
Abstract
Background
Glucose-6-phosphate dehydrogenase (G6PD) deficiency exhibits considerable allelic heterogeneity which manifests with variable biochemical and clinical penetrance. It has long been thought that G6PD deficiency confers partial protection against severe malaria, however prior genetic association studies have disagreed with regard to the strength and specificity of a protective effect, which might reflect differences in the host genetic background, environmental influences, or in the specific clinical phenotypes considered.
Methods
A case-control association study of severe malaria was conducted in The Gambia, a region in West Africa where there is considerable allelic heterogeneity underlying expression of G6PD deficiency trait, evaluating the three major nonsynonymous polymorphisms known to be associated with enzyme deficiency (A968G, T542A, and C202T) in a cohort of 3836 controls and 2379 severe malaria cases.
Results
Each deficiency allele exhibited a similar trend toward protection against severe malaria overall (15–26 % reduced risk); however, in stratifying severe malaria to two of its constituent clinical subphenotypes, severe malarial anaemia (SMA) and cerebral malaria (CM), the three deficiency alleles exhibited trends of opposing effect, with risk conferred to SMA and protection with respect to CM. To assess the overall effect of G6PD deficiency trait, deficiency alleles found across all three loci were pooled. G6PD deficiency trait was found to be significantly associated with protection from severe malaria overall (OR 0.83 [0.75–0.92], \(P = 0.0006\)), but this was limited to CM (OR 0.73 [0.61–0.87], \(P = 0.0005\)), with a trend toward increased risk for SMA, especially in fully-deficient individuals (OR 1.43 [0.99–2.08], \(P = 0.056\)). Sex-stratified testing largely comported with these results, with evidence suggesting that protection by G6PD deficiency trait is conferred to both males and females, though susceptibility to SMA may be restricted to fully-deficient male hemizygotes.
Conclusions
In a part of Africa where multiple alleles contribute to expression of G6PD deficiency trait, these findings clarify and extend previous work done in populations where a single variant predominates, and taken together suggest a causal role for G6PD deficiency trait itself with respect to severe malaria, with opposing effects seen on two major clinical subphenotypes.
Similar content being viewed by others


Background
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an X-linked enzyme disorder with a global distribution, affecting hundreds of millions of people worldwide [1]. It is a genetically heterogeneous trait, with hundreds of deficiency alleles of variable penetrance described in the literature [2], several having risen to polymorphic frequencies [3]. Prevalence of G6PD deficiency geographically correlates with the historical distribution of malaria [4], an infectious disease thought to have highly heritable differences in burden [5, 6], and a major contributor to childhood morbidity and mortality in endemic areas [7]. As such, G6PD deficiency has long been hypothesised to be a target of positive selection owing to partial protection afforded against malaria [8–11].
Genetic epidemiological evidence examining this hypothesis has been lacking in clarity, however, with past case-control association studies differing with respect to the strength and specificity of a protective effect [12–18], and even whether such an effect exists at all [19]. The difficulty in interpreting past studies lies not only in reconciling the different clinical phenotypes considered in each, but also in recognising different assumptions made in defining G6PD deficiency trait itself. With regard to the former, clinical phenotypes considered range from measures of malaria poorly correlated with disease burden, such as parasite rate or density [12, 20, 21], to more extreme phenotypes pathognomonic for severe, life-threatening malaria, including profound anaemia, respiratory distress, and cerebral manifestations such as convulsions or coma [13–15, 22]. Furthermore, World Health Organization criteria for diagnosis of severe malaria are broad, and include a wide range of symptoms and clinical findings, which vary substantially not only in terms of pathophysiology, but also with respect to prognostic value [23]. Interpreting trait carriage is also nontrivial in that G6PD deficiency is both a genetic and a biochemical trait. As a sex-linked genetic trait, hemizygous males and homozygous females fully express the deficiency trait, while heterozygous females exhibit incomplete or partial expression due to random X inactivation [24, 25]. As a biochemical trait, enzyme activity can be assayed quantitatively, but overlapping activity distributions for the different genotypic categories make qualitative assessment imperfect [16, 26, 27].
