
Abstract
Background
Chloroquine has been administered to the soldiers of the Republic of Korea as prophylaxis against vivax malaria. Recent increase in the number of chloroquine-resistant parasites has raised concern over the chemoprophylaxis and treatment of vivax malaria.
Methods
To monitor the development of chloroquine-resistant parasites in the Republic of Korea, analyses of single nucleotide polymorphisms (SNPs) of pvmdr1 and microsatellite markers were performed using samples collected from 55 South Korean soldiers infected with Plasmodium vivax.
Results
Four SNPs, F1076L, T529, E1233, and S1358, were identified. Among these, S1358 was detected for the first time in Korea. The microsatellite-based study revealed higher genetic diversity in samples collected in 2012 than in 2011.
Conclusions
Taken together, the results indicate that P. vivax with a newly identified SNP of pvmdr1 has been introduced into the Korean P. vivax population. Therefore, continuous monitoring for chloroquine-resistant parasites is required for controlling vivax malaria in the Republic of Korea.
Similar content being viewed by others


Background
Malaria caused by Plasmodium vivax is the most common human malaria infection, affecting 40 % of the world’s population [1, 2]. In the Republic of Korea, vivax malaria had been successfully eliminated by the late 1970s by an effective World Health Organization (WHO) programme. However, this infectious disease has re-emerged, since a soldier was diagnosed with P. vivax infection in 1993 [3, 4]. Since then, vivax malaria has been the only type of malaria detected in the Republic of Korea, accounting for 18,052 cases reported from 1994 to 2013 [5].
Vivax malaria endemic regions in the Republic of Korea are concentrated near the demilitarized zone that separates South Korea from the Democratic People’s Republic of Korea (DPRK or North Korea). Thus, military personnel and residents living in the demilitarized zone are under high risk of contracting vivax malaria. Military personnel accounted for 25.3 % (1029/4063) of all malaria cases reported from 2008 to 2010 [6]. This percentage rose to 44.7 % (1811/4063) when military personnel diagnosed with vivax malaria following discharge from the service were counted [7]. In this regard, mass chemoprophylaxis using chloroquine and primaquine has been administered to soldiers since the year 1997 to control vivax malaria infection. For the chemoprophylaxis, 300 mg chloroquine is administrated weekly to military personnel from June to August (for 12 weeks), and then, 30 mg primaquine is administrated daily for 2 weeks.
Chloroquine has been used to not only kill P. vivax in asexual blood stages and gametocytes, but also to prevent the spread of malaria in low-risk areas. However, its massive use in the treatment of vivax malaria and continuing long-term chemoprophylaxis could facilitate the acquisition of resistance to chloroquine [8]. Since the first report of chloroquine-resistant P. vivax in 1989 in Papua New Guinea, the number of chloroquine-resistant cases has increased in several countries, including Indonesia, Southeast Asia, India, and Central and South America [9–15]. Although cases of chloroquine-resistant malaria infections have been confirmed recently, chloroquine is still used as the therapeutic and chemoprophylactic drug for P. vivax infections in the Republic of Korea [16]. Thus, the administration of chloroquine to soldiers stationed near the demilitarized zone has raised the concern of accelerating the development of drug-resistant P. vivax.
