
Abstract
Background
RAS association domain family protein 1A (RASSF1A) promoter hypermethylation is suggested to be linked to hepatocellular carcinoma (HCC), but the results remained controversial.
Methods
We evaluated how RASSF1A promoter hypermethylation affects HCC risk and its clinicopathological characteristics through meta-analysis. Data on DNA methylation in HCC and relevant clinical data were also collected based on The Cancer Genome Atlas (TCGA) database to investigate the prognostic role of RASSF1A promoter hypermethylation in HCC.
Results
Forty-four articles involving 4777 individuals were enrolled in the pooled analyses. The RASSF1A promoter methylation rate was notably higher in the HCC cases than the non-tumor cases and healthy individuals, and was significantly related to hepatitis B virus (HBV) infection-positivity and large tumor size. Kaplan–Meier survival analysis revealed that HCC cases with RASSF1A promoter hypermethylation had worse outcomes. Receiver operating characteristic curves confirmed that RASSF1A promoter methylation may be a marker of HCC-related prognoses.
Conclusions
RASSF1A promoter hypermethylation is a promising biomarker for the diagnosis of HCC from tissue and peripheral blood, and is an emerging therapeutic target against HCC.
Similar content being viewed by others
Introduction
Liver cancer (LC) is the sixth leading cause of cancer-related morbidity, and the fourth major cause of cancer-related death, worldwide. Approximately 841,000 newly diagnosed LC cases and 782,000 LC-related deaths are reported annually [1]. Hepatocellular carcinoma (HCC) is a major histological subtype of LC, accounting for 70% to 85% of all LC cases, globally [2]. While significant progress has been made in the diagnosis and treatment HCC, patients with the disease still have unsatisfactory prognoses [3]. Consequently, new clinical strategies are needed to improve the efficacy of HCC treatment, including the development of novel diagnostic and prognostic biomarkers.
Recent emerging evidence suggests that the accumulation of epigenetic and genetic alterations has a role in the different stages of liver carcinogenesis [4]. Besides, CpG island methylation within gene promoters, key epigenetic regulatory factors, has an important role in HCC initiation and development [5]. Promoter hypermethylation may result in the silencing of some tumor suppressors that regulate the cell signaling pathways in tumor tissues [6,7,8]. Among them, the RAS association domain family protein 1A (RASSF1A) is an important tumor suppressor associated with multiple biological functions, and its promoter is frequently blocked due to promoter hypermethylation in numerous malignant tumors, including HCC [4, 9]. The promoter hypermethylation of RASSF1A may have potential screening value, and may serve as an attractive early diagnostic and prognostic biomarker in HCC.
While a number of individual studies are being performed in patients with HCC, results on the association between RASSF1A promoter hypermethylation and HCC risk or its clinicopathological features remain controversial [10]. Although a study focusing on the diagnostic accuracy of the same has been conducted, only seven articles focusing on RASSF1A methylation in peripheral blood have been enrolled for analysis [11]. Moreover, it remains to be systemically investigated whether RASSF1A promoter hypermethylation is related to the clinicopathological features of HCC and the associated prognoses. Accordingly, we aimed to more comprehensively evaluate the role of RASSF1A promoter hypermethylation in HCC.
Materials and methods
The study included two parts: meta-analysis and bioinformatic analysis. The meta-analysis was implemented in accordance with Preferred Reporting Items for Systematic Reviews and Meta-analyses guidelines [12]. The data from the TCGA are publicly available and open‐access; therefore, the local ethics committees did not need to approve the study because the current research follows the TCGA data access policies and publication guidelines.
Retrieval and screening of eligible studies
Electronic databases, including Cochrane Library, Web of Science, EMbase, and Pubmed were searched for the identification of English-language articles from inception till April 30, 2020. The search strategy of (HCC OR hepatocellular carcinoma OR liver cancer) AND (hypermethylation OR methylation OR epigenetics) AND (RASSF1A or RASSF1 or Ras association domain family 1 A) was utilized for retrieval. In addition, the reference lists in relevant reviews and included studies were also checked manually for the avoidance of omission.
The study inclusion criteria were: (1) studies that reported on the relationship of the promoter methylation of RASSF1A with HCC or the associated clinicopathological characteristics in patients with HCC; (2) studies that investigated the RASSF1A promoter methylation levels in both tissues and blood; (3) case–control studies that regarded people with HCC as cases (as confirmed from HCC tissues and peripheral blood) and people without HCC as controls (as confirmed using adjacent noncancerous tissues, benign lesions, normal tissues, and serum); and (4) studies that reported the exact RASSF1A methylation frequency in both cases and controls. Meanwhile, studies not conforming to the inclusion criteria were excluded. For duplicate studies, the most complete report was selected.