The most common G6PD deficiency allele in sub-Saharan Africa is the C202T (A-) variant, and studies of this mutation have driven most association studies in this region. However, while the C202T allele is predominant in east Africa [28], in West Africa there are several other deficiency variants (A968G, T542A, C680A) and The Gambia appears to be a notable exception to the general rule that 202T is the most prevalent allele. Recent data published by the MalariaGEN consortium has found opposing effects of the 202T allele on risk for specific severe malaria clinical subphenotypes, finding risk conferred with respect to severe malarial anaemia, and a trend toward protection with respect to cerebral malaria [29]. Given the allelic heterogeneity present in The Gambia and the robust clinical data that has been collected here over many decades [30], it represents a unique location to assess whether the effects of the 202T allele on modifying risk for specific severe malaria subphenotypes hold true for other functional alleles, and for G6PD deficiency trait as a whole, in an area where multiple alleles of variable penetrance contribute to overall prevalence of the trait [15, 31]. Presented here is a case-control association study of severe malaria and two of its major clinical presentations in The Gambia, examining all major functional alleles contributing to expression of G6PD deficiency trait in the region.
Methods
Ethics statement
Ethics approval was granted by The Gambia Government/Medical Research Council Joint Ethics Committee and by the Oxford Tropical Research Ethics Committee, and written informed consent was obtained from a parent or guardian prior to enrolment.
Study cohort
The case-control cohort studied is comprised of 3836 controls and 2379 severe malaria cases, recruited at the Royal Victoria Hospital in Banjul. Malaria cases were confirmed by thin blood smear, and designated as ‘severe’ following WHO criteria, with two partially overlapping clinical syndromes identified in the majority of cases: severe malarial anaemia and cerebral malaria. Severe malarial anaemia (SMA) is defined here as packed cell volume \(\le\)15 % or haemoglobin \(\le\)5 g/dl, while cerebral malaria (CM) is defined as having a Blantyre coma score \(\le\)3 in the absence of other obvious causes, e.g. hypoglycaemia or meningitis. A total of 556 (23 %) of these cases met the definition for SMA and 890 (37 %) met that for CM, with 129 (5 %) cases fitting both definitions (these were excluded in the analysis of each clinical subphenotype). Control samples were derived from cord blood obtained from local perinatal clinics. A demographic summary of the cohort is provided in Table 1.
Laboratory and statistical methods
Genotyping was performed on genomic DNA that was whole genome amplified via either primer extension pre-amplification or multiple displacement amplification reaction. Single nucleotide polymorphisms (SNPs) were then assayed via multiplexed genotyping on the Sequenom MASSarray platform. Automated genotype calls were manually curated via visual examination of cluster plots and mass spectra, with ambiguous genotypes re-coded as missing.
Data curation was performed using custom Perl scripts and the PLINK software package [32]. Gender designation was curated using additional X chromosome SNP data previously generated for these samples, using an inbreeding coefficient test that recodes gender based on observed heterozygosity. Any remaining heterozygous genotypes found in males were coded as missing, and individuals with ambiguous gender were excluded from the analysis. No significant frequency differences were found across genders and SNP genotype distribution in female controls did not significantly deviate from expected Hardy-Weinberg proportions (\(P > 0.05\), exact test).
Association testing was conducted using custom scripts written in R [33]. A logistic regression framework was employed, using a generalised linear model assuming a binomial error distribution. In the additive (trend) model, male genotypes were coded 0/2, with females coded as 0/1/2, as suggested by Clayton [34] and in keeping with biochemical penetrance data [16, 26]. In sex-stratified tests, male hemizygotes and female heterozygotes were coded 1, with individuals lacking deficiency alleles coded as 0.
