX, NIC-NW and NIC-NE
The haplotype network of P. vivax mdr1 comprising mutations at codons 500, 529–927, 958, 976, 1021, 1070 and 1076 for parasites from the study sites is shown in Fig. 2. All 13 haplotypes were found closely related; most haplotypes were at one mutational step from each other, and at 1–9 mutational steps among them. Haplotypes from SMX were only separated by 1–2 mutational among them. The most frequent haplotype (m1) in SMX was at one mutational step from the Sal I strain sequence. While haplotypes m2 and m3 were at about 6–9 mutational steps from m1; they were highly frequent in NIC-NE and NIC-NW, respectively.

Haplotype network of the P. vivax mdr1 from the study region. The network includes haplotypes from Southern Mexico (light brown), Northeast (green) and Northwest (dark red), Nicaragua. Each circle indicates a different haplotype, and lines are connecting them. The black square indicates a haplotype not sampled or extinct. All haplotypes were closely related and signs of gene diversification were detected. Sal I haplotype: XM001613678 correspond to haplotype m8. The network includes 141 sequences. The haplotypes showing 1076L and 976F/1076L are within a blue frame, respectively
Genetic diversity
Parasites from NIC-NE and mostly from the Mesoamerican region showed a nucleotide diversity (π = 0.0036) similar to that found at other geographic origins as well as the global level (π = 0.0028). Moreover, the minimal number of recombination events were from very low to cero (the value for Mesoamerica was Rm = 2) (Table 2).
Natural selection
Tajima’s D test statistical value was negative for SMX and NIC-NW, while being positive for NIC-NE and other sites, but no significant. The lowest value was for Brazil (−1.681) and the highest for Ecuador (0.949), but in either case, it was found to be no significant. Although a higher number of nonsynonymous than synonymous changes were detected, the dN–dS values showed no significant departure from neutrality. Parasites from Nepal had the highest dN–dS value with marginal significance (1.845; P = 0.067) (Table 2).
Haplotype relationships
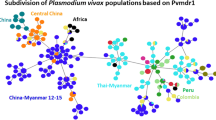
When using global sequences of P. vivax mdr1, the haplotype network evidenced the relationship of 19 global haplotypes (g1 → g19) identified in 610 sequences (Fig. 3). The most frequent haplotype g1 and probably the ancestral haplotype, it was predominant in parasites from Sri Lanka, India, Nepal, Pakistan and Madagascar and existed in few parasites from Latin America. In contrast, the haplotype g2 was highly frequent in parasites from Latin America (SMX, Brazil, Honduras and Ecuador). Haplotype g2 was at one mutational step from g5, the latter represent Sal I strain (the only haplotype with codon 958 → acg) which was found in parasites from Latin America and India. Moreover, from g2 emerged a branch of four descendant haplotypes (restricted to American parasites), one present in SMX and NIC-NE (connected to another haplotype present in parasites from SMX, NIC-NE and Brazil), and two exclusive to NIC-NE (Fig. 3). Whereas, haplotype g3 was included in one parasite from Brazil and another from NIC-NW, is at one mutational step from g4, the latter of which showed high frequency in NIC-NE and NIC-NW parasites and was found in one parasite from Brazil (Fig. 3). There were two different haplotypes (g1 and g6) showing polymorphisms T958M and F1076L. Haplotype g6 exclusive in NIC-NE emerged from a different branch of that detected in one parasite from Brazil and other geographic sites outside Latin America (g1).

Haplotype network of P. vivax mdr1 gene from different geographic origins. The network includes 610 sequences and 19 haplotypes. Parasites from Northeast Nicaragua (NIC-NE) and Brazil had the highest number of haplotypes (n = 7). Each circle indicates a different haplotype (n = 19) connected by lines. The empty squares indicate haplotypes not sampled or extinct. Each colour indicates a different country. Three frequent haplotypes were detected at different geographic sites, respectively. One haplotype was frequent in Latin America. Signs of gene diversification were found at the regional and at local level. Sal I haplotype: XM001613678. A gene fragment of 479 bp (nt 2772–3250) was used. NIC-NW, Northwest, Nicaragua. SMX, Southern Mexico
F ST values of P. vivax between Mesoamerica and other geographic origins
F ST values were mostly from low to moderate between sites inside Latin America, excluding NIC-NE and NIC-NW. F ST values, between SMX, Brazil, Honduras and Ecuador ranged from 0.015 to 0.194. The lowest value was between SMX and Brazil, and the highest between Honduras and Ecuador. In Asia (mostly the Indian subcontinent: Nepal, Iran, Pakistan, India and Sri Lanka), F ST values were from low to moderate (Table 3). Between NIC-NE and NIC-NW the value was also low (0.097), but higher between these sites and others inside and outside Latin America (0.4784–0.8039). In general, between Latin American and other sites outside this continent, F ST values were generally high (0.3813–0.9430).
