
Abstract
Background
The exosomal nucleic acid (exoNA) from the plasma and pleural fluid can potentially provide means to identify genomic changes in non-small cell lung cancer (NSCLC) patients who develop resistance to targeted epidermal growth factor receptor (EGFR) inhibitor therapy.
Methods
We compared the performance of the following tools to detect EGFR mutations in 54 plasma samples and 13 pleural fluid using cfDNA, combined TNA (exoTNA + cfTNA), or total cellular DNA: droplet digital PCR (ddPCR), the Cobas® EGFR Mutation Test v2 (Cobas) and NGS with Oncomine Pan-Cancer Cell-Free Assay.
Results
All three of these platforms demonstrated 100% specificity in the detection of EGFR mutations in the plasma. In the detection of an activating mutation (exon 19 deletion and L858R), Cobas using cfDNA, ddPCR using combined TNA, and NGS using combined TNA showed a sensitivity of 93, 95.3, and 93.8%, respectively. For T790M mutation detection, the Cobas, ddPCR, and NGS showed a sensitivity of 64.7, 88.2, and 93.3%, respectively. Pleural fluid analysis revealed enrichment of the T790M mutant copies in the exosomes. ddPCR using exoTNA showed higher sensitivity than did total cellular DNA from the pleural fluid.
Conclusion
These results demonstrated that combined TNA in the plasma and exoTNA in the pleural fluid can be used to evaluate low-abundant EGFR mutant copies in NSCLC.
Similar content being viewed by others
Background
The discovery of driver mutations in the epidermal growth factor receptor (EGFR) gene has led to a dramatic paradigm shift in the therapeutic strategies for advanced non-small-cell lung cancer (NSCLC). First- and second-generation tyrosine kinase inhibitors (TKIs), such as gefitinib, erlotinib, and afatinib, are standard therapies for patients with advanced NSCLC harboring mutations in EGFR. These mutations include deletion mutations in exon 19 and the L858R mutation in exon 21 [1, 2]. Unfortunately, acquired resistance to TKIs frequently develops within 1 or 2 years of initiating therapy. The most common mechanism of resistance, which accounts for ~ 60% of cases, is the acquired mutation T790M. MET amplification (5–7%) and ERBB2 amplification (5–13%) occur less frequently than do the T790M mutation [3,4,5].
Most commonly, a cancer’s molecular profile is assessed using the tumor tissue. Longitudinal surveillance of clonal evolution, including acquired resistance alterations following TKI therapy, is essential for precision medicine. This frequent surveillance cannot be effectively achieved using tissue biopsy specimens, however, due to the possibility of inappropriate or inadequate sampling that misses portions of the tumor that are developing treatment resistance or have acquired new driver mutations due to tumor heterogeneity [6]. Therefore, liquid biopsy samples are considered an alternative means of detecting clinically relevant mutations in NSCLC patients undergoing TKI therapy [7].
Circulating tumor DNA (ctDNA) is the most commonly analyzed DNA to detect EGFR mutations in NSCLC [8]. Many technical platforms targeting ctDNA are already being implemented in clinical practice. However, the low abundance of ctDNA is an obstacle for detecting EGFR mutations in NSCLC patients with acquired TKI-resistance. In particular, patients with a low T790M copy number (< 10 copies/mL) have similar responses to osimertinib than do those with a higher T790M copy number (≥ 10 copies/mL) [9]. In one study, there were more patients with low T790M copy numbers than there were with higher T790M copy numbers [10]. Therefore, the pre-analytical steps for maximizing the tumor-derived nucleic acid concentration and ultra-sensitive analytical techniques were essentially required to achieve the optimal sensitivity of the EGFR mutation.