Composite G6PD deficiency trait genotype was defined as the sum of deficiency alleles present at all deficiency loci (968G + 542A + 202T) for an individual. Thus, a male was coded as 0 or 2, based on the absence or presence of a single deficiency allele across the three loci, while a female with a total of one deficiency allele present was coded as 1, and two deficiency alleles as 2 (including 13 ‘compound heterozygote’ individuals possessing a single deficiency allele at two different loci). It should be noted that this composite genotype was never greater than two because this scenario would require two deficiency alleles to co-occur on the same chromosome. Full expression of the G6PD deficiency trait, i.e. complete enzyme deficiency, was a binary genotype (0 or 1) inferred according to the absence or presence of one (male hemizygotes) or two (female homozygotes and compound heterozygotes) deficiency alleles in total across the three loci. Thus, a 202T male hemizgygote, 968G/202T female compound heterozygote, and a 542AA female homozygote would all be scored as 1 for full G6PD deficiency trait expression.
Estimated odds ratios (OR) were adjusted by including the following covariates: HbS (sickle locus) genotype, self-reported ethnicity, and sex (omitted in sex-stratified tests). All odds ratios are presented here as ‘OR [95 % CI]’.
Results
Association testing of G6PD deficiency alleles
The case-control study population included a total of 2379 Gambian children with severe malaria and 3836 cord blood control samples drawn from the same region. Genotypes were obtained at four nonsynonymous polymorphic loci within G6PD known to be associated with enzyme deficiency, including A968G, T542A, C680A, and C202T (Table 2), in order to comprehensively survey the genetic diversity underlying G6PD deficiency in West Africa. Three of these four SNPs were found to be polymorphic, with C680A being the lone monomorphic variant. The 968G variant was found to be the most common deficiency allele in the data set, present at a frequency of 6.6 % in controls, with 202T and 542A found at frequencies of 3.2 and 1.2 %, respectively. Each deficiency allele was first tested for association with severe malaria overall, using an additive (trend) genetic model consistent with G6PD biochemical penetrance data [16, 26], with heterozygote females treated as intermediate between non-deficient and deficient individuals, the latter being comprised of hemizygote males and homozygote females. These tests revealed a similar signal of protection for all three deficiency alleles (Table 3), suggestive of 15–26 % reduced risk, with strong evidence for the higher-frequency variant 968G (OR 0.84 [0.74–0.96], \(P = 0.010\)), and milder evidence for 542A (OR 0.74 [0.53–1.03], \(P = 0.071\)) and 202T (OR 0.85 [0.70–1.03], \(P = 0.091\)).
Stratified testing of two major clinical subphenotypes
As severe malaria encompasses several clinical syndromes that differ with respect to pathophysiology and prognostic value, stratified association testing was performed on each of the two major clinical subphenotypes predominant in The Gambia, severe malarial anaemia (SMA) and cerebral malaria (CM). Although these subphenotype tests were not equally powered to detect signals of association due to sample size differences (\(N_{SMA} = 556\), \(N_{CM} = 890\)), they nonetheless suggested a differential direction of disease association with respect to SMA and CM for all three G6PD deficiency alleles (Table 3). While deficiency alleles seemed to confer protection against CM (968G: OR 0.73 [0.58–0.91]; 542A: OR 0.47 [0.23–0.99]; 202T: OR 0.82 [0.60–1.11]), they exhibited a modest, but consistent trend toward susceptibility for SMA (968G: OR 1.09 [0.88–1.35]; 542A: OR 1.36 [0.87–2.11]; 202T: OR 1.09 [0.79–1.49]). Interestingly, the 542A allele, which causes a more extreme enzyme deficiency state (WHO class 2, 1–2 % residual G6PD activity [35]), was associated with more extreme effect sizes for both subphenotypes– greater risk to SMA and greater protection from CM– than either 968G or 202T, which are of milder biochemical penetrance (WHO class 3, 10–20 % residual G6PD activity [36]).
