
Abstract
The Kirsten ras oncogene KRAS is a member of the small GTPase superfamily participating in the RAS/MAPK pathway. A single amino acid substitution in KRAS gene has been shown to activate the encoded protein resulting in cell transformation. This oncogene is involved in the malignant transformation in several tissues. Notably, numerous non-coding RNAs have been found to interact with KRAS protein. Such interaction results in a wide array of human disorders, particularly cancers. Orilnc1, KIMAT1, SLCO4A1-AS1, LINC01420, KRAS1P, YWHAE, PART1, MALAT1, PCAT-1, lncRNA-NUTF2P3-001 and TP53TG1 are long non-coding RNAs (lncRNAs) whose interactions with KRAS have been verified in the context of cancer. miR-143, miR-96, miR-134 and miR-126 have also been shown to interact with KRAS in different tissues. Finally, circITGA7, circ_GLG1, circFNTA and circ-MEMO1 are examples of circular RNAs (circRNAs) that interact with KRAS. In this review, we describe the interaction between KRAS and lncRNAs, miRNAs and circRNAs, particularly in the context of cancer.
Similar content being viewed by others


Introduction
The Kirsten ras oncogene KRAS is a homolog from the mammalian ras gene family [1]. The encoded protein by this gene has 88 amino acid residues [2] and is a member of the small GTPase superfamily participating in the RAS/MAPK pathway. In fact, KRAS protein serves as a switching device being turned on and off by the GTP and GDP molecules. Attachment of a GTP molecule to KRAS turns this switch on leading to signal transduction. When KRAS transforms the GTP to GDP, it will become inactivated. GDP binding with KRAS stops transmission of signals to the cell nucleus. RAS/MAPK signaling pathway instructs the cell to go through proliferation stages or to differentiate into mature cells with specialized function [2]. In addition, KRAS has inherent GTPase activity which is induced by GTPase-activating proteins, mediating the direct interaction of KRAS with the effector proteins [3]. Single amino acid substitutions in KRAS gene has been shown to activate the encoded protein [4], resulting in cell transformation as well as resistance to a wide array of chemotherapeutics and targeted therapies against epidermal growth factor receptors (EGFRs) [5].
Mutations in RAS have been detected in approximately 15% of acute myeloid leukemia (AML), more than 10% of adult T cell acute lymphoblastic leukemia and about one third of multiple myeloma cases [6]. In some AML cases, KRAS mutations are assumed to be commencing events in the course of disease. Moreover, these mutations can occur during progression of AML [6, 7]. The presence of KRAS mutations can negatively influence overall survival and complete remission rate of these patients. In fact, KRAS mutations predict poor prognosis of AML [8]. In breast cancer, KRAS is the most commonly mutated RAS protein. Mutations in KRAs are predictor of poor prognosis and higher rate of metastatic events [9]. In colorectal cancer, RAS mutations have been detected in 45% of patients, with KRAS being the most commonly mutated one [10]. The vast majority of KRAS mutations occur at codon 12 while codon 61 harbors very few mutations [11]. A comprehensive assessment of RAS mutations in different types of cancers, including those originated from adrenal gland, autonomic ganglia, biliary tract, bone, breast, central nervous system, cervix, endometrium and hematopoietic/lymphoid system has shown that the majority of cancer types favor mutation of a single isoform, this is usually KRAS [11].
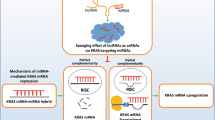
It has been recently evident that KRAS influence expression of a number of non-coding RNAs. Moreover, some non-coding RNAs have been found to participate in the pathogenesis of cancer through interacting with KRAS. In this review, we describe the interaction between KRAS and long non-coding RNAs (lncRNAs), microRNAs (miRNAs) and circular RNAs (circRNAs), particularly in the context of cancer. We summarized some of ncRNAs interacted with KRAS in Fig. 1.

A schematic representation of the interaction between non-coding RNAs and KRAS. The expression of different noncoding RNAs including lncRNAs, miRNAs, and circular RNAs could have an important effect on KRAS expression. Various noncoding RNAs via targeting KRAS could regulate the expression of anti- and pro-apoptotic genes, thus inhibiting or promoting cell apoptosis
Interaction between lncRNAs and KRAS
LncRNA are a group of non-coding RNAs with sizes more than 200 nucleotides. These transcripts can affect expression of genes at different levels. They have diverse types of interactions with mRNAs, DNA molecules, proteins, and miRNAs and accordingly control epigenetic events and transcription of genes. Moreover, they can affect gene expression at post-transcriptional level as well as translational and post-translational phases [12]. LncRNAs interact with DNA via triple-helix formation [13].