Exosomal nucleic acid (exoNA) has been studied as a target for cancer mutation testing. It has been shown to improve the sensitivity of detection in patients with limited T790M mutant copies of cell-free DNA (cfDNA), such as those with early-stage NSCLC or intrathoracic disease (M0/M1a) [7, 8, 11, 12]. Furthermore, body fluids such as bronchoalveolar lavage (BAL) and pleural fluids also have been tested to assess the feasibility of detecting EGFR mutations. These fluids have also been evaluated to determine which component (exosome, supernatant, and cell pellet) contained abundant tumor-derived nucleic acids in NSCLC patients [13, 14].
There is a constant need to improve the detection capability of clinically significant, but rare mutant alleles in clinical samples. Therefore, we compared the performance of different techniques (allele-specific real-time PCR, droplet digital PCR, and next-generation sequencing) using various sources of tumor-derived nucleic acids (cfDNA and exosome from plasma, exosome and total cellular DNA from pleural fluid). We demonstrated the optimal analytical method to improve the sensitivity of detecting EGFR mutations in different types of clinical samples.
Materials and methods
Patients and clinical specimens
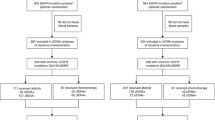
Patients were included in the two following criteria were all met: (1) they had a diagnosis of NSCLC and confirmed EGFR mutation on tissue genotyping, and (2) showed disease progression on first- or second-generation EGFR-TKI. Between November 2017 and September 2019, 64 consecutive NSCLC patients were prospectively enrolled. The EGFR genotyping result of tissue (n = 61) was obtained from specimens at initial diagnosis (n = 58) or from specimens matched with plasma (n = 3). Patients consented to the protocol approved by the Institutional Review Board of Gangnam Severance Hospital and Kangnam Sacred Heart Hospital. Fifty-four whole blood samples were obtained from 54 patients, and 13 pleural fluids was obtained from 13 patients. Three pleural fluid and blood samples were collected at the same time in three patients (P23, P25, and P54).
Preparation of nucleic acid from plasma and cDNA synthesis
The blood samples were collected in cell-free DNA Blood Collection Tubes (Roche, Pleasanton, CA, USA). Plasma isolation was performed using a two-step plasma separation procedure. The blood samples were centrifuged at 1200 g for 10 min, followed by high-speed centrifugation at 16,000 g for 10 min. The plasma aliquots were stored at − 80 °C [15].
Exosomes were isolated from the plasma using ExoQuick™ (System Biosciences, Mountain View, CA, USA). We extracted the plasma cfDNA and exosomal total nucleic acid (exoTNA) using the MagMAX™ Cell-Free DNA Isolation Kit (Thermo Fisher Scientific, Waltham, MA, USA) and MagMAX™ Total Nucleic Acid Isolation Kit (Thermo Fisher Scientific), respectively [11]. The concentrations of cfDNA and exoTNA were assessed with the Qubit™ 3.0 Fluorometer using the Qubit™ dsDNA HS Assay Kit (Thermo Fisher Scientific). cDNA synthesis was performed using a SuperScript™ VILO™ cDNA Synthesis Kit (Invitrogen, Carlsbad, CA, USA).
Preparation of nucleic acid from exosomes, and cell pellets and supernatants from pleural fluid
The pleural fluid was centrifuged at 3000 g for 15 min to remove cellular debris. The exosomes were isolated from the supernatants using ExoQuick™. A nucleic acid of exosomes was extracted with the MagMAX™ Total Nucleic Acid Isolation Kit. cDNA synthesis was performed using a SuperScript™ VILO™ cDNA Synthesis Kit. We extracted the total DNA from the cell pellet using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany). The sizes of the DNA fragments in the exosomes were assessed using a 2200 TapeStation Instrument (Agilent Technologies, Santa Clara, CA, USA) with the Genomic DNA ScreenTape System. The DNA concentration was assessed with the Qubit 3.0 Fluorometer (Thermo Fisher Scientific). The RNA yield and size distribution were analyzed using an Agilent 2100 Bioanalyzer with an RNA 6000 Pico kit (Agilent Technologies, Foster City, CA, USA).
