
Abstract
Background
In northern Papua New Guinea (PNG), most Plasmodium falciparum isolates proved resistant to chloroquine (CQ) in vitro between 2005 and 2007, and there was near-fixation of pfcrt K76T, pfdhfr C59R/S108N and pfmdr1 N86Y. To determine whether the subsequent introduction of artemisinin combination therapy (ACT) and reduced CQ-sulphadoxine-pyrimethamine pressure had attenuated parasite drug susceptibility and resistance-associated mutations, these parameters were re-assessed between 2011 and 2013.
Methods
A validated fluorescence-based assay was used to assess growth inhibition of 52 P. falciparum isolates from children in a clinical trial in Madang Province. Responses to CQ, lumefantrine, piperaquine, naphthoquine, pyronaridine, artesunate, dihydroartemisinin, artemether were assessed. Molecular resistance markers were detected using a multiplex PCR ligase detection reaction fluorescent microsphere assay.
Results
CQ resistance (in vitro concentration required for 50% parasite growth inhibition (IC50) >100 nM) was present in 19% of isolates. All piperaquine and naphthoquine IC50s were <100 nM and those for lumefantrine, pyronaridine and the artemisinin derivatives were in low nM ranges. Factor analysis of IC50s showed three groupings (lumefantrine; CQ, piperaquine, naphthoquine; pyronaridine, dihydroartemisinin, artemether, artesunate). Most isolates (96%) were monoclonal pfcrt K76T (SVMNT) mutants and most (86%) contained pfmdr1 N86Y (YYSND). No wild-type pfdhfr was found but most isolates contained wild-type (SAKAA) pfdhps. Compared with 2005–2007, the geometric mean (95% CI) CQ IC50 was lower (87 (71–107) vs 167 (141–197) nM) and there had been no change in the prevalence of pfcrt K76T or pfmdr1 mutations. There were fewer isolates of the pfdhps (SAKAA) wild-type (60 vs 100%) and pfdhfr mutations persisted.
Conclusions
Reflecting less drug pressure, in vitro CQ sensitivity appears to be improving in Madang Province despite continued near-fixation of pfcrt K76T and pfmdr1 mutations. Temporal changes in IC50s for other anti-malarial drugs were inconsistent but susceptibility was preserved. Retention or increases in pfdhfr and pfdhps mutations reflect continued use of sulphadoxine-pyrimethamine in the study area including through paediatric intermittent preventive treatment. The susceptibility of local isolates to lumefantrine may be unrelated to those of other ACT partner drugs.
Trial registration
Australian New Zealand Clinical Trials Registry ACTRN12610000913077.
Similar content being viewed by others

Background
Resistance of Plasmodium falciparum to anti-malarial drugs in Papua New Guinea (PNG) began with chloroquine (CQ) in the 1970s [1] and has since extended to amodiaquine [2] and sulphadoxine-pyrimethamine (SP) [3]. Because of this trend, together with efficacy data from a large-scale, multi-arm, treatment trial conducted in coastal PNG from 2005 to 2007 [4] and World Health Organization management guidelines at the time [5], artemisinin combination therapy (ACT) was introduced nationally as recommended therapy for uncomplicated malaria in 2010 [6]. Artemether (AM) plus lumefantrine (LM) is currently first-line and dihydroartemisinin (DHA) plus piperaquine (PQ) second-line treatment in PNG, but artemisinin plus naphthoquine (NQ) is also available in the private sector [7]. Resistance to artemisinin derivatives has, however, emerged in recent years in Southeast Asia [8], and is a concern for countries such as PNG in which ACT is now widely used.
Regular testing using economical, robust and sensitive in vitro anti-malarial drug susceptibility assays is an integral part of the surveillance for parasite resistance [9]. Of the different methods currently available, those based on fluorescence measurements of parasite growth using inexpensive intercalating DNA stains such as Sybr Green and Pico Green have proved efficient and inexpensive without loss of sensitivity [10,11]. Additional insight into mechanisms of resistance is provided by detection of single nucleotide polymorphisms in parasite genes determining drug response [12], including mutations in the P. falciparum CQ transporter (pfcrt), multidrug resistance 1 (pfmdr1), dihydrofolate reductase (pfdhfr), and dihydropteroate synthetase (pfdhps) genes.
The most recent parasite drug resistance data from PNG were collected as part of the comparative intervention trial conducted in coastal Madang and East Sepik Provinces between 2005 and 2007 [13,14]. Most of the isolates tested proved resistant to CQ in vitro but not to other ACT partner drugs or to the artemisinin derivatives themselves [13]. Consistent with this finding and previous heavy 4-aminoquinoline/SP use, there was near-fixation of pfcrt K76T, pfdhfr C59R and S108N, and pfmdr1 N86Y alleles, while multiple mutations were frequent [14].