Zhang et al. have designed an lncRNA microarray to find RAS-interacting lncRNAs. They have identified the lncRNA Orilnc1 as a downstream target of RAS that mediates oncogenic effects of RAS in cancer cell lines. They have also shown that expression of Orilnc1 is controlled by RAS/RAF/MEK/ERK axis through AP1 transcription factor. Over-expression of this lncRNA has been shown in BRAF-mutant cancer cells, including melanoma cell lines. Orilnc1 silencing has sufficiently prohibited proliferation and growth of cancer cells in vitro and in vivo. Furthermore, Orilnc1 silencing could reduce cyclin E1 levels leading to induction of cell cycle arrest at G1/S phase. Thus, Orilnc1 has been identified as non-protein regulator of RAS/RAF activity and a possible target for treatment of RAS/RAF-associated malignancies [14].
KIMAT1 has been identified as a KRAS-responsive lncRNA whose expression is correlated with expression level of KRAS in lung cancer cell lines as well as clinical samples. KIMAT1 has been found to be originated from Transposable Elements and is known to be induced by MYC. This lncRNA can interact with DHX9 and NPM1 and has a crucial role in enhancing stability of these proteins. Functionally, KIMAT1 is a known target for MYC that induces lung cancer through enhancement of the maturation of oncogenic miRNAs via increasing stability of DHX9 and NPM1. Moreover, this lncRNA can preclude synthesis of tumor suppressor miRNAs through MYC-related suppression of p21. KIMAT1 silencing could suppress expression of KRAS and inactivate KRAS downstream signaling. In fact, KIMAT1 and proteins which interact with this lncRNA regulate KRAS signaling. In vivo studies have confirmed the impact of KIMAT1 silencing in blocking growth of lung cancer. Cumulatively, KIMAT1 has a role in conserving a positive feedback circuit that maintains KRAS signaling in the course of lung carcinogenesis. Moreover, interference with KIMAT1 has been suggested as a strategy to impede KRAS-associated carcinogenesis [15].
LncRNA SLCO4A1-AS1 has been found as an up-regulated lncRNA in colorectal cancer tissues through in silico assessment of two sets of microarrays data of this cancer type. Further analyses have shown correlation between up-regulation of SLCO4A1-AS1 and poor prognosis of patients with colorectal cancer. Mechanistically, SLCO4A1-AS1 promotes proliferation, migration, and invasiveness of these neoplastic cells through regulation of EGFR/MAPK pathway. SLCO4A1-AS1 silencing has significantly reduced expression levels of EGFR, KRAS, BRAF and MAP3K1 through inhibition of phosphorylation [16].
LINC01420 is another KRAS-related lncRNA which is overexpressed in pancreatic cancer tissues and cell lines. LINC01420 silencing has reduced proliferation, epithelial-mesenchymal transition (EMT) and in vivo growth of pancreatic cancer. Notably, KRAS has been identified as the mediator of pro-proliferative effects of LINC01420 in pancreatic cancer. Moreover, expression of KRAS has been shown to be regulated by MYC. LINC01420 could enhance MYC binding with KRAS promoter in the nucleus of pancreatic cancer cells. Interestingly, LINC01420 has also increased MYC levels in the cytoplasm through sequestering miR-494-3p. Cumulatively, LINC01420 facilitates progression of pancreatic cancer via releasing MYC from inhibitory effects of miR-494-3p in cytoplasm and enhancing nuclear levels of MYC-activated KRAS [17].
Table 1 shows the interaction between lncRNAs and KRAS in the context of cancer.
Interaction between miRNAs and KRAS
miRNA are a group of non-coding RNAs that have about 22–24 nucleotides. These transcripts are single-stranded molecules that can inhibit protein synthesis through two different mechanisms. Mature miRNAs are produced via a two-step process through which primary miRNA is cleaved and loaded into the RNA-induced silencing complex. Base-pairing of miRNAs with target mRNAs can negatively regulate expression of target transcripts. Based on the degree of complementarity between miRNA and mRNA, the target mRNA is cleaved and degraded or its translation is inhibited [25].