Monitoring the genetic polymorphism that confers chloroquine resistance to malaria provides useful information regarding the efficacy of drugs in treating malaria. However, compared to P. falciparum, previous studies using genetic markers for chloroquine-resistant P. vivax did not conclude a strong correlation between the genetic markers and chloroquine-resistant phenotype in P. vivax, because the molecular mechanisms of chloroquine resistance in P. vivax is still elusive [17]. Among the genetic markers for chloroquine-resistant P. vivax, the multidrug resistance-1 gene of P. vivax (pvmdr1) has been identified as a possible genetic marker of chloroquine resistance with in vitro characterization of isolates [18]. In Southeast Asia and Papua New Guinea, the Y976F mutation (TAC→TTC) in pvmdr1 has been shown to be correlated with chloroquine resistance [19–21]. In addition, the association of severe malaria and expression levels of pvmdr1 with chloroquine resistance was reported by showing 2.4-fold increase in pvmdr1 expression levels in parasites from patients compared to the susceptible group of vivax malaria in the Brazilian Amazon [21]. This chloroquine resistance appears to be the result of a two-step mutation pathway, in which the F1076L mutation is followed by the Y976F mutation [22, 23]. The F1076L mutation is found in all Korean samples tested, and is unlikely to result in chloroquine treatment failure [24], while the Y976F mutation has not yet been reported in the Republic of Korea [25]. However, considering that the South Korean military has been performing mass chemoprophylaxis for more than 15 years, there likely is strong evolutionary pressure for selection of the double mutant.
Recently, the genetic diversity as well as intra- and inter-population relationships of P. vivax isolates obtained from the Republic of Korea (from 1994 to 2008) were analysed [26, 27]. By using microsatellite markers, this analysis provided an explanation for the genetic diversity observed among strains. In this study, the prevalence of five common non-synonymous single-nucleotide polymorphisms (SNPs) and four synonymous SNPs at the pvmdr1 locus, including the Y976F and F1076L, was examined in 55 P. vivax isolates obtained from military vivax malaria patients who had taken chemoprophylaxis near the demilitarized zone of the Republic of Korea. The population structure of these isolates was analysed using the microsatellite method with 10 microsatellite markers.
Methods
Ethics statements
This study was approved by the ethics committee of the Army Forces Medical Command (Approval No. AFMC-13-IRB-053, July 2011). An approval form was used to obtain written informed consent from each participant and all participants provided their informed consent for collecting a 5-mL blood sample.
Blood samples and DNA extraction
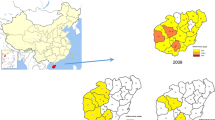
A total of 55 blood samples were collected from patients infected with malaria and admitted to the Armed Forces Hospitals near the demilitarized zone located in northern Gyeonggi-do Province in the northwest region of the Republic of Korea in 2011 and 2012 (Fig. 1). The admission and clinical management of the patients were undertaken independently of this study. Aliquots (200 μL) of venous blood samples were stored at −20 °C in ethylenediaminetetraacetic acid (EDTA)-coated bottles until extracting genomic DNA using a QIAamp® DNA Blood Mini Kit (Qiagen, USA) according to the manufacturer’s instructions.

Map of Gyeonggi-do Province in the Republic of Korea: endemic regions of vivax malaria near the demilitarized zone in which samples were collected
Identification of single nucleotide polymorphism (SNP) in the pvmdr1 gene
The pvmdr1 gene was amplified by nested PCR using pvmdr1 gene specific primers [25, 28]. Amplification of pvmdr1 gene fragments was performed applying a nested PCR approach and regents, primers, and cycling conditions as outlined in Table 1. The final PCR products were resolved by electrophoresis on a 1.5 % agarose gel stained with ethidium bromide, and visualized under ultraviolet illumination. The second PCR products were sequenced and the deduced amino acid sequences were compared to the amino acid sequence of pvmdr1 from the Sal I strain of P. vivax (Salvador I, GenBank accession no. AY571984). The amino acid sequence alignment and analyses were performed using Clustal Omega [29].