Extraction of data and evaluation of quality
Data were independently extracted by two reviewers following a pre-defined procedure. The data collected in this study included: author names, study design, year of publication, sample type, control sample, hypermethylated case number, hypomethylated case number, hypermethylated control number, hypomethylated control number, detection method and clinicopathologic parameters, such as age, sex, HBV infection, HCV infection, tumor number, tumor size, liver cirrhosis, AFP level, pathological grade, tumor differentiation and portal venous invasion.
Moreover, two reviewers independently evaluated the methodological quality of the enrolled studies according to the criteria stipulated in the NOS [13]. Each study was assigned a score ranging from 0 (poor quality) to 9 points (optimal quality) based on the selection, comparability and exposure of the cases and controls. Any disagreement between the two reviewers was settled by discussion.
Meta-analysis
Stata 12.0 software (Stata Corporation, College Station, Texas, USA) and R software (version 3.4.4) were adopted for the statistical analyses and plotting. The ORs of the cases versus controls in each study were calculated by 2 × 2 tables. A classic half-integer continuity correction was applied in studies that reported zero events in the treatment or control arm. The log ORs were then aggregated to obtain combined results. Then, the results for the controls were pooled in the non-tumor group (including adjacent non-cancerous tissues, benign lesions, and serum from patients with benign disease) and the normal group (including liver tissues and serum from healthy donors). Moreover, the heterogeneity between two studies was evaluated through the I2 statistic and Chi square tests. The level of heterogeneity was deemed significant at I2 > 50% and P < 0.10 for Chi square tests. This study adopted the random-effects model for all analyses to obtain conservative results [14]. Subgroup analyses stratified by sample type, detection method and sample size were performed for the investigation of possible heterogeneity sources. Additionally, the covariate impacts on those integrated results as well as the heterogeneity across different studies were evaluated by meta-regression analysis.
In the sensitivity analysis, one study was eliminated at a time for the evaluation of its influence on the pooled analysis. Egger’s test and Begg’s test were utilized for the identification of publication bias [14, 15]. For both tests, P < 0.05 indicated significant publication bias. Furthermore, the presence of potential publication bias was adjusted by the “trim and fill” approach [16], which estimated the potential studies omitted and then re-calculated the integrated results with these hypothetical studies. P < 0.05 (two-tailed) indicated statistical significance.
Extraction and analysis of TCGA data
Data on DNA methylation in HCC and corresponding clinical data were collected from the TCGA (Illumina Infinium Human Methylation 450 [HM450]) database including 485,577 probes. Then, the methylation levels of all the probes were determined by the β values. Empirical thresholds of 0.2 and 0.6 were adopted to distinguish between complete non-methylation, hypomethylation, and hypermethylation. Specifically, β ≤ 0.6 represented hypomethylation and β > 0.6 signified hypermethylation. Further, the Kaplan–Meier method was adopted for the construction of the OS and DFS curves for different RASSF1A methylation statuses, while the log-rank test was used for comparisons. In addition, this study also established time-dependent ROC curves, and determined the AUC values for the assessment of the predictive power of RASSF1A methylation status.
Results
Screening of studies
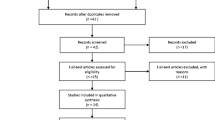
Totally, 479 articles were identified through the original search strategy (Fig. 1). Of them, 218 duplicate articles were ruled out, and an additional 61 were eliminated due to the lack of relevance to this study after their abstracts were read. Later, the full-texts of the remaining 59 studies were carefully read; 44 satisfied our study inclusion criteria and were enrolled for analyses. In detail, 12 studies presented data on the RASSF1A promoter methylation rate within HCC and assessed the association of this methylation with clinicopathological characteristics. Besides, 29 studies only assessed the frequency of RASSF1A promoter methylation, while 3 only evaluated the clinicopathological characteristics.