The interaction between miRNAs and KRAS has been appraised in the context of cancer as well as non-malignant conditions. In the context of cancer, several known tumor suppressor and oncogenic miRNAs have been found to interact with KRAS. For instance, miR-217 has been demonstrated to reduce expression of KRAS in pancreatic cancer cells. This miRNA has been downregulated in the majority of pancreatic ductal adenocarcinoma tissues and in all examined cell lines of this type of cancer compared with the equivalent controls. Up-regulation of miR-217 in these cells could inhibit tumor growth and suppress anchorage-independent colony forming ability of these cells. Up-regulation of miR-217 has also decreased expression levels of KRAS protein and reduced the constitutive phosphorylation of AKT [26]. miR-96 is another tumor suppressor miRNA which directly targets the KRAS in pancreatic cancer cells. Forced over-expression of miR-96 has effectively suppressed KRAS, diminished activity of Akt signaling, and induced cell apoptosis. In vitro and in vivo experiments have verified that the tumor suppressor role of miR-96 depends on its inhibitory effects on KRAS [27]. EVI1 as a universal oncoprotein in pancreatic cancer has been shown to up-regulate KRAS levels via suppression of miR-96 [28]. Consistent with these findings, resveratrol has been shown to prevent colorectal carcinogenesis in an animal model of Kras activated cancer possibly through up-regulation of miR-96 [29]. Another experiment in colorectal cancer has shown a panel of miRNAs that precisely discriminate KRAS-mutated colorectal cancer tissues from other samples [30].
Several studies have shown the functional link between miR-143 levels and KRAS in different settings. This tumor suppressive miRNA has been shown to target KRAS in colorectal [31] and pancreatic cancer cells [32]. Down-regulation of this miRNA has been associated with poor prognosis of patients with colorectal cancer and lower progression free survival of patients receiving EGFR-targeting therapy. Yet, it has not been related with objective response to EGFR-targeting therapies [33]. A novel synthetic miR-143 has been shown to interfere with KRAS signaling network and enhance effectiveness of EGFR inhibitors [34]. Finally, miR-143 has been shown to decrease proliferation and migratory aptitude of prostate cancer cells while enhancing the cytotoxic effects of docetaxel via inhibiting KRAS [35]. Table 2 shows the interaction between miRNAs and KRAS in the context of cancer.
The interaction between miRNAs and RAS pathway has also been appraised in the context of cardiac hypertrophy. Sayed et al. have reported that a group of miRNAs are differentially and temporally altered in the course of cardiac hypertrophy. Notably, the muscle-specific miRNA miR-1 has been shown to be decreased in very early phase of this process, continuing through day 7 following aortic constriction-associated hypertrophy of heart. This miRNA could inhibit expressions of RasGAP, Cdk9, fibronectin, and Rheb [69].
Interaction between circRNAs and KRAS
CircRNAs are a group of non-coding RNAs with an enclosed circular conformation that is shaped by either typical spliceosome-mediated or lariat-type splicing [70]. This circular configuration protects circRNAs from effects of RNases, thus circRNAs have more stability than linear RNAs [71]. Circ_GLG1 is a KRAS-related circRNA which is considerably over-expressed in colorectal tissues compared with nearby normal tissues. Silencing of circ_GLG1 in colorectal adenoma carcinoma cells could inhibit viability of tumor cells. Moreover, circ_GLG1 silencing reduces proliferation, invasiveness, and migratory potential of these cells. These processes could be reversed by transfection of miR-622 antagonist. Circ_GLG1 could promote KRAS expression through serving as a miR-622 sponge. Cumulatively, circ_GLG1/miR-622/KRAS axis has been found to participate in the pathogenesis of colorectal cancer [72].
CircITGA7 is another KRAS-related circRNA whose expression is considerably decreased in CRC tissues and cells in association with cancer progression. Forced over-expression of circITGA7 could suppress growth and metastatic potential of colorectal cancer cells. On the other hand, circITGA7 silencing could promote malignant behavior of these cells both in vitro and in vivo. Functionally, circITGA7 acts as a negative modulator of the Ras signaling pathway through binding with to miR-370-3p to antagonize its inhibitory effects on neurofibromin 1. Moreover, circITGA7 increases expression of ITGA7 via inhibiting RREB1 through the Ras pathway [73].
Another study has shown global down-regulation of circRNAs in DLD-1 and DKO-1 colorectal cancer cells (containing KRAS mutant allele) compared to DKs-8 cells (containing only wild type alleles of KRAS), representing an extensive influence of mutant KRAS on expression profile of circRNAs. Additional experiments in KRAS mutant HCT116 cells and KRAS wild type HKe3 cells have confirmed this observation. Notably, circRNAs have been detected in cancer-derived extracellular-vesicles in higher abundance than cells. This finding implies their potential as tumor biomarkers [74]. Table 3 shows the interaction between circRNAs and K-RAS in the context of cancer.
The interaction between circRNAs and KRAS has also been assessed in hyperglycemic conditions. A circRNA from human umbilical vein endothelial cell exosomes has been shown to affect senescence process in the vascular smooth muscle cells in hyperglycemic niche. CircRNA-0077930 has been found to serve as a sponge for miR-622 to increase expression of KRAS. Exosome-mediated transfer of circRNA-0077930 could induce senescence of smooth muscle cells through the above-mentioned mechanism. Besides, this circRNA could increase LDH activity and reduce superoxide dismutase activity in these cells [77].