Discussion
The Mesoamerican region has ecological and demographic conditions that favor malaria transmission. Chloroquine (CQ) and primaquine (PQ) have been the only drugs used in this region to treat patients infected with P. vivax and/or P. falciparum [19, 20, 30] and, so far, both Plasmodium species seem susceptible to CQ. According to this, mutations at pvmdr1 might not be associated to CQ resistance. Therefore, PvMDR1 976F and CQ resistance in vitro seemed to be a counterfeit association [8]. In fact, clinical studies have shown that polymorphisms at pvmdr1 e.g. 976F and others adjacent are not associated with CQ resistance [12, 14, 28], and there are no functional studies exposing pvmdr1 mutations participating in the development of CQ resistance. Schousboe et al. [11] suggested that pvmdr1 mutations 958M, 976F and 1076L arose independently and several times at different geographic sites. In Mesoamerica, the selective force of natural selection on pvmdr1 differed spatially, with lack of statistical significance. Besides, recent genomic studies have not revealed signature of selection on pvmdr1 in parasites from different geographic sites [31,32,33], including parasites from the Southeast Asia [33] where high levels of CQ resistance have been reported [8, 34].
Malaria has been historically hypo endemic in Mesoamerica. From 2000 to 2010 a reduction in more than 75% was recorded in several countries including Nicaragua and Mexico. Contrarily from 2012 to 2014, Nicaragua was the only country in Mesoamerica that reported a significant increase in malaria cases [2]. In this country, while intense and effective control strategies caused reduction in the number of malaria cases, climate changes and delay in the delivery of control measures have probably caused the increase in malaria transmission. Alike southern Mexico, in NIC-NW and NIC-NE several haplotypes exposing changes 976F and/or 1076L were exclusive and highly frequent. The haplotype networks suggest that these mutations resulted from a recent diversification in Mesoamerica. It is remarkable that haplotypes expressing 1076L, present in parasites from NIC-NE (haplotype g6) seemed the result of different evolutionary pathway than in other geographic sites (haplotype g1).
The presence of some pvmdr1 haplotypes in all sites (SMX, NE-NIC and NW-NIC), may be attributed to ancient parasite flow across the Mesoamerican region. It was notorious a predominance of blood infections with a single pvmdr1 haplotype, presumably monoclonal infections. A clonal transmission of P. vivax might be occurring in the region. It is supported by the lack of mixed genotype infections for pvmsp1 42 a gene fragment, of moderate diversity, reported in NIC-NE/NW [35]. In SMX, the rate of mixed genotype infections has been negligible during the last decade [16, 36].
In Mesoamerica, parasite flow might be controlled by different factors, including separation by distance, geographic barriers, cultural and language barriers, poor road infrastructure, the effectiveness of the malaria control interventions. There is a volcanic arc in between NIC-NE and NIC-NW. At either side of the arc exclusive pvmdr1 haplotypes were detected. Similarly, for the blood stage antigen pvmsp1 42 a divergent haplotype was exclusive in Chinandega municipality (NIC-NW) [35]. In NIC-NE, the most frequent pvmdr1 haplotypes in the region, so were at the municipality level. A similar spatial pattern of haplotypes was reported for pvmsp1 42 [35]. Yet, the presence of exclusive haplotypes suggests partial parasite flow between the Miskito and mining regions in NIC-NE. By using microsatellites, the analysis of P. falciparum exposed that in the mining region there was a genetic population distinct from that detected in the Miskito area [37]. The Miskito ethnic group (Waspam and Puerto Cabezas) live in multiple rural communities and it is partly inaccessible due to cultural and language barriers. Alike P. falciparum, P. vivax cause relapse episodes of long latency, an additional risk of parasite dispersion [38], and microsatellite markers provided higher resolution than single nucleotide polymorphism, and subtle genetic changes can be detected [39]. It is probable that some pvmdr1 mutations might have appeared at different sites in Mesoamerica as formerly suggested for other geographic sites [11].