Flow diagram of the study selection process
Characteristics of the enrolled articles
The features of the enrolled articles were shown in Table 1. Altogether, 44 case–control studies involving 4777 individuals published from 2002 to 2019 were enrolled in the analyses [10, 17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59]. Twenty-eight articles originated in Asia, consistent with the epidemiology of HCC. America produced the second highest number of enrolled papers (n = 8), followed by Africa (n = 5), while Italy and Germany published one article each. Three types of sample sources were predominantly investigated, including tissues (n = 31), peripheral blood (n = 11), and both tissues and peripheral blood (n = 2). In all our enrolled studies, HCC patients were regarded as ‘cases’; those without the disease were considered ‘controls’, and were assigned to the non-tumor group and normal group. Of those articles examining HCC risk, 11 used blood, 31 adopted tissues, and two examined both blood and tissues. Heterogeneous methods were adopted for the detection of the RASSF1A methylation status among the enrolled studies. The Newcastle-Ottawa scale (NOS) was adopted to assess the quality of the 41 articles that reported the RASSF1A methylation rates in the cases and controls, with scores ranging from 5 to 8, indicating a relatively high methodological quality (Additional file 1: Table S1). Another three studies that only reported on the disease’s clinicopathological characteristics were not eligible for NOS assessment, and were thus not evaluated.
Effect of RASSF1A promoter hypermethylation on HCC in the pooled analyses
Comparison of RASSF1A promoter hypermethylation between HCC and non-tumor groups
Data from 34 studies including 2075 HCC patients and 2276 non-tumor controls underwent meta-analyses for the evaluation of the effect of RASSF1A promoter hypermethylation on HCC risk (Fig. 2). We found that the frequency of RASSF1A gene promoter hypermethylation was remarkably related to a high HCC risk in the overall comparison (odds ratio [OR] = 6.87, 95% confidence interval [CI] = 4.98–9.50, P < 0.001), and moderate heterogeneity was present (I2 = 64.1%, P = 0.000).

Forest plot of the correlation between RASSF1A promoter hypermethylation with HCC in non-tumor groups. OR, odds ratio; CI, confidence interval
Further subgroup analyses stratified by sample type (blood and tissue), detection method (methylation-specific polymerase chain reaction [MSP] and others) and sample size (≥ 100 and < 100) were also performed to explore the possible heterogeneity sources across the various articles enrolled. Subgroup analyses stratified by sample type showed that RASSF1A gene promoter hypermethylation was significantly associated with HCC risk (blood: OR = 6.93, 95% CI = 4.12–11.65, P < 0.001; tissue: OR = 7.12, 95% CI = 4.78–10.59, P<0.001). In addition, in the subgroup analysis stratified by the detection method, RASSF1A gene promoter hypermethylation was evidently related to HCC risk (MSP: OR = 7.30, 95% CI = 5.17–10.29, P < 0.001; others: OR = 6.20, 95% CI = 3.13–12.30, P < 0.001). Similarly, the pooled results were consistent between the subgroups stratified by sample size (≥ 100: OR = 6.74, 95% CI = 4.28–10.61, P<0.001; < 100: OR = 6.67, 95% CI = 4.46–10.00, P < 0.001) (Fig. 3).

Forest plot of the subgroup analysis according to sample type (tissue and blood), detection method (MSP and others) and sample size (≥ 100 and < 100) for the correlation between RASSF1A promoter hypermethylation with HCC in non-tumor groups and normal groups. No., number; OR, odds ratio; CI, confidence interval
Comparison of RASSF1A promoter hypermethylation between HCC and normal groups
Totally, 26 studies enrolling 1898 HCC patients and 1002 normal controls were pooled for the assessment of how RASSF1A promoter hypermethylation affects HCC risk (Fig. 4). In the meta-analysis, the promoter methylation of RASSF1A was related to HCC risk in the cancer samples relative to the controls (OR = 31.05, 95% CI = 13.73–70.20, P < 0.001); in addition, a high heterogeneity level was detected across the various articles (I2 = 79.6%, P = 0.000).

Forest plot of the correlation between RASSF1A promoter hypermethylation with HCC in normal groups. OR, odds ratio; CI, confidence interval
Subgroup analyses revealed that the promoter methylation of RASSF1A was significantly correlated with the risk of HCC in all the subgroups stratified by sample type, detection method and sample size (Fig. 3).