Association of G6PD deficiency trait with severe malaria
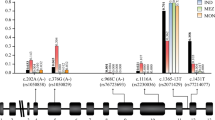
Since a consistent direction of association was observed with respect to SMA and CM for all three deficiency variants, and given that each contributes to the overall prevalence of G6PD deficiency trait in The Gambia, it made sense to consider them as a unit and examine the central hypothesis concerning the association of G6PD deficiency trait itself with severe malaria. Specifically, it was asked whether possession of one or more deficiency alleles at any of the three loci was associated with susceptibility to severe malaria. Thus, G6PD deficiency trait status was inferred using a composite genotype which represented the sum of deficiency alleles across all three loci, with this genotype scored in the same way as all other genotypes were, namely that hemizygous-deficient males were treated as equivalent to homozygous-deficient females, with heterozygous females being intermediate. Additionally, a binary composite genotype inferring presence or absence of full expression of the trait (i.e. full G6PD enzyme deficiency) was compiled, where presence of the trait required either one (male hemizygotes) or two (female homozygotes) deficiency alleles in total across the three loci. In these association tests (Table 3; Fig. 1), it was found that while G6PD deficiency trait exerted a strong protective effect against severe malaria overall (OR 0.83 [0.75–0.92], \(P = 0.0006\)), this protective effect was limited to CM (OR 0.73 [0.61–0.87], \(P = 0.0005\)), with a trend toward increased risk conferred to SMA, especially in fully-deficient individuals (OR 1.43 [0.99–2.08], \(P = 0.056\)), consistent with effects seen for the 202T deficiency allele alone in work recently published by the MalariaGEN consortium [29]. Importantly, this differential direction of association contrasts with the consistent strong protective effect across both SMA and CM clinical subphenotypes observed for sickle cell trait (Table 3; Fig. 1).

G6PD trait association tests—estimated odds ratios. Results from association tests using a composite G6PD genotype to infer G6PD deficiency trait status, where composite genotype is the total number of variant alleles present across all three deficiency SNPs (A968G, T542A, and C202T) in an individual (‘G6PD trait’). Presence or absence of full expression of the trait (i.e. full G6PD enzyme deficiency) was also tested, where presence of the ‘full’ expression of the trait required presence of one (male hemizygotes) or two (female homozygotes) deficiency alleles in total across the three loci (‘G6PD trait [full]’). Also shown are association test results for HbS (red). Estimated OR with 95 % CI is shown for association with severe malaria overall (ALL), as well as for each subphenotype (SMA, CM)
Sex-stratified association testing
As G6PD deficiency is an X-linked trait, and the subject of gender-specificity has been conflicting in previous association studies of severe malaria [12–15], sex-stratified association tests were also conducted to examine how signals of association might vary between hemizygous males and heterozygous females. It is important to note that these tests differ with respect to: (a) statistical power, as female heterozygotes are more common than male hemizygotes, and (b) interpretation with respect to biochemical penetrance, as hemizygous males are fully-deficient, while female heterozygotes are only partially-deficient. Such caveats notwithstanding, in these tests (Table 4), evidence of protection from severe malaria overall was found for both male hemizygotes (OR 0.75 [0.58–0.96], \(P = 0.023\)) and female heterozygotes (OR 0.74 (0.60–0.91) \(P = 0.0045\)), with this protective effect confined to CM for both males (OR 0.67 [0.45–1.02], \(P = 0.061\)) and females (OR 0.54 [0.38–0.77] \(P = 0.0006\)). The trend toward susceptibility observed for SMA was only observed in fully-deficient male hemizygotes (OR 1.41 [0.96–2.08], \(P = 0.083\)), especially those carrying severe deficiency variant 542A (OR 2.55 [1.00–6.52] \(P = 0.051\)), although the lack of a similar trend in female heterozygotes is likely a reflection of their intermediate deficiency state. Interestingly, the most common G6PD deficiency allele across most of Africa, 202T, was found to be protective against severe malaria overall and the CM subphenotype only in female heterozygotes. Association testing of fully-deficient female homozygotes was limited due to low frequencies, and provided inconclusive results.