Cobas EGFR Mutation Test v2
For the Cobas EGFR assay, 75 μL of DNA from each plasma sample (2 mL) was loaded into three reaction wells (25 μL DNA per well). Amplification and detection were performed using the Cobas z 480 analyzer (Roche Molecular Systems, Inc., Branchburg, NJ, USA). The data were interpreted using the Cobas z 480 software if the positive and negative controls showed valid results.
Droplet digital PCR (ddPCR)
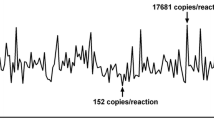
The ddPCR assays were performed using the PrimePCR™ ddPCR™ Mutation Detection Assay kit, and the PrimePCR™ ddPCR™ EGFR Exon 19 Deletions Screening Kit (Bio-Rad Laboratories, Hercules, CA, USA) [11]. Tumor-derived nucleic acids (cfDNA and exoTNA) were extracted from 1 to 2 mL of plasma or pleural fluid. The ddPCR assay for detecting EGFR mutations was validated using the Multiplex I cfDNA Reference Standards (Horizon Discovery, Cambridge, UK) and healthy control samples from a previous study [11]. Briefly, the amplifications were performed in a reaction volume of 20 μL on a QX100 Droplet Digital PCR System (Bio-Rad). The 20 μL of the PCR mixture was composed of 10 μL Bio-Rad Super mix TaqMan, 1–2 μL of each amplification primer/probe mix, and 8–9 μL of nucleic acids (NAs). Thermal cycling comprised an initial denaturing and polymerase hot-start activating step of 10 min at 95 °C, followed by 40 cycles of 95 °C for 30 s and 55 °C for 60 s. The results were analyzed with QuantaSoft v.1.7.2 software (Bio-Rad) and reported as copies per milliliter of plasma. The ddPCR assay was validated for detecting EGFR mutations and determining the limit of detection (LOD) in a previous study [11]. The assays were considered “positive” if the measured event rate was ≥ 2 events/assay and “negative” if the event rate within a gated region was < 2 events/assay.
Next-generation sequencing (NGS)
For NGS, a library was prepared using the Oncomine Pan-Cancer Cell-Free Assay (Thermo Fisher Scientific) targeting 52 cancer-associated genes (Additional file 1: Table S1). The NGS panel is designed to detect single nucleotide variations, small indels, copy-number alterations, and gene fusions. The libraries were prepared using > 5 ng nucleic acid input following the Oncomine Pan-Cancer Cell-Free Assay user guide. Templating and sequencing were performed using the Ion 540™ Kit on the Ion Chef™ and on the Ion S5 XL system (Thermo Fisher Scientific). Alignment to the hg19 human reference genome and variant calling were performed using the Torrent Suite™ and Ion Reporter™ software version 5.10, respectively. The Torrent Suite™ Software provided molecular coverage depth and read coverage depth at the target base; therefore, this software was able to increase the detection sensitivity for low-frequency variants [16, 17].
The average of median read coverage and median molecular coverage were 36,095 × and 1934 ×, respectively. Variants with an allele frequency of > 0.1% were reported. The measured allele frequency (%) was calculated as the mutant coverage depth divided by the total coverage depth.
Statistical analysis
The statistical analysis was performed using R (version 3.5.2, http://www.r-project.org) and MedCalc software (https://www.medcalc.org/). The nonparametric data were analyzed using the Kruskal–Wallis tests. Multiple comparisons were made using Dunn's test. A p-value < 0.05 was considered statistically significant.
Results
Patient and tumor characteristics
The baseline clinical and pathological characteristics of the 64 patients are described in Table 1. More women in our patient cohort than men (73.4% vs. 26.6%) might be reflected by the differential distribution of EGFR mutation between male and female patients with NSCLC, as reported by previous studies [18, 19]. Most patients had stage IV disease. Only two patients had M0 stage disease. Two patients had T790M mutations in the tissue from a repeat biopsy sample. All of the patients received gefitinib, erlotinib, and/or afatinib as first line treatment of their advanced or metastatic disease.