The results from this study and the epidemiological and demographic scenario suggest that a strong genetic drift might have influenced the spatial differences of the pvmdr1 haplotype frequencies in Mesoamerica. The Sal I type was only detected in SMX and at low frequency. In Honduras, parasites isolated from 2004 to 2009 exhibited haplotypes with PvMDR1 976F and/or 1076L changes at a frequency of <30% [13]; while in the Northeast Nicaragua area the frequency of those changes was almost 90%. These changes were also present in strains causing an outbreak in the Northwest Nicaragua in 2012, it corresponded to an exclusive haplotype. In South America, mutations 976F and 1076L were reported to have low frequency [12, 31].
Plasmodium vivax arriving at the American continent faced adaptive processes triggered by different local factors e.g. the specificity of the Anopheline species, the host immune pressure, the anti-malarial drugs employed, and the ecological conditions to mention some reasons [40,41,42]. Genomic studies have exposed that P. vivax exhibits low diversity and high differentiation in Latin America [42, 43]. Analysis of the mitochondrial genome exposed low F ST values between South America and Central America, the last ones corresponded to ten strains obtained in the 1970s [44]. Evidence based on the entire pvmdr1 gene also supports a high genetic differentiation between old and new world parasites [32]. In the present study, the F ST value using the pvmdr1 codons925–1083 matched partially the previous results, also suggesting a strong differentiation globally but low-moderate differentiation at regional level. Excepting parasites from NIC-NE/NW, these were highly differentiated from all other parasites including those from SMX and Honduras, possibly given by the recent expansion of pvmdr1 divergent haplotypes.
Regardless of its origin, P. vivax has been at some extent circulating in the American continent. Southern Mexico is a transit area for human migrations coming from South and Central America [45], and still a proportion of humans migrants entering North America can be potentially infected with P. vivax [46, 47]. Therefore, further genomic studies are necessary to uncover the evolution of P. vivax in the Mesoamerican region comparing to other sites of America and worldwide. Furthermore, molecular monitoring of P. vivax mdr1 at different sites in Mesoamerica would help to understand transmission dynamics. Because of its variability and spatial patterns, pvmdr1 is an important marker for epidemiological surveillance to support the current initiatives to control and eliminate malaria from Mesoamerica.
Conclusions
These results suggest a recent diversification of pvmdr1which likely occurred at a regional level, natural selection varied spatially and was mostly in a weakly to-mild manner. In this hypo-endemic region, a strong genetic drift might have caused the great differences in pvmdr1 haplotype frequency among the studied sites, and hence the high differentiation of parasites between NIC-NW/NIC-NE and SMX or other sites. These data are relevant to set a baseline level for epidemiological surveillance in the region.
Abbreviations
- Pvmdr1 :
-
P. vivax multidrug resistance 1 gene
- CQ:
-
chloroquine
- PQ:
-
primaquine
- SMX:
-
southern Chiapas, Mexico
- NIC-NE:
-
Northeast, Nicaragua (it is part of the North Caribbean Coast Autonomous Region)
- NIC-NW:
-
Northwest, Nicaragua (North Pacific Coast)
References
Guerra CA, Howes RE, Patil AP, Gething PW, Van Boeckel TP, Temperley WH, et al. The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS Negl Trop Dis. 2010;4:e774.
WHO. World malaria report. World Health Organization, Geneva. 2015. http://www.who.int/malaria/publications/world-malaria-report-2015/report/en/. Accessed 06 May 2017.
PAHO. Report on the situation of malaria in the Americas, 2011. Pan American Health Organization. http://www2.paho.org/hq/index.php?option=com_content&view=article&id=5175%3A2010-report-on-situation-malaria-americas-2011&catid=1841%3Amalaria-publications&Itemid=40360&lang=en. Accessed 06 May 2017.