Discussion
In examining multiple G6PD deficiency alleles in The Gambia, it was found that such alleles were associated with protection from severe malaria overall, but exhibited differential effects with respect to two important clinical presentations of severe malaria, conferring protection from cerebral malaria (CM) and increasing risk for severe malarial anaemia (SMA). Taken together with prior work from populations where a single variant, C202T (A-), predominates, there is compelling evidence for a direct, causal role for G6PD deficiency trait in modification of clinical malaria subphenotypes, confirming past results demonstrating protection from CM [14, 22], as well as the more recently elucidated association with SMA [29]. Indeed, while the allelic heterogeneity present in The Gambia presents a challenge with respect to achieving adequate statistical power to detect true effects among the multiple lower frequency variants present, in analysing these variants in tandem one not only reduces type I error, but also more directly assesses G6PD deficiency trait itself independent of the genetic background upon which any particular variant lies.
As this study was limited to just one country, with its own distinct genetic background and environmental exposure history, including its own unique pattern of exposure to malaria, it will be important that further investigation be conducted in as many study populations in as diverse a range of geographic areas as possible, and there is ongoing work toward this end being conducted by the MalariaGEN consortium (manuscript in preparation). As there is substantial variation in the biochemical penetrance of G6PD deficiency alleles, it will also be useful for future work to include enzyme activity assays when feasible.
The present study provides the somewhat paradoxical result that a single gene can be associated with protection from and susceptibility to disease from the same infectious agent. This highlights the fact that severe malaria comprises distinct clinical presentations, and the extent to which these syndromes differ from one another. These findings are not unexpected, then, given that G6PD deficiency is known to predispose individuals to haemolytic anaemia in the presence of oxidant stresses from infection, drugs, and diet [37]. As the stronger signal of association is associated with cerebral malaria, the subphenotype more predictive of severe morbidity and mortality [30, 38], these findings are consistent with the malaria protection hypothesis, and suggest that further association studies at G6PD should distinguish between the different presentations of severe malaria.
The differential association patterns seen here also suggest possible mechanistic differences between sickle cell trait (HbS) and G6PD deficiency in the modulation of malaria clinical phenotypes.
Sickle cell homozygous and heterozygous states have been variously reported to affect invasion and growth in vitro [39, 40], or to decrease parasite density in natural [41–43] and experimental [41, 44] settings. Modulation of acquired immunity [45], impaired rosetting [46] or reduced cytoadherence [47] may also reduce the survival of infected HbS erythrocytes and/or mitigate processes of inflammation, and thus might explain the protection seen against both forms of severe malaria in this study.
Studies of parasite phenotypes inside the G6PD-deficient erythrocyte have been more equivocal. While some have found evidence of invasion preferences [48] or aberrant in vitro growth phenotypes [49], others have not [50, 51]. Similarly, evidence of decreased parasite density in vivo is mixed for both Plasmodium vivax [11, 21] and Plasmodium falciparum [12, 14, 20]. This raises the possibility that the protection afforded by G6PD deficiency against cerebral malaria might depend on genetic background or operate through some mechanism other than parasite survival inside the enzyme-deficient red cell. Given that G6PD is an important housekeeping enzyme with a wide distribution in the body, it is interesting to consider, for example, how its role in endothelial function (via nitric oxide synthase [52, 53]), or in the respiratory burst of immune cells (via NADPH oxidase [54]), might contribute to malaria protection.
Importantly, differences in the protection afforded by HbS and G6PD deficiency highlight the need to study clinical biomarkers and conduct in vitro experiments relevant to severe manifestations of malaria. Ultimately, if knowledge gained from investigations surrounding the malaria protection hypothesis is to inform clinical treatment, a better understanding of the molecular mechanisms as they relate to severe disease will be crucial.
Conclusions
In a region of West Africa with significant functional allelic heterogeneity, these data confirm that G6PD deficiency state is protective against severe malaria, but in contrast to sickle cell trait, this protection is limited to the CM subphenotype, with possible increased risk conferred to SMA. These results support the malaria protection hypothesis, but also suggest important mechanistic differences between the malaria-protective mechanisms of G6PD deficiency versus sickle cell trait.