Relationship of the promoter hypermethylation of RASSF1A with the clinicopathological features
This study investigated a total of 11 characteristics from 15 studies that investigated the correlation of RASSF1A gene promoter hypermethylation with the clinicopathological features of HCC. The comprehensive data on the numerous clinicopathological features associated with HCC, and the association with the RASSF1A gene was presented in Table 2. As shown in the pooled analyses, RASSF1A promoter hypermethylation was remarkably related to tumor size (≥ 5 cm vs. < 5 cm, OR = 1.92, 95% CI = 1.07–3.42, P = 0.028) and hepatitis B virus (HBV) infection (positive vs. negative, OR = 1.50, 95% CI = 1.05–2.14, P = 0.026), but was not significantly associated with sex (male vs. female, OR = 1.36, 95% CI = 0.95–1.96, P = 0.094), age (≥ 50 vs. < 50, OR = 1.74, 95% CI = 0.82–3.69, P = 0.152), hepatitis C virus (HCV) infection (positive vs. negative, OR = 0.93, 95% CI = 0.20–4.26, P = 0.928), level of alpha fetoprotein (AFP) (≥ 20 μg/L vs. < 20 μg/L, OR = 1.25, 95% CI = 0.47–3.27, P = 0.657), tumor number (multiple vs. single, OR = 0.80, 95% CI = 0.47–1.36, P = 0.410), liver cirrhosis (presence vs. absence, OR = 1.06, 95% CI = 0.60–1.87, P = 0.834), histopathological stage (I + II vs. III + IV, OR = 1.84, 95% CI = 0.53–6.36, P = 0.338), tumor differentiation (poor vs. moderate or well, OR = 0.91, 95% CI = 0.41–2.02, P = 0.820) or portal venous invasion (presence vs. absence, OR = 0.61, 95% CI = 0.16–2.40, P = 0.481).
Meta-regression and sensitivity analyses
As for the results of the pooled meta-regression analysis on the correlation between the promoter hypermethylation of RASSF1A and HCC risk in both groups, a trend for sample type, detection method and sample size was demonstrated (Additional file 1: Table S2). Heterogeneity was detected in the pooled results; as a result, this study evaluated the contributions of diverse investigated features to heterogeneity. Nonetheless, there was no statistical significance (all P values > 0.05, Additional file 1: Table S2). The heterogeneity proportions in both groups ranged from − 9.70% to 8.14% (all P values > 0.05), with a high level of residual heterogeneity (τ2 range, 0.506–3.226). Owing to a lack of sufficient data in the enrolled articles, this study did not incorporate other factors that possibly contribute to heterogeneity into the meta-regression analyses.
To further investigate the robustness of the pooled results in both groups by sensitivity analyses, a random-effects model was adopted to eliminate one study at a time. None of the studies had a significant influence on the pooled results, indicating that our estimates were robust and reliable (Additional file 1: Figure S1).
Publication bias
With regards to the non-tumor group, the funnel plot appeared to be asymmetric (Additional file 1: Figure S2A), and statistical significance was observed in Begg’s test (P = 0.021), which raised the possibility of publication bias, although no significant publication bias was discovered in Egger’s test (P = 0.208). Subsequently, the “trim and fill” method was adopted for the evaluation of the possible impact of publication bias on the pooled effect. In consequence, the symmetric funnel plot was generated through the filling of 10 hypothetical negative articles (Additional file 1: Figure S2B). Typically, the adjusted OR obtained from the pooled analysis incorporating these hypothetical studies was still significant (OR = 5.14, 95% CI = 3.69–7.16, P < 0.001). Similarly, for the normal group, both Egger’s test (P < 0.001) and the funnel plot revealed the presence of potential publication bias (Additional file 1: Figure S2C), regardless of the absence of statistical significance in Begg’s test (P = 0.332). Thereafter, seven hypothetical negative studies were filled through the “trim and fill” approach, but RASSF1A promoter methylation was found to be significantly associated with HCC risk in the pooled analyses (OR = 15.71, 95% CI = 7.40–33.36, P < 0.001) (Additional file 1: Figure S2D).
Association of the promoter hypermethylation of RASSF1A with HCC-related prognoses
Baseline patient characteristics
Data on the promoter methylation of RASSF1A were identified within DNA methylation profiles from 380 The Cancer Genome Atlas (TCGA)-derived HCC as well as 50 non-carcinoma samples. Based on UCSC assembly-Dec.2013 (GRCh38/hg38), 11 probes situated at the promoter region of RASSF1A were selected (including cg13872831, cg24859722, cg04743654, cg00777121, cg08047457, cg12966367, cg21554552, cg25747192, cg06172942, cg25486143, cg27569446), and they contained the RASSF1A gene CpG island A (chr3: 50340373–50341109). In the TCGA cohort, the RASSF1A promoter methylation levels within the HCC samples significantly increased compared to those in the adjacent non-carcinoma liver tissues (Additional file 1: Figure S3). According to the probe methylated levels, all samples were classified into the hypomethylated (n = 196) and hypermethylated (n = 184) groups. Among the 380 TCGA-derived HCC samples, 349 had information available on overall survival (OS) and survival status, while 342 had data on disease-free survival (DFS) and recurrence status.