Analysis of ten microsatellite markers
To determine the relationships between the different pvmdr1 genotypes of P. vivax, 10 microsatellite markers were typed for 28 samples collected in the year 2011 and 27 collected in the year 2012. The 10 microsatellite markers used for this assay were as follows: MS1, MS3, MS5, MS8, MS10, MS12, MS16, MS20, Msp1F7, and Pv3,27. The primer sets and amplification conditions used for the PCRs have been described elsewhere [30, 31]. The fluorophore-labelled PCR products were quantified using an Applied Biosystems 3730 DNA Analyzer with the GeneMapper software Version 4.0 (Applied Biosystems, USA). In order to reduce potential artifacts from background noise or stutter, an arbitrary fluorescent intensity threshold of 50 relative florescence units was applied for peak detection. All electropherogram traces were additionally inspected manually. For each isolate, at each locus, the predominant allele, the highest intensity peak and any additional alleles with a peak height of at least one-third of the height of the predominant allele were scored [32]. Genotyping success was defined as the presence of at least one allele at a given locus in a given sample.
Population genetic analyses and statistical treatments
The major alleles of each locus were used for our population genetic analysis. The level of genetic diversity of the P. vivax population in Republic of Korea was assessed by allele number per locus (A) and expected heterozygosity (He). The He values for each locus were calculated using the formula He = [n/(n − 1)] [1 − ∑p 2 i ], where n is the number of isolates examined and p i is the frequency of the ith allele. The statistical significance of the differences in these values was evaluated by Welch’s t test.
Multilocus linkage disequilibrium (LD) was assessed using LIAN v3.6 based on the allelic data for the 10 microsatellite DNA loci [33]. This program computed the standardized index of association (I SA ), a measure of genotype-wide linkage. The P-values were determined by a Monte Carlo simulation process, performing 100,000 iterations. Only those samples for which a complete set of microsatellite alleles were scored were used for this analysis. Additionally, the multilocus genotypes found in multiple isolates were only counted once in the analysis [34].
Microsatellite genotypes of the isolates were determined based on a combination of the allelic data of the 10 loci. The relationships between the genotypes were determined by eBURST analysis [35].
Results and discussion
In the Republic of Korea, an extensive malaria chemoprophylaxis campaign using chloroquine and primaquine has been conducted annually since the year 1997. The cumulative numbers of the soldiers receiving this treatment exceeded approximately 1.8 million by 2011. Although this chemoprophylaxis has contributed to the containment of vivax malaria, the possibility of the emergence of chloroquine-resistant P. vivax strains has been a concern. Indeed, chemoprophylaxis failure has been reported in several cases, despite the attainment of sufficiently high plasma concentrations of hydroxychloroquine [6, 16]. Therefore, monitoring for chloroquine-resistant P. vivax is important for the control of malaria in Republic of Korea.
In vitro assays provide drug susceptibility estimates free from the effects of most factors that affect in vivo assays. However, the lack of a robust, standardized and widely applicable protocol for long-term in vitro culture hinders P. vivax malaria research. Therefore, short-term ex vivo assays have been successfully used to monitor the chloroquine resistance of P. vivax isolates [25, 36]. In addition to the ex vivo chloroquine susceptibility assays, molecular markers, such as pvmdr1, have been used to examine chloroquine resistance in P. vivax isolates. The pvmdr1 gene encoding for an ATP binding cassette transporter, has been shown to modulate the responses of P. vivax to chloroquine and other anti-malarial drugs [17, 28]. Therefore, the SNPs of pvmdr1 were evaluated in isolates obtained from patients residing in the demilitarized zone. Compared to the pvmdr1 gene of the reference P. vivax Sal I strain, SNPs at four loci of pvmdr1, one non-synonymous mutation (F1076L) and three synonymous SNPs (T529, E1233, and S1358; Table 2) were detected in the P. vivax isolated from patients. Mutant alleles at position 1076 (F1076L) were present in all isolates (100 %) obtained from samples collected in 2011 and 2012. The number of SNPs at position 529 (T529) and 1233 (E1233) was higher in the 2011 isolates (T529, 57.1 %; E1233, 42.9 %) than in 2012 (T529, 55.6 %; E1233, 33.3 %). Among these four SNPs, F1076L, T529, and E1233 have been reported previously [25]. The newly identified SNP was in codon 1358 (S1358). The S1358 SNP has been reported to be associated with the low chloroquine susceptibility of P. vivax in Thailand and Myanmar: this result could be considered to be an early sign of the increasing presence of the chloroquine-resistant P. vivax strains in the Republic of Korea [25]. However, none of the analysed samples harboured a mutation at codon 976 (Y976F), which has been identified as a possible genetic marker of chloroquine resistance in Southeast Asia and Papua New Guinea [28, 37–39]. This data suggests that chloroquine-resistant P. vivax may not currently be prevalent in the Republic of Korea. The dN/dS ratios, the ratio of the rate of non-synonymous substitutions to the rate of synonymous substitutions, were 0.935 (29/31) and 0.865 (32/37) for the isolates from 2011 and 2012, respectively. Although the dN/dS ratio of samples collected is below 1 and less than the dN/dS ratio of 2011 samples, these data cannot be concluded that the 2012 samples were under selective pressure.