At the time of progression, patients were tested for both the persistence of initial activating mutations (exon 19 deletion and L858R) and the onset of T790M resistance mutations using the Cobas assay in the plasma cfDNA. This testing was performed to determine the patients’ eligibility for osimertinib use. The Cobas assay showed the persistence of initial activating mutations in 41 patients. Among them, T790M resistance mutations were identified in 11 patients (11/41, 26.8%). Fourteen patients had negative results for both activating and resistance mutations.
Quantification of exoTNA and cfDNA in clinical samples
The cfDNA used in the Cobas® EGFR Mutation Test were extracted from plasma samples (2 mL). The exoTNA and cfDNA for the ddPCR and NGS were extracted from equal volumes of plasma samples from 54 patients. Among them, three samples were not available for NA quantification due to a lack of volume. The cfDNA amount for the Cobas® EGFR Mutation Test and both exoNA and cfDNA amount for the ddPCR assay are depicted in (Additional file 1: Fig. S1). The median amount level of cfDNA for the Cobas® EGFR Mutation Test was 16.8 ng. Those of cfDNA and exoNA for ddPCR were 10.3 ng and 11.41 ng, respectively. There was a significant difference between the NA yield of three groups (Kruskal–Wallis test, p = 0.0002). There were also significant differences in the NA yield between the cfDNA for Cobas® EGFR Mutation Test and ExoNA, and the cfDNA for the ddPCR assay (p < 0.05, Dunn's multiple comparisons test).
Comparison among Cobas, ddPCR and NGS using plasma from NSCLC patients who progressed under treatment with an EGFR-TKI
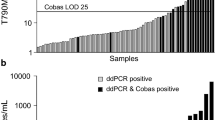
We compared the diagnostic performance of Cobas (cfDNA), ddPCR (cfDNA + exoTNA), and NGS (cfDNA + exoTNA) to detect an EGFR activating mutation (exon 19 deletion and L858R, Table 2) and the resistance T790M mutation (Table 3). NGS was possible in 38 of 54 cases with sufficient plasma samples. There were two patients who were confirmed to have T790M mutations on repeat tissue biopsy (P19 and P40, Additional file 1: Table S2). The plasma analysis of one patient (P19) revealed positive results from all three platforms (Cobas, ddPCR, and NGS). In contrast, plasma analysis in another patient (P40) showed negative results in all three platforms. Although most patients did not undergo tissue re-biopsy, all cases with T790M positivity detected from one or more liquid biopsy platforms (Cobas, ddPCR, or NGS) had disease progression after > 10 months of TKI treatment. Therefore, T790M positivity was defined as “true positive,” even if it was detected in only one liquid biopsy platform. An activating mutation was considered a “true positive” if there was positive detection from two or more liquid biopsy platforms, or positive detection from tissue genotyping and one or more liquid biopsy platforms. The sensitivities of Cobas, ddPCR, and NGS for detection activating mutations in the plasma were 93% (95% confidence interval, 91.7–94.3), 95.3% (94–96.6), and 93.8 (92.2–95.4), respectively (Table 2). Among three samples (P29, P40, P54) with false-negative activating mutations according to the Cobas assay, exon 19 deletion mutations were detected in two samples. These included one mutation from ddPCR (P54, 2 positive droplets/829 total droplets) and another from NGS (P29, variant allele frequency of 0.4%). The NGS assay failed to detect exon 19 deletion mutations in two samples (P20, P40). Only one exon 19 deletion of P20 was detected using both Cobas and ddPCR (6 mutant droplets) techniques.