RASSF1A promoter hypermethylation in the prediction of HCC-related prognoses
In the Kaplan–Meier survival analysis, HCC cases with RASSF1A gene promoter hypermethylation were found to have poorer OS (median OS: 3.90 years vs. 6.73 years; P = 0.0206) and DFS (median DFS: 1.38 years vs. 3.01 years; P = 0.0003) values than the hypomethylated cases (Fig. 5a, c). Additionally, receiver operating characteristic (ROC) curve analysis was conducted for the determination of the sensitivity and specificity of RASSF1A gene promoter hypermethylation in prognosis prediction. The areas under the curve (AUCs) pertaining to RASSF1A gene promoter hypermethylation in the prediction of the OS of HCC patients at 1, 2, 3 and 5 years were 0.51, 0.60, 0.60 and 0.58, respectively (Fig. 5b). Meanwhile, the time-dependent AUC values concerning RASSF1A gene promoter hypermethylation in the prediction of the OS of HCC patients at 1, 2, 3 and 5 years were 0.61, 0.69, 0.63 and 0.73, separately (Fig. 5d). Accordingly, we inferred that RASSF1A gene promoter methylation status exhibited high sensitivity and specificity.

Kaplan-Meier survival and time‐dependent ROC curves of different RASSF1A methylation statuses in the TCGA HCC cohort. a, b Prognostic influences of the promoter hypermethylation of RASSF1A on OS and DFS in the TCGA cohort; c, d Time-dependent ROC curve analysis of the predictive power of RASSF1A methylation statuses on OS and DFS
Discussion
In this study, we found that RASSF1A promoter hypermethylation is a promising biomarker for the diagnosis of HCC from tissue and peripheral blood. A number of factors participate in liver carcinogenesis, such as hepatitis virus infection, as well as environmental, genetics and epigenetic alterations [4, 60]. RASSF1A, a key tumor suppressor protein, controls cell cycle regulation and cell apoptosis [61, 62]. In 2002, Zhang et al. first reported that the promoter hypermethylation of RASSF1A was a major inactivating event in 85% (70/82) of HCC patients [18]. Since then, numerous studies with small sample sizes have demonstrated that the rate of RASSF1A promoter methylation is significantly increased within HCC tissues relative to non-carcinoma tissues [36, 44, 59], and the results of association analyses were consistent across studies [26, 35, 57]. Then, Zhao et al. conducted a meta-analysis that involved a total of seven case–control studies, which suggested that the promoter hypermethylation of RASSF1A within body fluids was significantly correlated with HCC risk [11]. However, some issues require further clarification, due to which we conducted the present updated study. A large number of studies investigating the association of RASSF1A promoter hypermethylation with HCC risk have been published since 2013. Further, the impact of the promoter hypermethylation of RASSF1A within cancer tissues on the risk and clinicopathological characteristics of HCC has not been summarized yet, and there was a need for the association of the promoter hypermethylation of RASSF1A with HCC prognoses to be analyzed. In our meta-analysis, in which we enrolled 44 articles and 9354 cases, RASSF1A promoter hypermethylation showed significant associations with HCC risk within tissues and peripheral blood samples, suggesting that it represents an early event in liver carcinogenesis. Data from the TCGA database indicated that RASSF1A gene promoter hypermethylation is significantly correlated with HCC risk. Additionally, two clinicopathological parameters, HBV infection and tumor size, were also found to be associated with RASSF1A promoter hypermethylation.
In at least 37 types of cancers, promoter hypermethylation is reportedly directly associated with absent RASSF1A gene expression [9, 63]. The RASSF1A gene was firstly recognized as a possible RAS-binding molecule in the promotion of apoptosis, due to the presence of an RAS-associated domain within the primary sequence [64]. Dammann et al. demonstrated that the RASSF1A gene has a role in tumor suppression, and that its functional loss results in the proliferation of cells and carcinogenesis [62]. Some studies suggest that the RASSF1A gene may also be involved in the stabilization of microtubules, regulation of DNA repair, and control of cell cycle and apoptosis [61, 65,66,67]. The methylation and inactivation of RASSF1A exert the most relevant cell protection functions via the inactivation of the Hippo and Wnt signaling pathways, as proven in HCC patients [68,69,70]. Compared to tumor tissues, RASSF1A promoter methylation is not commonly observed in normal tissues. In addition, high RASSF1A promoter hypermethylation rates are reported in many cancers, making them potential molecular markers for cancer diagnosis. In the present meta-analysis, the rate of RASSF1A promoter methylation within both the HCC tissues and peripheral blood samples apparently increased relative to that within the normal samples, and identical results were reported for non-tumor samples. This indicates that RASSF1A promoter hypermethylation may contribute to the entire HCC development process. Furthermore, this study also evaluated the prognostic value of RASSF1A promoter hypermethylation within HCC tissue samples. We found that RASSF1A promoter hypermethylation was related to poor OS and DFS values. Similar to our results, other studies have also demonstrated that the promoter hypermethylation of RASSF1A in peripheral blood has prognostic potential in HCC [53, 55, 71]. Taken together, these results suggest that the detection of RASSF1A promoter hypermethylation in tissue and peripheral blood samples may not only serve as a potential diagnostic biomarker for HCC, but also have essential prognostic value in HCC.