Following the identification of SNPs, the multiple clone infection pattern, genetic diversity as well as inter- and intra-population differences between the pvmdr1 groups was evaluated using 10 loci. Different alleles sizes observed in a single locus were classified as a multiplicity of infection (MOI). The MOI referred to multiple clone infection. Multiple clone infection was observed in some of the microsatellite loci in 24 of the 55 isolates (49.1 %). Multiple clone infection occurred more frequently in samples from 2011 (60.7 %, n = 17) than in those from 2012 (37.0 %, n = 10). The number of MOI loci per sample was also examined. The highest number of MOI loci per isolate was four, which we observed in a single isolate.
The major alleles of each locus were used for our population genetic analysis. As shown in Table 3, genetic diversity in P. vivax isolates from 2012 (A = 6.49 ± 0.49, He = 0.72 ± 0.14) was greater than in those from 2011 (A = 5.23 ± 0.76, He = 0.52 ± 0.29). Moreover, the level of multilocus LD (I S A ) was calculated using allelic data from all P. vivax isolates (Table 3). No significant multilocus LD (I S A = 0.040, P > 0.05) was observed in the P. vivax population. Notably, there was greater multilocus LD in the 2012 population (I S A = 0.052) than in the 2011 population (I S A = 0.028). The genetic diversity of P. vivax population in Korea was greater than that reported by Iwagami et al. [26, 27]. Although the sample size (n) was small, the increased genetic diversity and decreased multilocus LD levels of recent P. vivax isolates observed appears to be a trend in Korea [26, 40]. The multilocus LD levels were very low (I S A = 0.028 in 2011; I S A = 0.052 in 2012), suggesting a large possibility of outbreeding between different genotypes. These results are noteworthy since the number of vivax malaria cases is declining in Korea (1772 cases in 2010, 826 cases in 2011, and 542 cases in 2012), which suggests that genetically new P. vivax isolates may emerge in the Republic of Korea every year. However, because the isolates exhibited few genetic differences, it would be difficult to conclude that the new P. vivax isolates are from different high-risk areas of the Republic of Korea [41].
Microsatellite genotypes of the 55 isolates were determined based on a combination of the allelic data of the 10 microsatellite loci, and 30 genotypes (G1–G30; Table 4) were identified. Three major genotypes, G12, G16, and G17 were identified. Seven (12.7 %) of the 55 isolates belonged to genotype G16, whereas five isolates each belonged to genotypes G12 and G17. The relationship between the 30 genotypes was determined by eBURST analysis with the following criterion: when two isolates shared more than two identical loci (out of the three loci), these were connected with a branch (Fig. 2). The eBURST analysis revealed two major groups, Group 1 and Group 2. Group 1 contained 22 isolates (40.0 %), including the isolates of genotypes G12, G16, and G17. Group 2 contained three isolates (5.5 %), including the isolates of genotypes G5, G6, and G7. Five isolates of genotypes G4, G8, G29, and G30 were not included in the two major groups, nor were they connected to any other genotypes. Additionally, a single isolate with a new SNP at codon 1358 (S1358) was classified as genotype G24 and was located at the end of the branch, indicating that this genotype is newly introduced into the Republic of Korea P. vivax population.