Discussion
The data reviewed in the current manuscript show the close interaction between KRAS oncoprotein and several non-coding RNAs, particularly in the context of lung [15], pancreatic [17] and colorectal cancers [16]. In fact, these three types of cancer are the main types of malignancies in which KRAS has been found to be epigenetically modulated by non-coding RNAs. Glioma, retinoblastoma, osteosarcoma, bladder cancer, prostate cancer and esophageal cancer are other types of cancers in which the interaction between KRAS and non-coding RNAs has been verified.
The interaction between KRAS and non-coding RNAs not only affects cell proliferation and apoptosis [16], but also mediates EMT [17] and stemness [55]. LINC01420 [17], miR-134 [55] and miR-193a-3p [53] are examples of KRAS-interacting non-coding RNAs that partake in this process. KRAS-interacting transcripts also affect response of cancer cells to chemotherapeutics such as docetaxel [35] and cisplatin [75]. Most notably, a number of these transcripts have been found to determine prognosis and course of malignancy among affected individuals.
The data summarized in this review shows the combinatorial effect as well as balancing effects of different non-coding RNAs on KRAS regulation in cancers. In fact, KRAS is regulated by multiple non-coding RNAs, and many of the non-coding RNAs are relevant at a time in cancers. No study has revealed any organ or environment specificity in expression of these non-coding RNAs. Instead, most of above-mentioned non-coding RNAs have similar roles in the pathogenesis of several different cancers, indicating their universal effects in regulation of KRAS independent from tissue type.
LncRNAs that regulate expression of KRAS mostly exert this function through serving as sponges for miRNAs. MALAT1/miR-1, PCAT-1/miR-182/miR-217 and lncRNA-NUTF2P3-001/miR-3923 are examples of miRNA/lncRNAs that regulate expression of KRAS. Similarly, circRNAs can serve as molecular sponges for KRAS-associated miRNAs. Circ_GLG1/miR-622, circFNTA/miR-370-3p and circ-MEMO1/miR-101-3p axes have been shown to regulate expression of KRAS in colorectal, bladder and lung cancer cells. Therefore, a complex functional network between different classes of non-coding RNAs is involved in the regulation of KRAS levels in cancers. Identification of other elements of this multifaceted network can provide novel insight about the carcinogenesis and facilitate design of more appropriate targeted therapies.
Besides, it is worth mentioning that non-coding RNAs can act either upstream or downstream of KRAS. For instance, lncRNA Orilnc 1, circRNA FAT1 and HIPK3 are downstream targets of KRAS, but not the regulators of KRAS. Several other non-coding RNAs have been shown to regulate expression of KRAS.
Several mechanisms participate in KRAS regulation by lncRNAs. For instance, lncRNAs act as sponges for miRNAs that target KRAS. Moreover, lncRNAs have functional associations with numerous regulatory apparatuses, including chromatin remodeling elements, transcription factors, splicing apparatus and nuclear trafficking regulators [78]. Through these interactions, they can also regulate expression of KRAS. Modulation of establishment of G4 elements in the promoter region of KRAS is another possible mechanism by which lncRNAs can influence expression of KRAS. For instance, KRASIM, the microprotein coded by the lncRNA NCBP2-AS2 has been found to suppress expression of KRAS and inhibit ERK signaling in hepatocellular carcinoma cells [79].
The impact of KRAS-related non-coding RNAs on cellular activities has also been assessed in the context of hyperglycemia and cardiac hypertrophy. However, data regarding their impact on other non-malignant conditions is scarce.
Conclusion
Future studies are needed to find whether the presence of mutations in KRAS can affect the interaction between non-coding RNAs and this oncoprotein. Moreover, the impact of these non-coding RNAs on resistance to targeted therapies should be more clarified. Finally, the relative contribution of KRAS mutations and dysregulation of KRAS-related non-coding RNAs in the pathogenesis of human cancer should be clarified. This field will benefit from the development of new techniques, such as single cell sequencing and CRISPR-CAS9 gene editing.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Chang EH, Gonda MA, Ellis RW, Scolnick EM, Lowy DR. Human genome contains four genes homologous to transforming genes of Harvey and Kirsten murine sarcoma viruses. Proc Natl Acad Sci. 1982;79(16):4848–52.
McGrath JP, Capon DJ, Smith DH, Chen EY, Seeburg PH, Goeddel DV, et al. Structure and organization of the human Ki-ras proto-oncogene and a related processed pseudogene. Nature. 1983;304(5926):501–6.
Giglione C, Parrini MC, Baouz S, Bernardi A, Parmeggiani A. A new function of p120-GTPase-activating protein. J Biol Chem. 1997;272(40):25128–34.