WHO. Guidelines for the treatment of malaria. World Health Organization, Geneva. 2015. http://www.who.int/malaria/publications/atoz/9789241549127/en/. Accessed 06 May 2017.
Gonçalves LA, Cravo P, Ferreira MU. Emerging Plasmodium vivax resistance to chloroquine in South America: an overview. Mem Inst Oswaldo Cruz. 2014;109:534–9.
Orjuela-Sanchez P, de Santana Filho FS, Machado-Lima A, Chehuan YF, Costa MR, Alecrim M, et al. Analysis of single-nucleotide polymorphisms in the crt-o and mdr1 genes of Plasmodium vivax among chloroquine-resistant isolates from the Brazilian Amazon region. Antimicrob Agents Chemother. 2009;53:3561–4.
Brega S, Meslin B, de Monbrison F, Severini C, Gradoni L, Udomsangpetch R, et al. Identification of the Plasmodium vivax mdr-like gene (pvmdr1) and analysis of single-nucleotide polymorphisms among isolates from different areas of endemicity. J Infect Dis. 2005;191:272–7.
Suwanarusk R, Russell B, Chavchich M, Chalfein F, Kenangalem E, Kosaisavee V, et al. Chloroquine resistant Plasmodium vivax: in vitro characterisation and association with molecular polymorphisms. PLoS ONE. 2007;2:e1089.
Vargas-Rodríguez RDCM, da Silva Bastos M, Menezes MJ, Orjuela-Sánchez P, Ferreira MU. Single-nucleotide polymorphism and copy number variation of the multidrug resistance-1 locus of Plasmodium vivax: local and global patterns. Am J Trop Med Hyg. 2012;87:813–21.
Melo GC, Monteiro WM, Siqueira AM, Silva SR, Magalhães BML, Alencar ACC, et al. Expression Levels of pvcrt-o and pvmdr-1 are associated with chloroquine resistance and severe Plasmodium vivax malaria in patients of the Brazilian Amazon. PLoS ONE. 2014;9:e105922.
Schousboe ML, Ranjitkar S, Rajakaruna RS, Amerasinghe PH, Morales F, Pearce R, et al. Multiple origins of mutations in the mdr1 gene—a putative marker of chloroquine resistance in P. vivax. PLoS Negl Trop Dis. 2015;9:e0004196.
Gomes LR, Almeida-de-Oliveira NK, de Lavigne AR, de Lima SRF, de Pina-Costa A, Brasil P, et al. Plasmodium vivax mdr1 genotypes in isolates from successfully cured patients living in endemic and non-endemic Brazilian areas. Malar J. 2016;15:96.
Jovel IT, Mejia RE, Banegas E, Piedade R, Alger J, Fontecha G, et al. Drug resistance associated genetic polymorphisms in Plasmodium falciparum and Plasmodium vivax collected in Honduras. Cent Am Malar J. 2011;10:376.
Barnadas C, Ratsimbasoa A, Tichit M, Bouchier C, Jahevitra M, Picot S, et al. Plasmodium vivax resistance to chloroquine in Madagascar: clinical efficacy and polymorphisms in pvmdr1 and pvcrt-o genes. Antimicrob Agents Chemother. 2008;52:4233–40.
Sá JM, Nomura T. Neves Jd, Baird JK, Wellems TE, del Portillo HA. Plasmodium vivax: allele variants of the mdr1 gene do not associate with chloroquine resistance among isolates from Brazil, Papua, and monkey-adapted strains. Exp Parasitol. 2005;109:256–9.
Gonzalez-Ceron L, Rodriguez MH, Sandoval MA, Santillan F, Galindo-Virgen S, Betanzos AF, et al. Effectiveness of combined chloroquine and primaquine treatment in 14 days versus intermittent single dose regimen, in an open, non-randomized, clinical trial, to eliminate Plasmodium vivax in southern Mexico. Malar J. 2015;14:426.
Gonzalez-Ceron L, Martinez-Barnetche J, Montero-Solis C, Santillan F, Soto AM, Rodriguez MH, et al. Molecular epidemiology of Plasmodium vivax in Latin America: polymorphism and evolutionary relationships of the circumsporozoite gene. Malar J. 2013;12:243.