To determine whether there has been any recent change in P. falciparum drug resistance in the north coastal PNG area, the in vitro susceptibility of local P. falciparum isolates collected between 2011 and 2013 to artemisinin derivatives and ACT partner drugs were re-assessed, and the prevalence of drug resistance markers in the same parasite strains re-examined.
Methods
Study sites, patients and ethical approval
Venous blood samples were obtained from 52 children aged six months to five years with an uncomplicated P. falciparum mono-infection at a parasitaemia >0.5% who were recruited at Mugil (n = 43) and Alexishafen (n = 9) health centres in Madang Province to a randomized, comparative, efficacy trial of the ACT AM-LM and artemisinin-NQ (Australian New Zealand Clinical Trials Registry ACTRN12610000913077) [15]. The study received ethical approval from the Medical Research Advisory Committee of the PNG Department of Health (MRAC #10.39). In all cases, informed consent was obtained from the parents or legal guardians before recruitment and blood sampling.
Drug susceptibility assays
A Sybr Green fluorescence assay was used to assess drug susceptibility. All assays were carried out at the PNG Institute of Medical Research in Madang. The methodology used, a modified version of that first described by Smilkstein et al. [11], has been previously validated against tritium hypoxanthine incorporation, Pf lactate dehydrogenase (PfLDH), light microscopic schizont maturation, and flow cytometry-based drug susceptibility assays using the laboratory-adapted parasite strains 3D7, E8B and W2 [16]. For the present series of experiments, the 3D7 strain was used as reference with a mean CQ in vitro concentration required for 50% parasite growth inhibition (IC50) value of 14.3 nM. This compares with IC50 values of 18–20 nM for tritiated hypoxanthine isotopic assay and 23–33 nM for PfLDH assay using this strain in our laboratories at Fremantle Hospital in Australia (unpublished observations).
The anti-malarial compounds used in this assay were purchased from Sigma-Aldrich, St Louis, MI, USA (CQ diphosphate), Santa Cruz Biotechnologies, Santa Cruz, CA, USA (pyronaridine (PY) tetraphosphate), Hubei Onward Bio Development Co Ltd, Enshi City, Hubei, China (DHA, artesunate (AS), AM, LM) or kindly donated by Mangalam Pty Ltd, Bangalore, India (PQ phosphate and NQ phosphate). Solutions of 10 mM concentration were prepared for each drug in an appropriate solvent (CQ, PQ and PY in deionized water; AM in methanol; NQ in 50% v/v ethanol; LM in 1:1:1 v/v linoleic acid/Tween 80/ethanol; DHA in 70% v/v ethanol; AS in ethanol). These solutions were further diluted to a stock 1 mM concentration in deionized water. After sterile filtration, stock solutions were aliquoted into airtight microcentrifuge tubes and stored at −20°C. A fresh aliquot was used for each assay.
Red blood cells from slide-positive children were washed three times in standard RPMI 1640-based malaria cell culture medium [17] and, if necessary, diluted to 0.5-1.0% parasitaemia with red blood cells from a malaria-naïve donor of blood type O Rhesus negative. The culture medium consisted of RPMI 1640 HEPES (Sigma Aldrich, St Louis, MO) supplemented with 92.6 mg/L L-glutamine (Sigma Aldrich, St Louis, MO), 500 μg/L gentamicin (Sigma Aldrich, St Louis, MO), 50 mg/L hypoxanthine (Sigma Aldrich, St Louis, MO) and 0.5% w/v Albumax II lipid rich BSA (Life Technologies, Mulgrave, Victoria, Australia) [16]. Drug dilutions were set up in 96-well plates in triplicate, with eight dilutions for each drug. The haematocrit was set at 1% and the liquid volume per well was 200 μL. The assay plates were incubated for 48 hr in a candle jar using the method of Trager and Jensen [18], after which 50 μL of a red cell lysis buffer/Sybr green (Invitrogen, Carlsbad, CA, USA) mixture were added to each well. The plate was incubated for 15 min in the dark. Fluorescence was read on a microplate reader (Fluostar Optima, BMG Labtec, Offenburg, Germany) equipped with a 484 nm excitation filter and a 520 nm absorbance filter.
Molecular analysis
Parasite isolates where tested for genetic markers associated with drug resistance using a multiplex polymerase chain reaction ligase detection reaction fluorescent microsphere assay (PCR-LDR-FMA) assay as previously described [14,19]. In brief, PCR-LDR-FMA was performed using established primer sequences to detect single nucleotide polymorphisms in the known resistance loci of pfdhfr (codons 51, 58, 108, 164), pfdhps (codons 436–437, 540, 581, 613), pfcrt (codons 72–76) and pfmdr1 (codons 86, 184, 1034, 1042, 1246). Fluorescent products were detected using a Bio-Plex analyzer (Bio-Rad, Hercules, CA, USA). Data analysis was conducted as described previously [20,21].