Der CJ, Cooper GM. Altered gene products are associated with activation of cellular rasK genes in human lung and colon carcinomas. Cell. 1983;32(1):201–8.
Jančík S, Drábek J, Radzioch D, Hajdúch M. Clinical relevance of KRAS in human cancers. J Biomed Biotechnol. 2010. https://doi.org/10.1155/2010/150960.
Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood J Am Soc Hematol. 2012;120(17):3397–406.
Zhao S, Zhang Y, Sha K, Tang Q, Yang X, Yu C, et al. KRAS (G12D) cooperates with AML1/ETO to initiate a mouse model mimicking human acute myeloid leukemia. Cell Physiol Biochem. 2014;33(1):78–87.
Zhou J-D, Yao D-M, Li X-X, Zhang T-J, Zhang W, Ma J-C, et al. KRAS overexpression independent of RAS mutations confers an adverse prognosis in cytogenetically normal acute myeloid leukemia. Oncotarget. 2017;8(39):66087.
Galiè M. RAS as supporting actor in breast cancer. Front Oncol. 2019;9:1199.
Mustachio LM, Chelariu-Raicu A, Szekvolgyi L, Roszik J. Targeting KRAS in cancer: promising therapeutic strategies. Cancers. 2021;13(6):1204.
Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72(10):2457–67.
Zhang X, Wang W, Zhu W, Dong J, Cheng Y, Yin Z, et al. Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. Int J Mol Sci. 2019;20(22):5573.
Kuo CC, Hänzelmann S, Sentürk Cetin N, Frank S, Zajzon B, Derks JP, et al. Detection of RNA-DNA binding sites in long noncoding RNAs. Nucleic Acids Res. 2019;47(6):e32 (Epub 2019/01/31).
Zhang D, Zhang G, Hu X, Wu L, Feng Y, He S, et al. Oncogenic RAS regulates long noncoding RNA Orilnc1 in human cancer. Cancer Res. 2017;77(14):3745–57 (Epub 05/04).
Shi L, Magee P, Fassan M, Sahoo S, Leong HS, Lee D, et al. A KRAS-responsive long non-coding RNA controls microRNA processing. Nat Commun. 2021;12(1):2038.
Tang R, Chen J, Tang M, Liao Z, Zhou L, Jiang J, et al. LncRNA SLCO4A1-AS1 predicts poor prognosis and promotes proliferation and metastasis via the EGFR/MAPK pathway in colorectal cancer. Int J Biol Sci. 2019;15(13):2885–96.
Zhai H, Zhang X, Sun X, Zhang D, Ma S. Long non-coding RNA LINC01420 contributes to pancreatic cancer progression through targeting KRAS proto-oncogene. Dig Dis Sci. 2020;65(4):1042–52.
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465(7301):1033–8.
Bjeije H, Soltani BM, Behmanesh M, Zali MR. YWHAE long non-coding RNA competes with miR-323a-3p and miR-532-5p through activating K-Ras/Erk1/2 and PI3K/Akt signaling pathways in HCT116 cells. Hum Mol Genet. 2019;28(19):3219–31.
Chen SC, Diao YZ, Zhao ZH, Li XL. Inhibition of lncRNA PART1 chemosensitizes wild type but Not KRAS mutant NSCLC cells. Cancer Manag Res. 2020;12:4453–60 (Epub 2020/07/02).
Chang J, Xu W, Du X, Hou J. MALAT1 silencing suppresses prostate cancer progression by upregulating miR-1 and downregulating KRAS. Onco Targets Ther. 2018;11:3461–73.
Domvri K, Petanidis S, Anestakis D, Porpodis K, Bai C, Zarogoulidis P, et al. Exosomal lncRNA PCAT-1 promotes Kras-associated chemoresistance via immunosuppressive miR-182/miR-217 signaling and p27/CDK6 regulation. Oncotarget. 2020;11(29):2847.
Li X, Deng S-J, Zhu S, Jin Y, Cui S-P, Chen J-Y, et al. Hypoxia-induced lncRNA-NUTF2P3-001 contributes to tumorigenesis of pancreatic cancer by derepressing the miR-3923/KRAS pathway. Oncotarget. 2016;7(5):6000–14.
Zhang Y, Yang H, Du Y, Liu P, Zhang J, Li Y, et al. Long noncoding RNA TP53TG1 promotes pancreatic ductal adenocarcinoma development by acting as a molecular sponge of microRNA-96. Cancer Sci. 2019;110(9):2760–72.
Macfarlane L-A, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genom. 2010;11(7):537–61.
Zhao W-G, Yu S-N, Lu Z-H, Ma Y-H, Gu Y-M, Chen J. The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting KRAS. Carcinogenesis. 2010;31(10):1726–33.