Mexican Official Standard for epidemiological surveillance, prevention and control of vector-borne diseases, NOM-032-SSA2-2002. Health Secretary, Mexico. 2002. http://www.salud.gob.mx/unidades/cdi/nom/032ssa202.html. Accessed 06 May 2017.
Mexican Official Standard for epidemiological surveillance, prevention and control of vector-borne diseases, NOM-032-SSA2-2010. Health Secretary, Mexico. 2011. http://www.cenaprece.salud.gob.mx/programas/interior/vectores/descargas/pdf/nom_032_ssa2_2010_norma_petv.pdf. Accessed 06 May 2017.
National standard for the prevention, control and treatment of malaria: N-114, Nicaragua. http://www.minsa.gob.ni/index.php/repository/Descargas-MINSA/Direcci%C3%B3n-General-de-Regulaci%C3%B3n-Sanitaria/Normas-Protocolos-y-Manuales/Normas-2013/N–114-Norma-nacional-para-la-prevenci%C3%B3n-control-y-tratamiento-de-la-malaria/. Accessed 06 May 2017.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–9.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Wright S. The genetical structure of populations. Ann Eugenics. 1951;15:323–54.
Balloux F, Lugon-Moulin N. The estimation of population differentiation with microsatellite markers. Mol Ecol. 2002;11:155–65.
Shalini S, Chaudhuri S, Sutton PL, Mishra N, Srivastava N, David JK, et al. Chloroquine efficacy studies confirm drug susceptibility of Plasmodium vivax in Chennai, India. Malar J. 2014;13:129.
Ganguly S, Saha P, Guha SK, Das S, Bera DK, Biswas A, et al. In vivo therapeutic efficacy of chloroquine alone or in combination with primaquine against vivax malaria in Kolkata, West Bengal, India, and polymorphism in pvmdr1 and pvcrt-o genes. Antimicrob Agents Chemother. 2013;57:1246–51.
Ranjitkar S, Schousboe ML, Thomsen TT, Adhikari M, Kapel CM, Bygbjerg IC, et al. Prevalence of molecular markers of anti-malarial drug resistance in Plasmodium vivax and Plasmodium falciparum in two districts of Nepal. Malar J. 2011;10:75.
PAHO. Report on the situation of malaria in the Americas. Pan American Health Organization. 2008. http://www1.paho.org/hq/dmdocuments/2010/Informe-Situacion-Paludismo-Americas–2008-Nicaragua.pdf. Accessed 06 May 2017.
Winter DJ, Pacheco MA, Vallejo AF, Schwartz RS, Arevalo-Herrera M, Herrera S, et al. Whole genome sequencing of field isolates reveals extensive genetic diversity in Plasmodium vivax from Colombia. PLoS Negl Trop Dis. 2015;9:e0004252.
Hupalo DN, Luo Z, Melnikov A, Sutton PL, Rogov P, Escalante A, et al. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat Genet. 2016;48:953–8.
Pearson RD, Amato R, Auburn S, Miotto O, Almagro-Garcia J, Amaratunga C, et al. Genomic analysis of local variation and recent evolution in Plasmodium vivax. Nat Genet. 2016;48:959–64.
Ratcliff A, Siswantoro H, Kenangalem E, Wuwung M, Brockman A, Edstein MD, et al. Therapeutic response of multidrug-resistant Plasmodium falciparum and P. vivax to chloroquine and sulfadoxine–pyrimethamine in southern Papua, Indonesia. Trans R Soc Trop Med Hyg. 2007;101:351–9.
Gutiérrez S, González-Cerón L, Montoya A, Sandoval MA, Tórres ME, Cerritos R. Genetic structure of Plasmodium vivax in Nicaragua, a country in the control phase, based on the carboxyl terminal region of the merozoite surface protein-1. Infect Genet Evol. 2016;40:324–30.
Flores-Alanis A, González-Cerón L, Santillán F, Ximenez C, Sandoval MA, Cerritos R. Temporal genetic changes in Plasmodium vivax apical membrane antigen 1 over 19 years of transmission in southern Mexico. Parasit Vectors. 2017;10:217.