Data analysis
Concentrations of anti-malarial drugs for each isolate and anti-malarial were log-transformed and the fluorescence values were normalized such that the smallest value in each dataset represented 0 and the largest value (drug-free control) unity. The dose–response curve Y = 100/(1 + 10k (logIC50-logX)) was then fitted to each dataset, where Y corresponds to the percentage of growth at drug concentration X, and k is the Hill slope. For calculations of means and 95% confidence intervals (CI) as well as for analysis of associations between pairs of different anti-malarial drugs, the log10 IC50 values were used as these were normally distributed by Kolmogorov-Smirnov test. Associations between IC50 values were determined using Pearson’s correlation coefficient, and significant pairwise correlations (P < 0.05) were considered moderate for 0.3 ≤ r ≤0.50 or strong for r >0.5.
Factor analysis was conducted after testing the cross-correlation matrix for sphericity using Bartlett’s Test and using the Kaiser-Maier-Olkin statistic to determine the appropriateness of the data for this analysis. The distribution of the Eigenvalues of the cross-correlation matrix indicated that factoring into two components was the most appropriate approach. Since it was hypothesized that the underlying factors relate to the mechanisms of drug action and are thus related to each other, a non-orthogonal (direct-oblimin) rotation was applied to the solution.
Results
In vitro drug susceptibility measurements
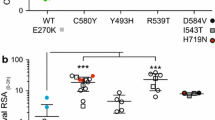
From a total of 416 drug assays (52 isolates and 8 drugs), 379 (91.1%) provided a valid dose–response curve that could be used for analysis. Numbers of successful assays per drug were 47 (NQ), 44 (LM), 50 (DHA), 50 (AS), 48 (AT), 45 (PY), 47 (PQ) and 48 (CQ). The growth responses of the parasite isolates to the panel of anti-malarial drugs used in the present study are summarized in Figure 1. CQ resistance (IC50 > 100 nM [22,23]) was present in nine out of 48 isolates (19%). For alternative resistance thresholds of 87 nM, 70 nM and 25 nM as recommended by other authors [24-26], the percentages of resistant strains were 26, 42 and 88%, respectively. Although there is no recommended threshold for PQ or NQ, 100 nM has been suggested for PQ and may also be appropriate for NQ [27]. All isolates had an IC50 value for PQ and NQ <100 nM, but two strains had IC50 values that were close to this value (85 nM for PQ in one case and 96 nM for NQ in the other). Logarithmic mean IC50 values and their 95% CIs were 87 (71–107) nM for CQ, 21.0 (16.9-26.1) nM for PQ, 4.2 (3.1-5.8) nM for NQ, 8.0 (6.0-10.6) nM for PY, 1.5 (1.1-2.1) nM for LM, 5.2 (4.2-6.5) nM for DHA, 6.1 (4.9-7.6) nM for AS and 5.7 (4.2-7.9) nM for AM.

50% inhibitory concentrations (IC 50 ) for the anti-malarial compounds investigated in the present study. Chloroquine (CQ), piperaquine (PQ), naphthoquine (NQ), pyronaridine (PY), lumefantrine (LM), dihydroartemisinin (DHA), artesunate (AS) artemether (AM). Panel A shows the logarithmic means and 95% CIs and Panel B shows measurements for single isolates.
Table 1 shows the cross-correlation analysis for each pair of anti-malarial drugs. The strongest correlations were observed between the IC50 values for i) CQ, PQ, NQ, and PY (0.31 < r < 0.55) with an exception being that between CQ and PY (P > 0.05), and ii) between DHA, AM, AS, and PY (0.45 < r < 0.66) with an exception being that between PY and AM (P > 0.05). The IC50 values for LM showed no significant correlations with those of any of the other drugs. Factor analysis indicated two underlying components, which may explain most of the variation in drug responses (see Figure 2). Within this two-component space, the eight drugs clustered into three distinct groups (LM on its own; CQ, PQ and NQ; PY, DHA, AM, and AS).

Factor analysis of IC 50 values. Kaiser-Maier-Olkin (KMO) statistics and Bartlett’s Test for sphericity indicated the appropriateness of the data for factor analysis (KMO statistic: 0.62, Bartlett’s test p-value: <0.0001). The distribution of the Eigenvalues indicated that a separation into two components was most appropriate. Within the two-component space, three distinct clusters of drugs were observed (1: lumefantrine (LM) 2: chloroquine (CQ), piperaquine (PQ), naphthoquine (NQ); 3: pyronaridine (PY), artesunate (AS), dihydroartemisinin (DHA), artemether (AM)).