Regarding the association of RASSF1A promoter hypermethylation with the clinicopathological features of HCC, we discovered that the former is related to HBV infection. Several recent studies have suggested the presence of a relationship between DNA methylation and HCC in association with HBV infection in patients with HCC [72,73,74,75]. Some recent studies have suggested that chronic inflammation may be associated with aberrant gene promoter methylation and silencing in ulcerative colitis and gastritis [76, 77]. Moreover, other internal or environmental stimulating factors, including viral infection and hypoxia, may also cause the spread of epigenetic alterations as silent events [9]. It has been demonstrated in certain studies that HBV X protein (HBx) plays an essential role in HBV-related HCC genesis [78, 79]. HBx upregulates the expression of DNA-methyltransferase1 (DNMT1) as well as DNMT3b, thus inducing tumor suppressor gene (TSG) hypermethylation [6, 80]. Additionally, Schagdarsurengin et al. investigated the role of RASSF1A during liver carcinogenesis in vitro, they analysed the methylation status of the RASSF1A promoter in HBV-positive human hepatocellular carcinoma cell line (Hep3B) and found RASSF1A promoter CpG island was hypermethylated [20]. The same result was also revealed by Zhang and his colleagues [18]. They also demonstrated that hypermethylation of RASSF1A was detected in Hep3B cells but not in HepG2 cells [18]. The similar results have been yielded in nasopharyngeal carcinoma (NPC) with Epstein–Barr virus (EBV) infection [81]. Lo et al. found that promoter hypermethylation and transcription silencing of RASSF1A were consistently detected in all EBV-positive NPC cell lines [82]. The expression of EBV latent proteins will constitutively activate multiple signaling pathways, enhance genetic instability, induce epigenetic changes, modulate microenvironment and erase host immune response during early stage of cancer development [81].
Nonetheless, a larger number of studies in vitro are warranted to clarify the effect of HBV infection on DNA methylation in the induction of HCC development. Additionally, Okamoto et al. revealed that HBV and HCV infection activates the innate immune response dependent on the natural killer cells to induce DNA methylation, including the RASSF1A gene [75]. In this study, only four enrolled studies investigated the association of HCV infection with RASSF1A methylation; nevertheless, the results of our pooled analysis were not statistically significant. Future studies should examine the possible biological mechanisms underlying hepatitis virus-caused DNA methylation within the context of HCC. Interestingly, the promoter hypermethylation of RASSF1A was significantly related to tumor size, suggesting that the level of RASSF1A methylation may reflect tumor load, consistent with previous studies [51, 58, 71]. RASSF1A, the TSG associated with cell cycle, inhibits cyclin D1 accumulation and induces cell cycle arrest at the G1 phase [83, 84]. RASSF1A promoter hypermethylation also promotes the cell cycle process in those impaired hepatocytes through the escape from arrest at the G1 phase [10]. Nevertheless, statistical significance was not noted in the additional clinicopathological features. This is possibly due to the small sample size and high heterogeneity. Further studies are warranted to examine the association of RASSF1A gene promoter hypermethylation with the clinicopathological features of HCC.