Abbreviations
- G6PD:
-
glucose-6-phosphate dehydrogenase
- SMA:
-
severe malarial anaemia
- CM:
-
cerebral malaria
- WHO:
-
World Health Organisation
- SNP:
-
single nucleotide polymorphism
- HbS:
-
sickle cell trait
- OR:
-
odds ratio
- CI:
-
confidence interval
References
Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371:64–74.
Beutler E, Vulliamy TJ. Hematologically important mutations: glucose-6-phosphate dehydrogenase. Blood Cells Mol Dis. 2002;28:93–103.
Beutler E. G6PD: population genetics and clinical manifestations. Blood Rev. 1996;10:45–52.
Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol Dis. 2009;42:267–78.
Mackinnon MJ, Mwangi TW, Snow RW, Marsh K, Williams TN. Heritability of malaria in Africa. PLoS Med. 2005;2:1253–9.
Sørensen TI, Nielsen GG, Andersen PK, Teasdale TW. Genetic and environmental influences on premature death in adult adoptees. N Engl J Med. 1988;318:727–32.
Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. 2005;77:171–92.
Haldane J. The rate of mutation of human genes. In: Proceedings of the Eighth International Congress of Genetics. 1949. p 267–273.
Tishkoff SA, Varkonyi R, Cahinhinan N, Abbes S, Argyropoulos G, Destro-Bisol G, et al. Haplotype diversity and linkage disequilibrium at human G6PD: recent origin of alleles that confer malarial resistance. Science. 2001;293:455–62.
Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–7.
Louicharoen C, Patin E, Paul R, Nuchprayoon I, Witoonpanich B, Peerapittayamongkol C, et al. Positively selected G6PD-Mahidol mutation reduces Plasmodium vivax density in Southeast Asians. Science. 2009;326:1546–9.
Bienzle U, Ayeni O, Lucas A, Luzzatto L. Glucose-6-phosphate dehydrogenase and malaria. Greater resistance of females heterozygous for enzyme deficiency and of males with non-deficient variant. Lancet. 1972;1:107–10.
Ruwende C, Khoo SC, Snow RW, Yates SN, Kwiatkowski D, Gupta S, et al. Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature. 1995;376:246–9.
Guindo A, Fairhurst RM, Doumbo OK, Wellems TE, Diallo DA. X-linked G6PD deficiency protects hemizygous males but not heterozygous females against severe malaria. PLoS Med. 2007;4:516–22.
Clark T, Fry A, Auburn S, Campino S, Diakite M, Green A, et al. Allelic heterogeneity of G6PD deficiency in West Africa and severe malaria susceptibility. Eur J Hum Genet. 2009;17:1080–5.
Johnson MK, Clark TD, Njama-Meya D, Rosenthal PJ, Parikh S. Impact of the method of G6PD deficiency assessment on genetic association studies of malaria susceptibility. PLoS One. 2009;4:e7246.
Manjurano A, Sepulveda N, Nadjm B, Mtove G, Wangai H, Maxwell C, et al. African glucose-6-phosphate dehydrogenase alleles associated with protection from severe malaria in heterozygous females in Tanzania. PLoS Genet. 2015;11:e1004960.
Sirugo G, Predazzi IM, Bartlett J, Tacconelli A, Walther M, Williams SM. G6PD A- deficiency and severe malaria in The Gambia: heterozygote advantage and possible homozygote disadvantage. Am J Trop Med Hyg. 2014;90:856–9.
Martin SK, Miller LH, Alling D, Okoye VC, Esan GJ, Osunkoya BO, et al. Severe malaria and glucose-6-phosphate-dehydrogenase deficiency: a reappraisal of the malaria/G-6-P.D. hypothesis. Lancet. 1979;1:524–6.
Allison AC, Clyde DF. Malaria in African children with deficient erythrocyte glucose-6-phosphate dehydrogenase. Br Med J. 1961;1:1346–9.
Kruatrachue M, Charoenlarp P, Chongsuphajaisiddhi T, Harinasuta C. Erythrocyte glucose-6-phosphate dehydrogenase and malaria in Thailand. Lancet. 1962;2:1183–6.