Yu S, Lu Z, Liu C, Meng Y, Ma Y, Zhao W, et al. miRNA-96 suppresses KRAS and functions as a tumor suppressor gene in pancreatic cancer. Cancer Res. 2010;70(14):6015–25.
Tanaka M, Suzuki HI, Shibahara J, Kunita A, Isagawa T, Yoshimi A, et al. EVI1 oncogene promotes KRAS pathway through suppression of microRNA-96 in pancreatic carcinogenesis. Oncogene. 2014;33(19):2454–63.
Saud SM, Li W, Morris NL, Matter MS, Colburn NH, Kim YS, et al. Resveratrol prevents tumorigenesis in mouse model of Kras activated sporadic colorectal cancer by suppressing oncogenic Kras expression. Carcinogenesis. 2014;35(12):2778–86 (Epub 10/03).
Milanesi E, Dobre M, Bucuroiu AI, Herlea V, Manuc TE, Salvi A, et al. miRNAs-based molecular signature for KRAS mutated and wild type colorectal cancer: an explorative study. J Immunol Res. 2020;2020:4927120.
Chen X, Guo X, Zhang H, Xiang Y, Chen J, Yin Y, et al. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene. 2009;28(10):1385–92.
Xie F, Li C, Zhang X, Peng W, Wen T. MiR-143-3p suppresses tumorigenesis in pancreatic ductal adenocarcinoma by targeting KRAS. Biomed Pharmacother. 2019;119: 109424.
Pichler M, Winter E, Stotz M, Eberhard K, Samonigg H, Lax S, et al. Down-regulation of KRAS-interacting miRNA-143 predicts poor prognosis but not response to EGFR-targeted agents in colorectal cancer. Br J Cancer. 2012;106(11):1826–32 (Epub 05/01).
Akao Y, Kumazaki M, Shinohara H, Sugito N, Kuranaga Y, Tsujino T, et al. Impairment of K-Ras signaling networks and increased efficacy of epidermal growth factor receptor inhibitors by a novel synthetic miR-143. Cancer Sci. 2018;109(5):1455–67 (Epub 04/14).
Xu B, Niu X, Zhang X, Tao J, Wu D, Wang Z, et al. miR-143 decreases prostate cancer cells proliferation and migration and enhances their sensitivity to docetaxel through suppression of KRAS. Mol Cell Biochem. 2011;350(1):207–13.
Kang M, Li Y, Zhu S, Zhang S, Guo S, Li P. MicroRNA-193b acts as a tumor suppressor gene in human esophageal squamous cell carcinoma via target regulation of KRAS. Oncol Lett. 2019;17(4):3965–73.
Mokhlis HA, Bayraktar R, Kabil NN, Caner A, Kahraman N, Rodriguez-Aguayo C, et al. The modulatory role of microRNA-873 in the progression of KRAS-driven cancers. Mole Ther Nucleic Acids. 2019;14:301–17 (Epub 2019/01/18).
Lundberg IV, Wikberg ML, Ljuslinder I, Li X, Myte R, Zingmark C, et al. MicroRNA expression in KRAS- and BRAF-mutated colorectal cancers. Anticancer Res. 2018;38(2):677–83.
Shi L, Middleton J, Jeon Y-J, Magee P, Veneziano D, Laganà A, et al. KRAS induces lung tumorigenesis through microRNAs modulation. Cell Death Dis. 2018;9(2):219.
Shen H, Xing C, Cui K, Li Y, Zhang J, Du R, et al. MicroRNA-30a attenuates mutant KRAS-driven colorectal tumorigenesis via direct suppression of ME1. Cell Death Differ. 2017;24(7):1253–62.
Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68(20):8535–40.
Mosakhani N, Sarhadi VK, Borze I, Karjalainen-Lindsberg M-L, Sundström J, Ristamäki R, et al. MicroRNA profiling differentiates colorectal cancer according to KRAS status. Genes Chromosom Cancer. 2012;51(1):1–9.
Tsang WP, Kwok TT. The miR-18a* microRNA functions as a potential tumor suppressor by targeting on K-Ras. Carcinogenesis. 2009;30(6):953–9.
Pugh S, Thiébaut R, Bridgewater J, Grisoni M-L, Moutasim K, Rousseau F, et al. Association between miR-31-3p expression and cetuximab efficacy in patients with KRAS wild-type metastatic colorectal cancer: a post-hoc analysis of the new EPOC trial. Oncotarget. 2017;8(55):93856–66.
Kent OA, Mendell JT, Rottapel R. Transcriptional regulation of miR-31 by oncogenic KRAS mediates metastatic phenotypes by repressing RASA1. Mol Cancer Res. 2016;14(3):267–77 (Epub 01/08).