Larrañaga N, Mejía RE, Hormaza JI, Montoya A, Soto A, Fontecha GA. Genetic structure of Plasmodium falciparum populations across the Honduras-Nicaragua border. Malar J. 2013;12:354.
Contacos PG, Collins WE, Jeffery GM, Krotoski WA, Howard WA. Studies on the characterization of Plasmodium vivax strains from Central America. Am J Trop Med Hyg. 1972;21:707–12.
Schlotterer C. The evolution of molecular markers—just a matter of fashion? Nat Rev Genet. 2004;5:63–9.
Joy DA, Gonzalez-Ceron L, Carlton JM, Gueye A, Fay M, McCutchan TF, et al. Local adaptation and vector-mediated population structure in Plasmodium vivax malaria. Mol Biol Evol. 2008;25:1245–52.
Sinka ME, Bangs MJ, Manguin S, Coetzee M, Mbogo CM, Hemingway J, et al. The dominant Anopheles vectors of human malaria in Africa, Europe and the Middle East: occurrence data, distribution maps and bionomic precis. Parasit Vectors. 2010;3:117.
Neafsey DE, Galinsky K, Jiang RH, Young L, Sykes SM, Saif S, et al. The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat Genet. 2012;44:1046–50.
Chan ER, Menard D, David PH, Ratsimbasoa A, Kim S, Chim P, et al. Whole genome sequencing of field isolates provides robust characterization of genetic diversity in Plasmodium vivax. PLoS Negl Trop Dis. 2012;6:e1811.
Taylor JE, Pacheco MA, Bacon DJ, Beg MA, Machado RL, Fairhurst RM, et al. The evolutionary history of Plasmodium vivax as inferred from mitochondrial genomes: parasite genetic diversity in the Americas. Mol Biol Evol. 2013;30:2050–64.
Betanzos AF, Gonzalez-Ceron L, Rodriguez MH, Torres-Monzon JA. Seroepidemiología del paludismo en un grupo de migrantes en tránsito (Chiapas, 2008). Salud Publica Mex. 2012;54:523–9.
Cullen KA, Mace KE, Arguin PM. (CDC). CfDCaP. Malaria Surveillance—United States, 2013. MMWR Surveill Summ. 2016;65:1–22.
Hwang J, Cullen KA, Kachur SP, Arguin PM, Baird JK. Severe morbidity and mortality risk from malaria in the United States, 1985–2011. Open Forum Infect Dis. 2014;1:ofu034.
Authors’ contributions
LGC, participate in all components of the study; conception and design, data analysis and interpretation of data and the major contribution in writing the manuscript. AM, sample collection in Nicaragua and drafted the manuscript. JCG and FS performed molecular techniques and analysed data. RC, revise the genetic analysis and drafted the manuscript. MAS, performed molecular techniques. All authors read and approved the final manuscript.
Acknowledgements
We are grateful to the lab personnel at the sentinel sites in Nicaragua for collaborating by preparing the P. vivax samples on filter paper. We give special thanks to Martha Lopez in Rosita and Dexter Manzanares in Waspam, as well as the local authorities in charge of the facilities. Thanks to Rafael Avendaño and Olga L. Palomeque at the Regional Center for Research in Public Health in Mexico, and to Maritza Torres at the National Centre for Diagnosis and Reference, Ministry of Health in Nicaragua for their technical assistance.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article. The DNA sequences and the data related were deposited in the GenBank database with Accession Numbers KX180164–KX180638.
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the National Institute of Public Health in Mexico and the Ethics Committee of the National Center for Diagnosis and Reference (CNDR) of the Ministry of Health in Nicaragua. Written consent was obtained from all individuals participating in this study. In the case of minors, the parents or guardians gave consent prior to enrollment.
Funding
This work was funded by CONACyT-Mexico project CB-2009-01-131247, AMI/RAVEDRA-OPS/OMS-Nicaragua and the global fund-malaria component-Nicaragua (2012–2013).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
González-Cerón, L., Montoya, A., Corzo-Gómez, J.C. et al. Genetic diversity and natural selection of Plasmodium vivax multi-drug resistant gene (pvmdr1) in Mesoamerica. Malar J 16, 261 (2017). https://doi.org/10.1186/s12936-017-1905-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-017-1905-x