Gilles HM, Fletcher KA, Hendrickse RG, Lindner R, Reddy S, Allan N. Glucose-6-phosphate-dehydrogenase deficiency, sickling, and malaria in African children in South Western Nigeria. Lancet. 1967;1:138–40.
Marsh K, Forster D, Waruiru C, Mwangi I, Winstanley M, Marsh V, et al. Indicators of life-threatening malaria in African children. N Engl J Med. 1995;332:1399–404.
Beutler E, Yeh M, Fairbanks V. The normal human female as a mosaic of X-chromosome activity: studies using the gene for G-6-PD-deficiency as a marker. Proc Natl Acad Sci USA. 1962;48:9–16.
Gall JC, Brewer GJ, Dern RJ. Studies of glucose-6-phosphate dehydrogenase activity of individual erythrocytes: the methemoglobin-elution test for identification of females heterozygous for G6PD deficiency. Am J Hum Genet. 1965;17:359–68.
May J, Meyer CG, Grossterlinden L, Ademowo OG, Mockenhaupt FP, Olumese PE, et al. Red cell glucose-6-phosphate dehydrogenase status and pyruvate kinase activity in a Nigerian population. Trop Med Int Health. 2000;5:119–23.
Shah SS, Diakite SAS, Traore K, Diakite M, Kwiatkowski DP, Rockett KA, et al. A novel cytofluorometric assay for the detection and quantification of glucose-6-phosphate dehydrogenase deficiency. Sci Rep. 2012;2:299.
Shah SS, Macharia A, Makale J, Uyoga S, Kivinen K, Craik R, et al. Genetic determinants of glucose-6-phosphate dehydrogenase activity in Kenya. BMC Med Genet. 2014;15:93.
Network Malaria Genomic Epidemiology. Reappraisal of known malaria resistance loci in a large multicenter study. Nat Genet. 2014;46:1197–204.
Jallow M, Casals-Pascual C, Ackerman H, Walther B, Walther M, Pinder M, et al. Clinical features of severe malaria associated with death: a 13-year observational study in the Gambia. PLoS One. 2012;7:e45645.
Araujo CD, Migot-Nabias F, Guitard J, Pelleau S, Vulliamy T, Ducrocq R. The role of the G6PD AEth376G/968C allele in glucose-6-phosphate dehydrogenase deficiency in the Seerer population of Senegal. Haematologica. 2006;91:262–3.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. 2008.
Clayton D. Testing for association on the X chromosome. Biostatistics. 2008;9:593–600.
Beutler E, Kuhl W, Sáenz GF, Rodríguez W. Mutation analysis of glucose-6-phosphate dehydrogenase (G6PD) variants in Costa Rica. Hum Genet. 1991;87:462–4.
Beutler E, Kuhl W, Vives-Corrons JL, Prchal JT. Molecular heterogeneity of glucose-6-phosphate dehydrogenase A. Blood. 1989;74:2550–5.
Mason PJ, Bautista JM, Gilsanz F. G6PD deficiency: the genotype-phenotype association. Blood Rev. 2007;21:267–83.
Mishra SK, Newton CRJC. Diagnosis and management of the neurological complications of falciparum malaria. Nat Rev Neurol. 2009;5:189–98.
Friedman M. Erythrocytic mechanism of sickle cell resistance to malaria. Proc Natl Acad Sci. 1978;75:1994–7.
Pasvol G, Weatherall DJ, Wilson RJ. Cellular mechanism for the protective effect of haemoglobin S against P. falciparum malaria. Nature. 1978;274:701–3.
Allison A. Protection afforded by sickle-cell trait against subtertian malarial infection. Br Med J. 1954;1:290–4.
Williams TN, Mwangi TW, Wambua S, Alexander ND, Kortok M, Snow RW, et al. Sickle cell trait and the risk of Plasmodium falciparum malaria and other childhood diseases. J Infect Dis. 2005;192:178–86.