Population structure of the 30 genotypes (n = 55) of P. vivax in Republic of Korea were analysed by eBURST. H1-H30 are the microsatellite genotypes. *Genotype includes the newly identified SNP on codon S1358 (TCC/TCT)
Conclusions
In conclusion, the pvmdr1 gene was analysed in samples collected from South Korean soldiers. The results showed that an isolate with a new SNP (S1358) of pvmdr1 has been introduced into the Korean P. vivax population and that the genetic diversity of the Korean P. vivax population is likely to be greater in 2012 than in 2011. Therefore, further continuous monitoring for the presence of chloroquine resistant parasites using molecular markers is needed for the control of vivax malaria in the Republic of Korea.
References
Price RN, Tjitra E, Guerra CA, Yeung S, White NJ, Anstey NM. Vivax malaria: neglected and not benign. Am J Trop Med Hyg. 2007;77:79–87.
Guerra CA, Howes RE, Patil AP, Gething PW, Van Boeckel TP, Temperley WH, et al. The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS Negl Trop Dis. 2010;4:e774.
Chai IH, Lim GI, Yoon SN, Oh WI, Kim SJ, Chai JY. Occurrence of tertian malaria in a male patient who has never been abroad. Korean J Parasitol. 1994;32:195–200.
Yeom JS, Jun G, Kim JY, Lee WJ, Shin EH, Chang KS, et al. Status of Plasmodium vivax malaria in the Republic of Korea, 2008–2009: decrease followed by resurgence. Trans R Soc Trop Med Hyg. 2012;106:429–36.
Korea Centers for Disease Control and Prevention Infectious Disease Statistics System. http://is.cdc.go.kr/
Jeong S, Yang HW, Yoon YR, Lee WK, Lee YR, Jha BK, et al. Evaluation of the efficacy of chloroquine chemoprophylaxis for vivax malaria among Republic of Korea military personnel. Parasitol Int. 2013;62:494–6.
Park JW, Jun G, Yeom JS. Plasmodium vivax malaria: status in the Republic of Korea following reemergence. Korean J Parasitol. 2009;47(Suppl):S39–50.
von Seidlein L, Greenwood BM. Mass administrations of antimalarial drugs. Trends Parasitol. 2003;19:452–60.
Schuurkamp GJ, Spicer PE, Kereu RK, Bulungol PK, Rieckmann KH. Chloroquine-resistant Plasmodium vivax in Papua New Guinea. Trans R Soc Trop Med Hyg. 1992;86:121–2.
Rieckmann H, Davis DR, Hutton DC. Plasmodium vivax resistance to chloroquine? Lancet. 1989;2:1183–4.
Schwartz IK, Lackritz EM, Patchen LC. Chloroquine-resistant Plasmodium vivax from Indonesia. N Engl J Med. 1991;324:927.
Baird JK. Chloroquine resistance in Plasmodium vivax. Antimicrob Agents Chemother. 2004;48:4075–83.
Ruebush TK 2nd, Zegarra J, Cairo J, Andersen EM, Green M, Pillai DR, et al. Chloroquine-resistant Plasmodium vivax malaria in Peru. Am J Trop Med Hyg. 2003;69:548–52.
Soto J, Toledo J, Gutierrez P, Luzz M, Llinas N, et al. Plasmodium vivax clinically resistant to chloroquine in Colombia. Am J Trop Med Hyg. 2001;65:90–3.
Marlar-Than Myat-Phone-Kyaw, Aye-Yu-Soe Khaing-Khaing-Gyi, Ma-Sabai Myint-Oo. Development of resistance to chloroquine by Plasmodium vivax in Myanmar. Trans R Soc Trop Med Hyg. 1995;89:307–8.