Forzati F, De Martino M, Esposito F, Sepe R, Pellecchia S, Malapelle U, et al. miR-155 is positively regulated by CBX7 in mouse embryonic fibroblasts and colon carcinomas, and targets the KRAS oncogene. BMC Cancer. 2017;17(1):170.
Fan Q, Hu X, Zhang H, Wang S, Zhang H, You C, et al. MiR-193a-3p is an important tumour suppressor in lung cancer and directly targets KRAS. Cell Physiol Biochem. 2017;44(4):1311–24.
Tsunoda T, Takashima Y, Yoshida Y, Doi K, Tanaka Y, Fujimoto T, et al. Oncogenic KRAS regulates miR-200c and miR-221/222 in a 3D-specific manner in colorectal cancer cells. Anticancer Res. 2011;31(7):2453–9.
Inoue A, Mizushima T, Wu X, Okuzaki D, Kambara N, Ishikawa S, et al. A miR-29b byproduct sequence exhibits potent tumor-suppressive activities via inhibition of NF-κB signaling in KRAS-mutant colon cancer cells. Mol Cancer Ther. 2018;17(5):977–87.
Hara T, Jones MF, Subramanian M, Li XL, Ou O, Zhu Y, et al. Selective targeting of KRAS-mutant cells by miR-126 through repression of multiple genes essential for the survival of KRAS-mutant cells. Oncotarget. 2014;5(17):7635–50.
Fiala O, Pitule P, Hosek P, Liska V, Sorejs O, Bruha J, et al. The association of miR-126-3p, miR-126-5p and miR-664-3p expression profiles with outcomes of patients with metastatic colorectal cancer treated with bevacizumab. Tumor Biol. 2017;39(7):1010428317709283.
Ebrahimi F, Gopalan V, Wahab R, Lu C-T, Anthony Smith R, Lam AK-Y. Deregulation of miR-126 expression in colorectal cancer pathogenesis and its clinical significance. Exp Cell Res. 2015;339(2):333–41.
Mamoori A, Wahab R, Islam F, Lee K, Vider J, Lu C-T, et al. Clinical and biological significance of miR-193a-3p targeted KRAS in colorectal cancer pathogenesis. Hum Pathol. 2018;71:145–56.
Ota T, Doi K, Fujimoto T, Tanaka Y, Ogawa M, Matsuzaki H, et al. KRAS up-regulates the expression of miR-181a, miR-200c and miR-210 in a three-dimensional-specific manner in DLD-1 colorectal cancer cells. Anticancer Res. 2012;32(6):2271–5.
Zhang Y, Kim J, Mueller AC, Dey B, Yang Y, Lee DH, et al. Multiple receptor tyrosine kinases converge on microRNA-134 to control KRAS, STAT5B, and glioblastoma. Cell Death Differ. 2014;21(5):720–34.
Liu Y, Zhang M, Qian J, Bao M, Meng X, Zhang S, et al. miR-134 functions as a tumor suppressor in cell proliferation and epithelial-to-mesenchymal transition by targeting KRAS in renal cell carcinoma cells. DNA Cell Biol. 2015;34(6):429–36 (Epub 03/26).
Zhao Y, Pang D, Wang C, Zhong S, Wang S. MicroRNA-134 modulates glioma cell U251 proliferation and invasion by targeting KRAS and suppressing the ERK pathway. Tumor Biol. 2016;37(8):11485–93.
Guo L, Bai Y, Ji S, Ma H. MicroRNA-98 suppresses cell growth and invasion of retinoblastoma via targeting the IGF1R/k-Ras/Raf/MEK/ERK signaling pathway. Int J Oncol. 2019;54(3):807–20.
Ruzzo A, Graziano F, Vincenzi B, Canestrari E, Perrone G, Galluccio N, et al. High let-7a microRNA levels in KRAS-mutated colorectal carcinomas may rescue anti-EGFR therapy effects in patients with chemotherapy-refractory metastatic disease. Oncologist. 2012;17(6):823–9 (Epub 05/14).
Jin X, Sun Y, Yang H, Li J, Yu S, Chang X, et al. Deregulation of the MiR-193b-KRAS axis contributes to impaired cell growth in pancreatic cancer. PLoS ONE. 2015;10(4): e0125515.
Keklikoglou I, Hosaka K, Bender C, Bott A, Koerner C, Mitra D, et al. MicroRNA-206 functions as a pleiotropic modulator of cell proliferation, invasion and lymphangiogenesis in pancreatic adenocarcinoma by targeting ANXA2 and KRAS genes. Oncogene. 2015;34(37):4867–78.
Hatley ME, Patrick DM, Garcia MR, Richardson JA, Bassel-Duby R, van Rooij E, et al. Modulation of K-Ras-dependent lung tumorigenesis by microRNA-21. Cancer Cell. 2010;18(3):282–93.