Drug resistance markers and their association with in vitro drug susceptibility
Most isolates (96%) were monoclonal pfcrt K76T mutants of haplotype SVMNT (codons 72 to 76). One isolate contained a mix of CVMNK (wild-type) and SVMNT (mutant). In only one isolate was CVMNK (wild-type) detected as the only clone. This isolate exhibited an IC50 of 12.1 nM against CQ, which was the lowest in IC50 for CQ found in the study. The polyclonal CVMNK/SVMNT isolate exhibited a CQ IC50 of 30.0 nM, which was also amongst the lowest values determined in the present study.
Two mutant haplotypes of pfmdr1 were identified, the most prevalent being the YYSND type associated with CQ resistance (86% of isolates, N86Y mutation). The NFSND haplotype was found in 5% of isolates (Y184F mutation). The wild-type NYSND occurred in only 9% of isolates. The presence of NYSND was associated with reduced CQ IC50 values (P < 0.05, see Table 2).
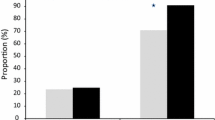
No wild-type pfdhfr was found in the isolates studied. All isolates carried the C59R mutation and the S108N mutation (with absence of mutation at codons 51 and 164, Figure 3). Among them, 13% of isolates were polyclonal with the S108T mutation detected as well (haplotype NRTI). The SAKAA wild-type haplotype of the pfdhps gene was found as the only haplotype in 60% of all isolates and in a further 9% in polyclonal infections. The SGKAA and the SGEAA mutant haplotypes occurred in 9 and 14% as monoclonal infections, respectively, and in a further 9% of polyclonal infections. An additional haplotype FGEAA was found in 6% of infections, mixed with the SGEAA haplotype.

Prevalence of drug resistance-associated gene mutations in parasite isolates from north coastal PNG. Numbers in parentheses denote the number of samples that resulted in interpretable genotyping results. Panel A shows data collected in the present study and panel B shows data collected between 2005 and 2007 [14]. Percentages of wild-type genotypes (■), mutants (□) and mixed infections (grey square) are shown.
Comparison of the present findings with those of similar surveillance in 2005–2007
The geometric mean and 95% CIs for parasite isolates obtained from children with uncomplicated malaria in a study carried out in Madang and East Sepik Provinces between 2005 and 2007, as well as the present drug susceptibility data, are shown in Table 3 [13]. The method used in the earlier study was the PfLDH assay [28]. The IC50 values were lower for the present isolates than those obtained an average of six years earlier for CQ, NQ and LM, but higher for PQ and DHA.
The P. falciparum polymorphisms associated with anti-malarial drug resistance for the isolates obtained between 2005 and 2007 [14], as well as those of the present study, are shown graphically in Figure 3. There had been no change in the near-fixation of pfcrt K76T in the six years between studies and the prevalence of pfmdr1 mutations was also similar. The NRTI haplotype was not reported in the previous study using the same methodology [14] but it was detected by the present genotyping. The pfdhps wild-type gene (SAKAA) was found as the only haplotype in 60% of all isolates and in a further 9% of polyclonal infections, a lower prevalence than the ~100% reported previously [14].
Discussion
The present data demonstrate that there have been changes in the drug resistance characteristics of parasite isolates collected between 2005–2007 and 2011–2013 from areas of north coastal PNG with intense malaria transmission. Although a different methodology was used to assess in vitro sensitivity in the present study, and notwithstanding limitations in assigning thresholds for in vitro drug sensitivity [29], more strains appeared CQ-sensitive than in 2005–2007 [13,14] despite the majority retaining the mutant pfcrt K76T allele over time. There were also apparent temporal reductions in the IC50s of LM and NQ, while those for PQ and DHA increased albeit still within relatively low nM ranges. The proportion of parasites carrying the wild-type pfdhps gene had fallen over time and more mutations had appeared in pfdhps in the 2011–2013 isolates, consistent with continued use of SP in the study area. Factor analysis suggested that the in vitro susceptibilities of PNG P. falciparum strains to LM and PY may be unrelated to those of other long half-life ACT partner drugs, with the IC50 of PY clustering with those of the artemisinin derivatives. Interpretation of the present and previous data needs to take into account several factors. These comprise i) temporal changes in anti-malarial drug use in the study areas, ii) potential effects of the introduction of non-pharmacological strategies to reduce malaria transmission, and iii) differences in assay methodology between 2005–2007 and 2011–2013.
Recommendations regarding replacement of regimens based on CQ and SP by ACT for treatment of uncomplicated malaria in PNG children were implemented in 2010 [6], but translation of this policy into practice has been slow. In addition, the use of CQ and SP as first-line intermittent preventive therapy (IPT) in pregnancy has continued, and an IPT trial in infants involving SP was conducted in the Mugil area (from where most of the present isolates were collected) between 2006 and 2010 [30]. Therefore, CQ-SP drug pressure had been reduced, but not eliminated, over a period of two to three years leading up to isolate collection in the study area.