The sensitivities of Cobas, ddPCR, and NGS to detect T790M mutations in plasma were 64.7% (95% confidence interval, 63.4–66), 88.2% (86.9–89.5), and 93.3 (91.7–94.9), respectively (Table 3). Among 17 patients with T790M mutations that were confirmed by tissue genotyping and on one additional liquid biopsy platform, the Cobas assay did not detect the T790M mutation in six patients, although it identified coexisting activating mutations in five patients. These false negative T790M cases from the Cobas assay consisted of cases with very few copies of T790M in the plasma according to the results of the ddPCR (3–6 mutant droplets) and NGS assays (0.4–1.2% variant allele frequency). However, one case (P40) in which T790M was only identified on tissue re-biopsy was excluded (Additional file 1: Table S2). In addition, the detection rates of T790M with ddPCR (cfDNA + exoTNA) or NGS (cfDNA + exoTNA) were as much as 20–30% higher than were those using Cobas (cfDNA) (Table 3).
The EGFR T790M mutation is the most frequent mutation associated with the development of acquired resistance to EGFR-TKIs. The T790M was detected in the plasma of 17 patients (31.5%) who progressed despite treatment with an EGFR-TKI (n = 54). The T790M positivity rate was 27.8% using ddPCR (15/54), but only 20.4% using Cobas (11/54). The genetic alterations related to EGFR-TKI tolerance were further explored using NGS assay. Seven patients (7/38, 18.4%) were found to have resistance mutations other than EGFR T790M. KRAS mutations (G12D and G13D) were identified in two patients (P28 and P39), and PIK3CA mutations (E545K) were found in three patients (P30, P33, and P37). Gene amplifications that are associated with TKI resistance were identified in two patients (EGFR amplification in P8 and EGFR/CDK4 amplifications in P52).
Furthermore, six patients (6/38, 15.8%) harbored various copies of T790M mutations (2–2098 positive droplets in ddPCR) along with other genetic mutations that are known to be associated with TKI resistance (including CDK6/KRAS/CCND1 amplifications in P19, PIK3CA in P20 and P43, EGFR/CDK6 amplifications in P22, EGFR amplification in P23, and BRAF in P50).
Assessment of T790M mutant alleles according to different sources of tumor-derived nucleic acids from pleural fluid
Three pleural fluid samples were used to analyze different sources of tumor-derived nucleic acids (exosomes, cell pellets, and supernatants) and determine which compartment was suitable for the analysis. The event numbers of T790M positive and wild type cases are depicted in (Additional file 1: Fig. S2). Analysis of three samples revealed the largest number of mutant copies in exoTNA, followed by exoDNA and total DNA from the supernatants. In one sample (Sample 2), the total DNA from the cell pellet demonstrated the largest number of mutant copies compared to that in the exoTNA, exoDNA, and total DNA from the supernatants. Therefore, we selected the exoTNA, total cellular DNA and tested the pleural fluid samples from 13 NSCLC patients with disease progression after EGFR-TKI treatment (Additional file 1: Table S3). The ddPCR, using the total DNA extracted from the cell pellet, revealed T790M mutations in 38.5% of tested pleural fluid samples (5/13) (Table 4). Using the exoTNA extracted from the supernatant, two more cases (P56 and P61) also demonstrated T790M positivity (53.8%, 7/13). Two samples showed very low mutant copies in ddPCR (including four positive droplets among 4595 total droplets, and two positive droplets among 3900 total droplets). Therefore, the ddPCR using the total cellular DNA showed 71.4% sensitivity compared with the exoTNA from the supernatant.
The distribution of isolated nucleic acids is shown in (Additional file 1: Fig. S3). The high molecular weight DNA (10–15 kb long) was more abundant in exosomes from pleural fluids than it was from other samples.
Discussion
In this study, we assessed the performance of three liquid biopsy platforms (including the Cobas assay, ddPCR, and NGS) on different sources of tumor-derived nucleic acids (cfDNA, exoTNA, and total DNA) and clinical specimens (plasma and pleural fluid). The plasma analysis indicated that a combination of cfDNA and exoTNA showed higher sensitivity than that did exoTNA or cfDNA alone. In the pleural fluid analysis, exoTNA from supernatants showed enrichment of the T790M mutant copies by other sources of nucleic acids from supernatants or cell pellets.