Yuan P, He X-H, Rong Y-F, Cao J, Li Y, Hu Y-P, et al. KRAS/NF-κB/YY1/miR-489 signaling axis controls pancreatic cancer metastasis. Cancer Res. 2017;77(1):100–11.
Wang P, Zhu C-F, Ma M-Z, Chen G, Song M, Zeng Z-L, et al. Micro-RNA-155 is induced by K-Ras oncogenic signal and promotes ROS stress in pancreatic cancer. Oncotarget. 2015;6(25):21148–58.
Liu X, Wang Y, Zhao J. MicroRNA-337 inhibits colorectal cancer progression by directly targeting KRAS and suppressing the AKT and ERK pathways. Oncol Rep. 2017;38(5):3187–96.
Zhang X, Guo Q, Chen J, Chen Z. Quercetin enhances cisplatin sensitivity of human osteosarcoma cells by modulating microRNA-217-KRAS axis. Mol Cells. 2015;38(7):638–42 (Epub 06/10).
Seviour EG, Sehgal V, Mishra D, Rupaimoole R, Rodriguez-Aguayo C, Lopez-Berestein G, et al. Targeting KRas-dependent tumour growth, circulating tumour cells and metastasis in vivo by clinically significant miR-193a-3p. Oncogene. 2017;36(10):1339–50 (Epub 09/26).
Subramani A, Alsidawi S, Jagannathan S, Sumita K, Sasaki AT, Aronow B, et al. The brain microenvironment negatively regulates miRNA-768-3p to promote K-ras expression and lung cancer metastasis. Sci Rep. 2013;3(1):2392.
Sayed D, Hong C, Chen I-Y, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100(3):416–24.
Zhao X, Cai Y, Xu J. Circular RNAs: biogenesis, mechanism, and function in human cancers. Int J Mol Sci. 2019;20(16):3926.
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141–57.
Hao S, Qu R, Hu C, Wang M, Li Y. A circular RNA derived from golgi glycoprotein 1 mRNA regulates KRAS expression and promotes colorectal cancer progression by targeting microRNA-622. Onco Targets Ther. 2020;13:12637–48 (Epub 2020/12/19).
Li X, Wang J, Zhang C, Lin C, Zhang J, Zhang W, et al. Circular RNA circITGA7 inhibits colorectal cancer growth and metastasis by modulating the Ras pathway and upregulating transcription of its host gene ITGA7. J Pathol. 2018;246(2):166–79.
Dou Y, Cha DJ, Franklin JL, Higginbotham JN, Jeppesen DK, Weaver AM, et al. Circular RNAs are down-regulated in KRAS mutant colon cancer cells and can be transferred to exosomes. Sci Rep. 2016;6(1):37982.
Chen J, Sun Y, Ou Z, Yeh S, Huang C-P, You B, et al. Androgen receptor-regulated circFNTA activates KRAS signaling to promote bladder cancer invasion. EMBO Rep. 2020;21(4): e48467.
Ding C, Xi G, Wang G, Cui D, Zhang B, Wang H, et al. Exosomal circ-MEMO1 promotes the progression and aerobic glycolysis of non-small cell lung cancer through targeting MiR-101–3p/KRAS axis. Front Genet. 2020;11:962.
Wang S, Zhan J, Lin X, Wang Y, Wang Y, Liu Y. CircRNA-0077930 from hyperglycaemia-stimulated vascular endothelial cell exosomes regulates senescence in vascular smooth muscle cells. Cell Biochem Funct. 2020;38(8):1056–68.
Saliani M, Mirzaiebadizi A, Javadmanesh A, Siavoshi A, Ahmadian MR. KRAS-related long noncoding RNAs in human cancers. Cancer Gene Ther. 2021. https://doi.org/10.1038/s41417-021-00381-x.
Xu W, Deng B, Lin P, Liu C, Li B, Huang Q, et al. Ribosome profiling analysis identified a KRAS-interacting microprotein that represses oncogenic signaling in hepatocellular carcinoma cells. Sci China Life Sci. 2020;63(4):529–42.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
SGF wrote the manuscript and revised it. MT designed and supervised the study. ZSF, RJK and BMH collected the data and designed the tables and figures. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent forms were obtained from all study participants. Informed consent forms were obtained from all study participants. The study protocol was approved by the ethical committee of Shahid Beheshti University of Medical Sciences. All methods were performed in accordance with the relevant guidelines and regulations.
Consent of publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ghafouri-Fard, S., Shirvani-Farsani, Z., Hussen, B.M. et al. Emerging role of non-coding RNAs in the regulation of KRAS. Cancer Cell Int 22, 68 (2022). https://doi.org/10.1186/s12935-022-02486-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-022-02486-1