The dynamics governing repopulation by CQ-sensitive strains in areas in which CQ treatment pressure has been removed completely are not well understood, but the time-scale is probably approaching a decade [24,31,32]. The fact that the present isolates were collected after a short period of incomplete removal of CQ-SP pressure is reflected in the present molecular analyses which showed no reduction or an increase in parasites carrying genetic markers that correlate with CQ-SP resistance. However, CQ resistance mutations are frequently found in isolates that show in vitro susceptibility [24,25,33], and there is also evidence that CQ IC50 values can fall relatively quickly (within a few years) after reduction in drug pressure [34,35].
The introduction of long-lasting insecticide-impregnated bed nets (LLINs), such as was started on a large scale in PNG in 2004 [36], could theoretically also attenuate drug pressure by reducing malaria transmission. The evidence for this effect on molecular resistance markers in studies from sub-Saharan Africa is conflicting [37,38]. However, these studies were relatively short-term compared with the time needed for re-establishment of full sensitivity after drug withdrawal [24,31,32] and no in vitro susceptibility data were presented. It remains possible that increasing LLIN use in coastal PNG between 2005–2007 and 2011–2013, together with the partial replacement of CQ-SP by ACT, both contributed to the lower IC50 for CQ in the present study.
There is no accepted standardized protocol for determining in vitro anti-malarial drug susceptibilities and the results may differ according to the methodology employed. There is evidence that the PfLDH assay generates higher IC50 values than other methodologies including the Sybr Green assay used in the present study [16,39,40]. However, the reported differences are typically modest (typically 10–30 nM across a range of IC50 values, as seen with our own data for the 3D7 strain) compared with the substantial reduction in CQ IC50 observed between 2005–2007 and 2011–2013. The increase in PQ IC50 over time in north coastal PNG might appear paradoxical given than CQ and PQ susceptibility have both been considered to reflect pfcrt mutations [41]. Nevertheless, not all studies show this relationship [42], while the PQ IC50 values in both time periods were both well below the conventional 100 nM cut-point for resistance in all but one isolate in the present study. The small temporal increase in DHA IC50 may also be of no clinical significance given that the IC50 value in all isolates was in the very low nM range. Standard drug susceptibility assays do not detect early stage artemisinin resistance defined by a slow parasite clearance time for which there is now a molecular marker [43], but the isolates were obtained between 2011 and 2013 from patients in a clinical trial [15] in which there was no evidence of longer parasite clearance times after AM-LM than in the equivalent trial conducted from 2005 to 2007 [4].
Several studies have shown moderate to strong correlations between in vitro parasite responses to CQ and PY [44,45] while in others, including the present study, there has been no such association [46,47]. The future of PY-containing ACT is uncertain because of hepatotoxicity [48]. However, it does not appear to exhibit cross-resistance with CQ in the present parasite isolates, which would be an advantage if PY-based ACT became available for repeated use in PNG. The present data confirm the moderate associations between CQ, PQ and NQ which were also observed previously in north coastal PNG [13]. Although the IC50s for the latter two compounds are relatively low, their significant association with CQ susceptibility may have implications for the longevity of ACT formulations incorporating them. The lack of clustering of LM susceptibility with other longer half-life anti-malarial drugs suggests that it may have an independent mechanism of action. As has been done by other groups [49-51], we included the three artemisinin drugs in common clinical use, even though DHA is the active metabolite of both AS and AM in vivo, since there is evidence that their activity against P. falciparum in vitro is not uniform [52]. Consistent with this latter observation, the association between AM and DHA was only moderate while those between AS and both AM and DHA were the strongest observed.
Conclusion
Although the prevalence of molecular markers of anti-malarial drug resistance has not fallen in north coastal PNG over the six years between the present study and a previous cross-sectional survey, CQ susceptibility has increased even allowing for different methods of in vitro parasite drug sensitivity testing. This may reflect attenuation of drug pressure through changes in national treatment policy and the roll-out of LLINs in the study areas. Although there are no parasite strains showing definite in vitro resistance to PQ and NQ, the association of PQ and CQ IC50s suggests that future susceptibility testing should include these ACT partner drugs which are currently available in PNG as alternatives to LM. Since LM drug susceptibility appears independent of other available and potential ACT partner drugs, the use of AM-LM as first-line treatment of uncomplicated malaria in PNG may not lead to clinically significant cross-resistance.