Lee SW, Lee M, Lee DD, Kim C, Kim YJ, Kim JY, et al. Biological resistance of hydroxychloroquine for Plasmodium vivax malaria in the Republic of Korea. Am J Trop Med Hyg. 2009;81:600–4.
Sá JM, Nomurab T, Nevesc JD, Baird K, Wellems TE, Portillo HA. Plasmodium vivax: allele variants of the mdr1 gene do not associate with chloroquine resistance among isolates from Brazil, Papua, and monkey-adapted strains. Exp Parasitol. 2005;109:256–9.
Suwanarusk R, Russell B, Chavchich M, Chalfein F, Kenangalem E, Kosaisavee V, et al. Chloroquine resistant Plasmodium vivax: in vitro characterisation and association with molecular polymorphisms. PLoS One. 2007;2:e1089.
Rungsihirunrat K, Muhamad P, Chaijaroenkul W, Kuesap J, Na-Bangchang K. Plasmodium vivax drug resistance genes; Pvmdr1 and Pvcrt-o polymorphisms in relation to chloroquine sensitivity from a malaria endemic area of Thailand. Korean J Parasitol. 2015;53:43–9.
Lin JT, Patel JC, Kharabora O, Sattabongkot J, Muth S, Ubalee R, et al. Plasmodium vivax isolates from Cambodia and Thailand show high genetic complexity and distinct patterns of P. vivax multidrug resistance gene 1 (pvmdr1) polymorphisms. Am J Trop Med Hyg. 2013;88:1116–23.
Melo GC, Monteiro WM, Siqueira AM, Silva SR, Magalhaes BM, Alencar AC, et al. Expression levels of pvcrt-o and pvmdr-1 are associated with chloroquine resistance and severe Plasmodium vivax malaria in patients of the Brazilian Amazon. PLoS One. 2014;9:e105922.
Brega S, Meslin B, de Monbrison F, Severini C, Gradoni L, Udomsangpetch R, et al. Identification of the Plasmodium vivax mdr-like gene (pvmdr1) and analysis of single-nucleotide polymorphisms among isolates from different areas of endemicity. J Infect Dis. 2005;191:272–7.
Orjuela-Sanchez P, Karunaweera ND, da Silva-Nunes M, da Silva NS, Scopel KK, Goncalves RM, et al. Single-nucleotide polymorphism, linkage disequilibrium and geographic structure in the malaria parasite Plasmodium vivax: prospects for genome-wide association studies. BMC Genet. 2010;11:65.
Kim YK, Kim C, Park I, Kim HY, Choi JY, Kim JM. Therapeutic efficacy of chloroquine in Plasmodium vivax and the pvmdr1 polymorphisms in the Republic of Korea under mass chemoprophylaxis. Am J Trop Med Hyg. 2011;84:532–4.
Lu F, Lim CS, Nam DH, Kim K, Lin K, Kim TS, et al. Genetic polymorphism in pvmdr1 and pvcrt-o genes in relation to in vitro drug susceptibility of Plasmodium vivax isolates from malaria-endemic countries. Acta Trop. 2011;117:69–75.
Iwagami M, Fukumoto M, Hwang SY, Kim SH, Kho WG, Kano S. Population structure and transmission dynamics of Plasmodium vivax in the Republic of Korea based on microsatellite DNA analysis. PLoS Negl Trop Dis. 2012;6:e1592.
Iwagami M, Hwang SY, Kim SH, Park SJ, Lee GY, Matsumoto-Takahashi EL, et al. Microsatellite DNA analysis revealed a drastic genetic change of Plasmodium vivax population in the Republic of Korea during 2002 and 2003. PLoS Negl Trop Dis. 2013;7:e2522.
Suwanarusk R, Chavchich M, Russell B, Jaidee A, Chalfein F, Barends M, et al. Amplification of pvmdr1 associated with multidrug-resistant Plasmodium vivax. J Infect Dis. 2008;198:1558–64.