May J, Evans JA, Timmann C, Ehmen C, Busch W, Thye T, et al. Hemoglobin variants and disease manifestations in severe falciparum malaria. JAMA. 2007;297:2220–6.
Beutler E, Dern R, Flanagan C. Effect of sickle-cell trait on resistance to malaria. Br Med J. 1955;1:1189–91.
Williams TN, Mwangi TW, Roberts DJ, Alexander ND, Weatherall DJ, Wambua S, et al. An immune basis for malaria protection by the sickle cell trait. PLoS Med. 2005;2:e128.
Carlson J, Nash GB, Gabutti V, al Yaman F, Wahlgren M. Natural protection against severe Plasmodium falciparum malaria due to impaired rosette formation. Blood. 1994;84:3909–14.
Cholera R, Brittain NJ, Gillrie MR, Lopera-Mesa TM, Diakité SAS, Arie T, et al. Impaired cytoadherence of Plasmodium falciparum-infected erythrocytes containing sickle hemoglobin. Proc Natl Acad Sci USA. 2008;105:991–6.
Luzzatto L, Usanga FA, Reddy S. Glucose-6-phosphate dehydrogenase deficient red cells: resistance to infection by malarial parasites. Science. 1969;164:839–42.
Roth EF, Raventos-Suarez C, Rinaldi A, Nagel RL. Glucose-6-phosphate dehydrogenase deficiency inhibits in vitro growth of Plasmodium falciparum. Proc Natl Acad Sci USA. 1983;80:298–9.
Friedman MJ. Oxidant damage mediates variant red cell resistance to malaria. Nature. 1979;280:245–7.
Cappadoro M, Giribaldi G, O’Brien E, Turrini F, Mannu F, Ulliers D, et al. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood. 1998;92:2527–34.
Leopold JA, Walker J, Scribner AW, Voetsch B, Zhang YY, Loscalzo AJ, et al. Glucose-6-phosphate dehydrogenase modulates vascular endothelial growth factor-mediated angiogenesis. J Biol Chem. 2003;278:32100–6.
Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, et al. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007;13:189–97.
Agudelo-Flórez P, Costa-Carvalho BT, López JA, Redher J, Newburger PE, Olalla-Saad ST, et al. Association of glucose-6-phosphate dehydrogenase deficiency and X-linked chronic granulomatous disease in a child with anemia and recurrent infections. Am J Hematol. 2004;75:151–6.
Authors’ contributions
SS, KR, TEW and DPK designed the study. MJ, FSJ, KAB and MP managed patient enrolment as well as clinical data and sample collection. SS, AR, RC and CH validated and conducted genotyping assays. SS conducted the statistical analyses. SS, KR, TEW and DPK wrote the paper. All authors read and approved the final manuscript.
Acknowledgments
We thank the all participants and communities in The Gambia who made this study possible and the healthcare workers who assisted with this work. MalariaGEN is supported by the Wellcome Trust (077383/Z/05/Z) and by the Foundation for the National Institutes of Health (566) as part of the Bill and Melinda Gates Grand Challenges in Global Health Initiative. The Resource Centre for Genomic Epidemiology of Malaria is supported by the Wellcome Trust (090770/Z/09/Z). Support was also provided by the Medical Research Council (G0600718). Dominic Kwiatkowski receives support from the Medical Research Council (G19/9). The Wellcome Trust also provides core awards to The Wellcome Trust Centre for Human Genetics (075491/Z/04; 090532/Z/09/Z) and the Wellcome Trust Sanger Institute (077012/Z/05/Z). This study was also supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases at the National Institutes of Health, and Shivang Shah is also supported by the Medical Scientist Training Program at the NIH.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Consortia
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Shah, S.S., Rockett, K.A., Jallow, M. et al. Heterogeneous alleles comprising G6PD deficiency trait in West Africa exert contrasting effects on two major clinical presentations of severe malaria. Malar J 15, 13 (2016). https://doi.org/10.1186/s12936-015-1045-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-015-1045-0