Abbreviations
- ACT:
-
Artemisinin combination therapy
- AM:
-
Artemether
- AS:
-
Artesunate
- CI:
-
Confidence interval
- CQ:
-
Chloroquine
- DHA:
-
Dihydroartemisinin
- IC50 :
-
in vitro concentration required for 50% parasite growth inhibition
- IPT:
-
Intermittent preventive therapy
- LLIN:
-
Long-lasting insecticide-impregnated bed net
- LM:
-
Lumefantrine
- NQ:
-
Naphthoquine
- PCR-LDR-FMA:
-
Multiplex polymerase chain reaction ligase detection reaction fluorescent microsphere assay
- PNG:
-
Papua New Guinea
- PQ:
-
Piperaquine
- PY:
-
Pyronaridine
- SP:
-
Sulphadoxine-pyrimethamine
References
Saint-Yves IF. The alleged resistance of Plasmodium falciparum to chloroquine in the Milne Bay District. P N G Med J. 1971;14:77–8.
al-Yaman F, Genton B, Mokela D, Narara A, Raiko A, Alpers MP. Resistance of Plasmodium falciparum malaria to amodiaquine, chloroquine and quinine in the Madang Province of Papua New Guinea, 1990–1993. P N G Med J. 1996;39:16–22.
Casey GJ, Ginny M, Uranoli M, Mueller I, Reeder JC, Genton B, et al. Molecular analysis of Plasmodium falciparum from drug treatment failure patients in Papua New Guinea. Am J Trop Med Hyg. 2004;70:251–5.
Karunajeewa HA, Mueller I, Senn M, Lin E, Law I, Gomorrai PS, et al. A trial of combination antimalarial therapies in children from Papua New Guinea. N Engl J Med. 2008;359:2545–57.
WHO. Guidelines for the treatment of malaria. Geneva: World Health Organization; 2006.
Papua New Guinea Department of Health. National Malaria Treatment Protocol. 7th ed. Port Moresby: Government of Papua New Guinea; 2009.
Benjamin J, Moore B, Lee ST, Senn M, Griffin S, Lautu D, et al. Artemisinin-naphthoquine combination therapy for uncomplicated pediatric malaria: a tolerability, safety, and preliminary efficacy study. Antimicrob Agents Chemother. 2012;56:2465–71.
Wongsrichanalai C, Sibley CH. Fighting drug-resistant Plasmodium falciparum: the challenge of artemisinin resistance. Clin Microbiol Infect. 2013;19:908–16.
Noedl H, Wongsrichanalai C, Wernsdorfer WH. Malaria drug-sensitivity testing: new assays, new perspectives. Trends Parasitol. 2003;19:175–81.
Quashie N, de Koning H, Ranford-Cartwright L. An improved and highly sensitive microfluorimetric method for assessing susceptibility of Plasmodium falciparum to antimalarial drugs in vitro. Malar J. 2006;5:95.
Smilkstein M, Sriwilaijaroen N, Kelly J, Wilairat P, Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother. 2004;48:1803–6.
Petersen I, Eastman R, Lanzer M. Drug-resistant malaria: molecular mechanisms and implications for public health. FEBS Lett. 2011;585:1551–62.
Wong RP, Lautu D, Tavul L, Hackett SL, Siba P, Karunajeewa HA, et al. In vitro sensitivity of Plasmodium falciparum to conventional and novel antimalarial drugs in Papua New Guinea. Trop Med Int Health. 2010;15:342–9.
Wong RP, Karunajeewa H, Mueller I, Siba P, Zimmerman PA, Davis TM. Molecular assessment of Plasmodium falciparum resistance to antimalarial drugs in Papua New Guinea using an extended ligase detection reaction fluorescent microsphere assay. Antimicrob Agents Chemother. 2011;55:798–805.
Laman M, Moore BR, Benjamin JM, Yadi G, Bona C, Warrel J, et al. Artemisinin-naphthoquine versus artemether-lumefantrine for uncomplicated malaria in Papua New Guinean children: An open-label randomized trial. PLoS Med. 2014;11:e100177.
Karl S, Wong RP, St Pierre TG, Davis TM. A comparative study of a flow-cytometry-based assessment of in vitro Plasmodium falciparum drug sensitivity. Malar J. 2009;8:294.
Karl S, Davis TME, St Pierre TG. Parameterization of high magnetic field gradient fractionation columns for applications with Plasmodium falciparum infected human erythrocytes. Malar J. 2010;9:116.
Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–5.
Barnadas C, Senn N, Iga J, Timinao L, Javati S, Malau E, et al. Plasmodium falciparum and Plasmodium vivax genotypes and efficacy of intermittent preventive treatment in Papua New Guinea. Antimicrob Agents Chemother. 2014;58:6958–61.
DaRe JT, Kouri DP, Zimmerman PA, Thomas PJ. Differentiating Plasmodium falciparum alleles by transforming Cartesian X,Y data to polar coordinates. BMC Genet. 2010;11:57.