The advantage of ddPCR is that it can sensitively detect low-abundant target molecules. Therefore, ddPCR is often used for ctDNA testing [20]. Currently, most liquid biopsy methods used in clinical research and practice only used cfDNA to detect cancer mutations. Even with ddPCR, which is currently considered the most sensitive technique, the sensitivity for detecting EGFR T790M mutations is only 70–77% of that when using tissue testing if cfDNA is used alone [20, 21]. Therefore, it is crucial to increase the sensitivity of the liquid biopsy technique to detect resistant mutations and identify patients who could potentially benefit from osimertinib therapy. In this study, we found that the sensitivity of ddPCR also depends on the type of specimens and the sources of nucleic acid. The use of combined TNA (cfDNA + exoTNA) in the plasma and exoTNA in the pleural fluid allowed for the detection of target mutations more sensitively than that using cfDNA or total DNA alone.
Both ddPCR and real-time PCR (Cobas) are sensitive and relatively easy and fast tests. However, only targeted hot-spot mutations are detectable using these techniques. The advantage of NGS, in contrast, is that many panel genes can be examined at once to detect known resistance mutations of EGFR-TKI other than EGFR hot-spot mutations. For instance, KRAS, EGFR, and PIK3CA mutations are known to be acquired resistance mechanisms to EGFR-TKIs [22, 23]. Amplifications in KRAS, EGFR, and cell cycle genes (including CCND1, CDK4, and CDK6) have also been reported as TKI resistance mechanisms [24, 25]. In this study, we detected mutations in KRAS and PIK3CA, and amplifications in EGFR and CDK4 in patients who did not have the EGFR T790M mutation.
In addition, some patients had other resistance mutations that were found in addition to EGFR T790M. The phenomenon by which T790M and other resistant mutations occur simultaneously is clonal heterogeneity [26]. For instance, the BRAF and PIK3CA mutations and EGFR amplification can coexist with T790M in patients who progressed despite EGFR-TKI therapy [27,28,29]. The clinical significance of this co-occurring mutation is that it can affect the treatment outcomes [26].
The size distribution of tumor-derived nucleic acids in the exosome may be a critical consideration for selecting an extraction method. Our group previously showed that a short-length exoTNA (200 bp) contained detectable tumor-derived nucleic acids in the exosome from a NSCLC patient’s plasma [11]. However, the high molecular weight DNA (10–15 kb long) was more abundant in the exosomes from pleural fluids in this study than it was from the plasma. Therefore, the target size of the exoNAs depends upon the clinical sample types, and should be considered in the detection of EGFR mutations in NSCLC patients.
One limitation of our study is that matched tissue sampling was not performed in most cases. This might be the reason for the lower frequency of T790M mutations in our study than the previously reported prevalence of 40–55% [29]. However, in 22 of 64 patients (34.4%) who progressed despite EGFR-TKI therapy, the T790M mutation was identified in one or more liquid biopsy platforms (using the plasma or pleural fluid). We identified differences in the analytical performances of each type of specimen and nucleic acid using data from these patients.
Previous reports suggested that exoNA could be a novel DNA source for genetic testing [30]. In our study, the distribution of nucleic acid from exosomes was different than that from a cell pellet or supernatant. The nucleic acid from exosomes was enriched, high molecular weight DNA. This finding was consistent with that from previous reports [13, 30]. Further studies with larger cohorts are needed to substantiate our findings.