Clustal Omega. http://www.ebi.ac.uk/Tools/msa/clustalo/
Koepfli C, Mueller I, Marfurt J, Goroti M, Sie A, Oa O, et al. Evaluation of Plasmodium vivax genotyping markers for molecular monitoring in clinical trials. J Infect Dis. 2009;199:1074–80.
Karunaweera ND, Ferreira MU, Hartl DL, Wirth DF. Fourteen polymorphic microsatellite DNA markers for the human malaria parasite Plasmodium vivax. Mol Ecol Notes. 2007;7:172–5.
Hudson RR. Analytical results concerning linkage disequilibrium in models with genetic transformation and conjugation. J Evol Biol. 1994;7:535–48.
Anderson TJ, Su XZ, Bockarie M, Lagog M, Day KP. Twelve microsatellite markers for characterization of Plasmodium falciparum from finger-prick blood samples. Parasitology. 1999;119:113–25.
Anderson TJ, Haubold B, Williams JT, Estrada-Franco JG, Richardson L, Mollinedo R, et al. Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol. 2000;17:1467–82.
Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186:1518–30.
Chotivanich K, Udomsangpetch R, Chierakul W, Newton PN, Ruangveerayuth R, Pukrittayakamee S, et al. In vitro efficacy of antimalarial drugs against Plasmodium vivax on the western border of Thailand. Am J Trop Med Hyg. 2004;70:395–7.
Marfurt J, de Monbrison F, Brega S, Barbollat L, Muller I, Sie A, et al. Molecular markers of in vivo Plasmodium vivax resistance to amodiaquine plus sulfadoxine-pyrimethamine: mutations in pvdhfr and pvmdr1. J Infect Dis. 2008;198:409–17.
Imwong M, Pukrittayakamee S, Pongtavornpinyo W, Nakeesathit S, Nair S, Newton P, et al. Gene amplification of the multidrug resistance 1 gene of Plasmodium vivax isolates from Thailand, Laos, and Myanmar. Antimicrob Agents Chemother. 2008;52:2657–9.
Vargas-Rodriguez RDC, da Silva Bastos M, Menezes MJ, Orjuela-Sanchez P, Ferreira MU. Single-nucleotide polymorphism and copy number variation of the multidrug resistance-1 locus of Plasmodium vivax: local and global patterns. Am J Trop Med Hyg. 2012;87:813–21.
Choi YK, Choi KM, Park MH, Lee EG, Kim YJ, Lee BC, et al. Rapid dissemination of newly introduced Plasmodium vivax genotypes in South Korea. Am J Trop Med Hyg. 2010;82:426–32.
Kim JY, Suh EJ, Yu HS, Jung HS, Park IH, Choi YK, et al. Longitudinal and cross-sectional genetic diversity in the Korean Peninsula based on the P. vivax merozoite surface protein gene. Public Health Res Perspect. 2011;2:158–63.
Author’s contributions
DIC, YH, and YKG designed the study; SJ, SDDB and HWY conducted the experiments; DIC, YH, and YKG were involved in data analysis; SJ provided the materials; YH and YKG contributed to writing the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by the Korean Military Medical Research Project funded by the Republic of Korea Ministry of National Defense (ROK-MND-2012-KMMRP-15) and Kyungpook National University Research Fund, 2012. We thank the medical officers in the Republic of Korea Armed Forces Medical Command for providing the patient data.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Dong-Il Chung and Sookwan Jeong contributed equally to this work
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chung, DI., Jeong, S., Dinzouna-Boutamba, SD. et al. Evaluation of single nucleotide polymorphisms of pvmdr1 and microsatellite genotype in Plasmodium vivax isolates from Republic of Korea military personnel. Malar J 14, 336 (2015). https://doi.org/10.1186/s12936-015-0845-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-015-0845-6