Barnadas C, Kent D, Timinao L, Iga J, Gray LR, Siba P, et al. A new high-throughput method for simultaneous detection of drug resistance associated mutations in Plasmodium vivax dhfr, dhps and mdr1 genes. Malar J. 2011;10:282.
Bacon DJ, Jambou R, Fandeur T, Le Bras J, Wongsrichanalai C, Fukuda MM, et al. World Antimalarial Resistance Network (WARN) II: In vitro antimalarial drug susceptibility. Malar J. 2007;6:120.
Basco L, Ringwald P. Drug-resistant malaria: problems with its definition and technical approaches. Sante. 2000;10:47–50.
Mwai L, Ochong E, Abdirahman A, Kiara SM, Ward S, Kokwaro G, et al. Chloroquine resistance before and after its withdrawal in Kenya. Malar J. 2009;8:106.
Thomas SM, Ndir O, Dieng T, Mboup S, Wypij D, Maguire JH, et al. In vitro chloroquine susceptibility and PCR analysis of pfcrt and pfmdr1 polymorphisms in Plasmodium falciparum isolates from Senegal. Am J Trop Med Hyg. 2002;66:474–80.
Mbaisi A, Liyala P, Eyase F, Achilla R, Akala H, Wangui J, et al. Drug susceptibility and genetic evaluation of Plasmodium falciparum isolates obtained in four distinct geographical regions of Kenya. Antimicrob Agents Chemother. 2004;48:3598–601.
Basco LK, Ringwald P. In vitro activities of piperaquine and other 4-aminoquinolines against clinical isolates of Plasmodium falciparum in Cameroon. Antimicrob Agents Chemother. 2003;47:1391–4.
Makler MT, Ries JM, Williams JA, Bancroft JE, Piper RC, Gibbins BL, et al. Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am J Trop Med Hyg. 1993;48:739–41.
Ringwald P, Bickii J, Basco LK. In vitro activity of antimalarials against clinical isolates of Plasmodium falciparum in Yaounde, Cameroon. Am J Trop Med Hyg. 1996;55:254–8.
Senn N, Rarau P, Stanisic DI, Robinson L, Barnadas C, Manong D, et al. Intermittent preventive treatment for malaria in Papua New Guinean infants exposed to Plasmodium falciparum and P. vivax: a randomized controlled trial. PLoS Med. 2012;9:e1001195.
Laufer MK, Thesing PC, Eddington ND, Masonga R, Dzinjalamala FK, Takala SL, et al. Return of chloroquine antimalarial efficacy in Malawi. N Engl J Med. 2006;355:1959–66.
Frosch A, Venkatesan M, Laufer M. Patterns of chloroquine use and resistance in sub-Saharan Africa: a systematic review of household survey and molecular data. Malar J. 2011;10:116.
Chen N, Russell B, Staley J, Kotecka B, Nasveld P, Cheng Q. Sequence polymorphisms in pfcrt are strongly associated with chloroquine resistance in Plasmodium falciparum. J Infect Dis. 2001;183:1543–5.
Huong NM, Hewitt S, Davis TM, Dao LD, Toan TQ, Kim TB, et al. Resistance of Plasmodium falciparum to antimalarial drugs in a highly endemic area of southern Viet Nam: a study in vivo and in vitro. Trans R Soc Trop Med Hyg. 2001;95:325–9.
Thanh NV, Cowman AF, Hipgrave D, Kim TB, Phuc BQ, Cong LD, et al. Assessment of susceptibility of Plasmodium falciparum to chloroquine, quinine, mefloquine, sulfadoxine-pyrimethamine and artemisinin in southern Viet Nam. Trans R Soc Trop Med Hyg. 2001;95:513–7.
Hetzel MW, Gideon G, Lote N, Makita L, Siba PM, Mueller I. Ownership and usage of mosquito nets after four years of large-scale free distribution in Papua New Guinea. Malar J. 2012;11:192.
Alifrangis M, Lemnge MM, Ronn AM, Segeja MD, Magesa SM, Khalil IF, et al. Increasing prevalence of wildtypes in the dihydrofolate reductase gene of Plasmodium falciparum in an area with high levels of sulfadoxine/pyrimethamine resistance after introduction of treated bed nets. Am J Trop Med Hyg. 2003;69:238–43.
Shah M, Kariuki S, Vanden Eng J, Blackstock AJ, Garner K, Gatei W, et al. Effect of transmission reduction by insecticide-treated bednets (ITNs) on antimalarial drug resistance in western Kenya. PLoS One. 2011;6:e26746.