Conclusion
In conclusion, combined TNA from the plasma and exoTNA from the pleural fluid could be used as a feasible target for low-abundant EGFR mutant copies in NSCLC. We demonstrated the optimal analytical method to improve the sensitivity of detecting EGFR mutations in different types of clinical samples.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Abbreviations
- EGFR:
-
Epidermal growth factor receptor
- NSCLC:
-
Non-small-cell lung cancer
- TKIs:
-
Tyrosine kinase inhibitors
- ctDNA:
-
Circulating tumor DNA
- exoNA:
-
Exosomal nucleic acid
- exoTNA:
-
Exosomal total nucleic acid
- cfDNA:
-
Cell-free DNA
- BAL:
-
Bronchoalveolar lavage
- ddPCR:
-
Droplet digital PCR
- NAs:
-
Nucleic acids
- LOD:
-
Limit of detection
- NGS:
-
Next-generation sequencing
References
Tan DS, Yom SS, Tsao MS, Pass HI, Kelly K, Peled N, et al. the international association for the study of lung cancer consensus statement on optimizing management of egfr mutation-positive non-small cell lung cancer: status in 2016. J Thorac Oncol. 2016;11:946–63.
Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–46.
Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–7.
Fujiwara Y, Goto Y, Kanda S, Horinouchi H, Yamamoto N, Sakiyama N, et al. Efficacy and safety of osimertinib in a Japanese compassionate use program. Jpn J Clin Oncol. 2017. https://doi.org/10.1093/jjco/hyx050.1-5.
Yu HA, Suzawa K, Jordan E, Zehir A, Ni A, Kim R, et al. Concurrent alterations in EGFR-mutant lung cancers associated with resistance to EGFR kinase inhibitors and characterization of MTOR as a mediator of resistance. Clin Cancer Res. 2018;24:3108–18.
Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108–12.
Molina-Vila MA. Liquid biopsy in lung cancer: present and future. Transl Lung Cancer Res. 2016;5:452–4.
Calabuig-Farinas S, Jantus-Lewintre E, Herreros-Pomares A, Camps C. Circulating tumor cells versus circulating tumor DNA in lung cancer-which one will win? Transl Lung Cancer Res. 2016;5:466–82.
Buder A, Hochmair MJ, Schwab S, Bundalo T, Schenk P, Errhalt P, et al. Cell-free plasma DNA-guided treatment with osimertinib in patients with advanced EGFR-mutated NSCLC. J Thorac Oncol. 2018;13:821–30.
Buder A, Setinek U, Hochmair MJ, Schwab S, Kirchbacher K, Keck A, et al. EGFR mutations in cell-free plasma DNA from patients with advanced lung adenocarcinoma: improved detection by droplet digital PCR. Target Oncol. 2019;14:197–203.
Kim Y, Shin S, Kim B, Lee KA. Selecting short length nucleic acids localized in exosomes improves plasma EGFR mutation detection in NSCLC patients. Cancer Cell Int. 2019;19:251.
Krug AK, Enderle D, Karlovich C, Priewasser T, Bentink S, Spiel A, et al. Improved EGFR mutation detection using combined exosomal RNA and circulating tumor DNA in NSCLC patient plasma. Ann Oncol. 2018;29:700–6.
Hur JY, Kim HJ, Lee JS, Choi CM, Lee JC, Jung MK, et al. Extracellular vesicle-derived DNA for performing EGFR genotyping of NSCLC patients. Mol Cancer. 2018;17:15.
Shin S, Kim J, Kim Y, Cho SM, Lee KA. Assessment of real-time PCR method for detection of EGFR mutation using both supernatant and cell pellet of malignant pleural effusion samples from non-small-cell lung cancer patients. Clin Chem Lab Med. 2017. https://doi.org/10.1515/cclm-2016-0851.
Meddeb R, Pisareva E, Thierry AR. Guidelines for the preanalytical conditions for analyzing circulating cell-free DNA. Clin Chem. 2019;65:623–33.
Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods. 2014;11:163–6.
Shugay M, Britanova OV, Merzlyak EM, Turchaninova MA, Mamedov IZ, Tuganbaev TR, et al. Towards error-free profiling of immune repertoires. Nat Methods. 2014;11:653–5.
Lee SH, Kim WS, Choi YD, Seo JW, Han JH, Kim MJ, et al. Analysis of mutations in epidermal growth factor receptor gene in korean patients with non-small cell lung cancer: summary of a nationwide survey. J Pathol Transl Med. 2015;49:481–8.
Pinto JA, Vallejos CS, Raez LE, Mas LA, Ruiz R, Torres-Roman JS, et al. Gender and outcomes in non-small cell lung cancer: an old prognostic variable comes back for targeted therapy and immunotherapy? ESMO Open. 2018;3:e000344.
Sacher AG, Paweletz C, Dahlberg SE, Alden RS, O’Connell A, Feeney N, et al. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016;2:1014–22.
Oxnard GR, Thress KS, Alden RS, Lawrance R, Paweletz CP, Cantarini M, et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non-small-cell lung cancer. J Clin Oncol. 2016;34:3375–82.
Ortiz-Cuaran S, Scheffler M, Plenker D, Dahmen L, Scheel AH, Fernandez-Cuesta L, et al. Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin Cancer Res. 2016;22:4837–47.
Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26.
Nagano T, Tachihara M, Nishimura Y. Mechanism of resistance to epidermal growth factor receptor-tyrosine kinase inhibitors and a potential treatment strategy. Cells. 2018;7:212.
Nakatani K, Yamaoka T, Ohba M, Fujita KI, Arata S, Kusumoto S, et al. KRAS and EGFR amplifications mediate resistance to rociletinib and osimertinib in acquired afatinib-resistant NSCLC harboring exon 19 Deletion/T790M in EGFR. Mol Cancer Ther. 2019;18:112–26.
Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815.
Ko R, Kenmotsu H, Serizawa M, Koh Y, Wakuda K, Ono A, et al. Frequency of EGFR T790M mutation and multimutational profiles of rebiopsy samples from non-small cell lung cancer developing acquired resistance to EGFR tyrosine kinase inhibitors in Japanese patients. BMC Cancer. 2016;16:864.
Wu SG, Chang YL, Yu CJ, Yang PC, Shih JY. The role of PIK3CA mutations among lung adenocarcinoma patients with primary and acquired resistance to EGFR tyrosine kinase inhibition. Sci Rep. 2016;6:35249.
Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 2018;29:i10–9.
Qu X, Li Q, Yang J, Zhao H, Wang F, Zhang F, et al. Double-stranded dna in exosomes of malignant pleural effusions as a novel DNA source for EGFR mutation detection in lung adenocarcinoma. Front Oncol. 2019;9:931.
Acknowledgements
Not applicable.
Funding
This research was conducted with funding from AstraZeneca.
Author information
Authors and Affiliations
Contributions
Designed and supervised the research, interpreted data, and reviewed and edited the manuscript: KAL; performed experiments and analysis of all samples, interpreted experimental data and wrote manuscript: YK and SS. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All human samples are collected with the approval of the Institutional Review Board of Gangnam Severance Hospital (Number: 3-2017-0211) and Kangnam Sacred Heart Hospital (Number: 2018-07-029).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12935_2021_1761_MOESM1_ESM.docx
Additional file 1: Fig. S1. Nucleic acid yield plots from exoTNA and cfDNA; Fig. S2. Assessment of T790M mutant alleles according to different sources of tumor-derived nucleic acids (exosomes, cell pellet and supernatant) from the pleural fluid and extraction methods; Fig. S3. The distribution of isolated nucleic acids; Table S1. List of genes included in Oncomine Pan-Cancer Cell-Free Assay; Table S2. Clinical characteristics and plasma mutation test results from 54 patients; Table S3. Clinical characteristics and EGFR mutation results identified in the pleural fluid from 13 patients.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kim, Y., Shin, S. & Lee, KA. Exosome-based detection of EGFR T790M in plasma and pleural fluid of prospectively enrolled non-small cell lung cancer patients after first-line tyrosine kinase inhibitor therapy. Cancer Cell Int 21, 50 (2021). https://doi.org/10.1186/s12935-021-01761-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-021-01761-x