Kaddouri H, Nakache S, Houzé S, Mentré F, Le Bras J. Assessment of the drug susceptibility of Plasmodium falciparum clinical isolates from Africa by using a Plasmodium lactate dehydrogenase immunodetection assay and an inhibitory maximum effect model for precise measurement of the 50-percent inhibitory concentration. Antimicrob Agents Chemother. 2006;50:3343–9.
Brockman A, Singlam S, Phiaphun L, Looareesuwan S, White NJ, Nosten F. Field evaluation of a novel colorimetric method - double-site enzyme-linked lactate dehydrogenase immunodetection assay - to determine drug susceptibilities of Plasmodium falciparum clinical isolates from northwestern Thailand. Antimicrob Agents Chemother. 2004;48:1426–9.
Muangnoicharoen S, Johnson DJ, Looareesuwan S, Krudsood S, Ward SA. Role of known molecular markers of resistance in the antimalarial potency of piperaquine and dihydroartemisinin in vitro. Antimicrob Agents Chemother. 2009;53:1362–6.
Mwai L, Kiara SM, Abdirahman A, Pole L, Rippert A, Diriye A, et al. In vitro activities of piperaquine, lumefantrine, and dihydroartemisinin in Kenyan Plasmodium falciparum isolates and polymorphisms in pfcrt and pfmdr1. Antimicrob Agents Chemother. 2009;53:5069–73.
Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–5.
Elueze EI, Croft SL, Warhurst DC. Activity of pyronaridine and mepacrine against twelve strains of Plasmodium falciparum in vitro. J Antimicrob Chemother. 1996;37:511–8.
Pradines B, Tall A, Parzy D, Spiegel A, Fusai T, Hienne R, et al. In-vitro activity of pyronaridine and amodiaquine against African isolates (Senegal) of Plasmodium falciparum in comparison with standard antimalarial agents. J Antimicrob Chemother. 1998;42:333–9.
Kurth F, Pongratz P, Belard S, Mordmuller B, Kremsner P, Ramharter M. In vitro activity of pyronaridine against Plasmodium falciparum and comparative evaluation of anti-malarial drug susceptibility assays. Malar J. 2009;8:79.
Pradines B, Briolant S, Henry M, Oeuvray C, Baret E, Amalvict R, et al. Absence of association between pyronaridine in vitro responses and polymorphisms in genes involved in quinoline resistance in Plasmodium falciparum. Malar J. 2010;9:339.
Assessment report. Pyramax (pyronaridine tetraphosphate/artesunate). Procedure No.: EMEA/H/W/002319 [http://www.ema.europa.eu/docs/en_GB/document_library/Other/2012/06/WC500129290.pdf] (accessed November 2014).
Jambou R, Legrand E, Niang M, Khim N, Lim P, Volney B, et al. Resistance of Plasmodium falciparum field isolates to in-vitro artemether and point mutations of the SERCA-type PfATPase6. Lancet. 2005;366:1960–3.
Pradines B, Bertaux L, Pomares C, Delaunay P, Marty P. Reduced in vitro susceptibility to artemisinin derivatives associated with multi-resistance in a traveller returning from South-East Asia. Malar J. 2011;10:268.
Yang H, Liu D, Yang Y, Fan B, Yang P, Li X, et al. Changes in susceptibility of Plasmodium falciparum to artesunate in vitro in Yunnan Province, China. Trans R Soc Trop Med Hyg. 2003;97:226–8.
Skinner TS, Manning LS, Johnston WA, Davis TM. In vitro stage-specific sensitivity of Plasmodium falciparum to quinine and artemisinin drugs. Int J Parasitol. 1996;26:519–25.
Acknowledgements
We acknowledge staff of the PNG Institute of Medical Research for assistance in conducting the field and laboratory studies, and Mangalam Pty Ltd, Bangalore, India for provision of anti-malarial drug samples. We would also like to thank Dr James Balmer from BMG Labtec for helpful discussion. This study was funded by a National Health and Medical Research Council (NHMRC) of Australia grant (#634343). TK was supported by an Esso-Highlands PNG Institute of Medical Research scholarship, SK by a NHMRC Early Career Fellowship (#1052760), ML by an Australian Award PhD Scholarship, BRM by a NHMRC Early Career Fellowship (#1036951), IM by a NHMRC Senior Research Fellowship (#1043345) and TMED by an NHMRC Practitioner Fellowship (#572561).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors contributed to the design of this study, collection of data, analysis of data and/or the interpretation of the results, and to the writing of the manuscript. All authors edited and approved the final version of the manuscript.
Tamarah Koleala and Stephan Karl contributed equally to this work.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Koleala, T., Karl, S., Laman, M. et al. Temporal changes in Plasmodium falciparum anti-malarial drug sensitivity in vitro and resistance-associated genetic mutations in isolates from Papua New Guinea. Malar J 14, 37 (2015). https://doi.org/10.1186/s12936-015-0560-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-015-0560-3