Given the moderate to high heterogeneity detected in the relationship of RASSF1A gene promoter hypermethylation with HCC risk, this study further conducted meta-regression and subgroup analyses. However, we were unable to identify any factor that significantly contributed to the heterogeneity level in the aforementioned two analyses. Therefore, further studies are needed to examine the effects of those factors. Typically, the clinical and methodological heterogeneities detected across all the enrolled articles represent an essential issue. In this meta-analysis, a large variety of assay methods was used in each study, including a total of seven diverse techniques. Moreover, different thresholds were applied for the assessment of DNA methylation in each study. Non-quantitative methods such as MSP were utilized for the detection of DNA methylation, which precluded the necessity to determine a cut-off point. Previous studies reported different RASSF1A hypermethylation rates in HCC using different sets of CpGs [18, 26, 44, 85]. Apart from the selection of the detection technique and thresholds, the determination of the precise genomic positions of those CpG dinucleotides analyzed is also of great importance [86]. With regards to clinical heterogeneity, only 20.5% (9/44) of our enrolled articles mentioned the presence or absence of preoperative treatment [38, 40, 41, 45, 47, 50, 53, 55]. Some studies suggested that both radiotherapy and chemotherapy can alter a patient’s DNA methylation status; therefore, the type of preoperative treatment performed should be clarified [87, 88]. Moreover, four studies investigated the relationship between RASSF1A hypermethylation in peripheral blood and the risk of HCC, and the diagnosis of HCC was confirmed by imaging techniques and serum AFP levels, rather than through pathological examination [56,57,58,59]. The diagnosis of HCC based on imaging techniques is recommended by guidelines; however, histopathological evaluation remains the gold standard for HCC diagnosis [89, 90]. This is because imaging is not always specific, and there is limited expertise and lack of advanced imaging in many medical centers. Additionally, some studies enrolled in the present study had a retrospective design with a small sample size, which may have led to selection bias [91]. Future studies must examine the effects of the aforementioned factors. Furthermore, normalization of the methods used for the analysis of methylation status, use of uniform definitions, and presence of cooperation among different research groups to obtain a large sample size may be beneficial to studies focusing on the role of methylation marker alterations in cancer.
Reactivation of the TSG that is silenced epigenetically is considered a promising anti-tumor treatment strategy. Over the last few decades, different inhibitors of DNA methylation and natural compounds have been tested in different cancers [92]. In particular, 5-aza-2′-deoxycytidine (Dacogen or Decitabin) and 5-aza-cytidine (Azadine or Vidaza) can lead to RASSF1A promoter demethylation and the reactivation of RASSF1A expression in diverse types of tumor cells [62, 93,94,95]. Our study further demonstrated that the promoter hypermethylation of RASSF1A was not only a prognostic indicator but also an emerging therapeutic target against HCC.
Nonetheless, several limitations should be noted in this study. First, the funnel plots of both the non-tumor group and normal group were slightly asymmetric, indicating the presence of potential publication bias. However, the results were not significantly changed by the “trim and fill” method, suggesting that the relationship of RASSF1A promoter methylation with HCC was meaningful, but not an artifact caused by unpublished negative studies. Second, heterogeneity was present in the current study, which may have been a result of numerous factors. We did not identify any factors that made significant contributions to heterogeneity in the meta-regression and subgroup analyses. Finally, only some of our enrolled studies investigated the relationship of RASSF1A promoter methylation with HCC-related prognoses. In this regard, the prognostic role of RASSF1A was only investigated among patients from the TCGA HCC cohort. Consequently, a larger number of high-quality studies are warranted to resolve the limitations mentioned above.
Conclusions
In the present study, the significance of the promoter hypermethylation of RASSF1A in HCC diagnoses and prognoses was examined. We found that the rate of RASSF1A promoter hypermethylation was increased among HCC patients compared to healthy people and those without HCC. Moreover, RASSF1A promoter hypermethylation was significantly related to HBV infection and tumor size, and showed associations with worse prognoses in HCC. Therefore, in addition to its diagnostic value, RASSF1A promoter hypermethylation may also be used as a valuable prognostic marker and an emerging target for anti-HCC treatment; further high-quality, well-designed prospective studies are needed to confirm the same.
Availability of data and materials
All the original data of the current study are available from the corresponding author on reasonable request.
Abbreviations
- RASSF1A:
-
RAS association domain family protein 1A
- MSP:
-
Methylation-specific polymerase chain reaction
- OR:
-
Odds ratio
- CI:
-
Confidence interval
- OS:
-
Overall survival
- DFS:
-
Disease-free survival
- TCGA:
-
The Cancer Genome Atlas
- NOS:
-
Newcastle-Ottawa scale
- LC:
-
Liver cancer
- HCC:
-
Hepatocellular carcinoma
- AUC:
-
Area under the curve
- ROC:
-
Receiver operating characteristic
- HBV:
-
Hepatitis B virus
- HBx:
-
Hepatitis B virus X protein
- HCV:
-
Hepatitis C virus
- AFP:
-
Alpha fetoprotein
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
Njei B, Rotman Y, Ditah I, Lim JK. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology. 2015;61(1):191–9.
Khan FS, Ali I, Afridi UK, Ishtiaq M, Mehmood R. Epigenetic mechanisms regulating the development of hepatocellular carcinoma and their promise for therapeutics. Hep Int. 2017;11(1):45–53.
Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8(4):286–98.
Park IY, Sohn BH, Yu E, Suh DJ, Chung YH, Lee JH, Surzycki SJ, Lee YI. Aberrant epigenetic modifications in hepatocarcinogenesis induced by hepatitis B virus X protein. Gastroenterology. 2007;132(4):1476–94.
Weng X, Wu J, Lv Z, Peng C, Chen J, Zhang C, He B, Tong R, Hu W, Ding C, et al. Targeting Mybbp1a suppresses HCC progression via inhibiting IGF1/AKT pathway by CpG islands hypo-methylation dependent promotion of IGFBP5. EBioMedicine. 2019;44:225–36.
Tian Y, Wong VW, Chan HL, Cheng AS. Epigenetic regulation of hepatocellular carcinoma in non-alcoholic fatty liver disease. Semin Cancer Biol. 2013;23(6):471–82.
Malpeli G, Innamorati G, Decimo I, Bencivenga M, Nwabo Kamdje AH, Perris R, Bassi C. Methylation dynamics of RASSF1A and its impact on cancer. Cancers. 2019;11(7):959.
Um TH, Kim H, Oh BK, Kim MS, Kim KS, Jung G, Park YN. Aberrant CpG island hypermethylation in dysplastic nodules and early HCC of hepatitis B virus-related human multistep hepatocarcinogenesis. J Hepatol. 2011;54(5):939–47.
Zhao ZH, Fan YC, Yang Y, Wang K. Association between Ras association domain family 1A promoter methylation and hepatocellular carcinoma: a meta-analysis. World J Gastroenterol. 2013;19(41):7189–96.
Moher D, Shamseer L, Clarke M, Ghersi D, Liberati A, Petticrew M, Shekelle P, Stewart LA. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev. 2015;4(1):1.
Higgins J GS. Cochrane handbook for systematic reviews of interventions version 5.1.0. http://handbook-5-1cochrane.org. 2011.
Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics. 1994;50(4):1088–101.
Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315(7109):629–34.
Duval S, Tweedie R. Trim and fill: a simple funnel-plot-based method of testing and adjusting for publication bias in meta-analysis. Biometrics. 2000;56(2):455–63.
Yu J, Ni M, Xu J, Zhang H, Gao B, Gu J, Chen J, Zhang L, Wu M, Zhen S, et al. Methylation profiling of twenty promoter-CpG islands of genes which may contribute to hepatocellular carcinogenesis. BMC cancer. 2002;2:29.
Zhang YJ, Ahsan H, Chen Y, Lunn RM, Wang LY, Chen SY, Lee PH, Chen CJ, Santella RM. High frequency of promoter hypermethylation of RASSF1A and p16 and its relationship to aflatoxin B1-DNA adduct levels in human hepatocellular carcinoma. Mol Carcinog. 2002;35(2):85–92.
Lee S, Lee HJ, Kim JH, Lee HS, Jang JJ, Kang GH. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am J Pathol. 2003;163(4):1371–8.
Schagdarsurengin U, Wilkens L, Steinemann D, Flemming P, Kreipe HH, Pfeifer GP, Schlegelberger B, Dammann R. Frequent epigenetic inactivation of the RASSF1A gene in hepatocellular carcinoma. Oncogene. 2003;22(12):1866–71.
Zhong S, Yeo W, Tang MW, Wong N, Lai PB, Johnson PJ. Intensive hypermethylation of the CpG island of Ras association domain family 1A in hepatitis B virus-associated hepatocellular carcinomas. Clin Cancer Res. 2003;9(9):3376–82.
Lehmann U, Berg-Ribbe I, Wingen LU, Brakensiek K, Becker T, Klempnauer J, Schlegelberger B, Kreipe H, Flemming P. Distinct methylation patterns of benign and malignant liver tumors revealed by quantitative methylation profiling. Clin Cancer Res. 2005;11(10):3654–60.
Park HJ, Yu E, Shim YH. DNA methyltransferase expression and DNA hypermethylation in human hepatocellular carcinoma. Cancer Lett. 2006;233(2):271–8.
Yeo W, Wong N, Wong WL, Lai PB, Zhong S, Johnson PJ. High frequency of promoter hypermethylation of RASSF1A in tumor and plasma of patients with hepatocellular carcinoma. Liver Int. 2005;25(2):266–72.
Calvisi DF, Ladu S, Gorden A, Farina M, Conner EA, Lee JS, Factor VM, Thorgeirsson SS. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130(4):1117–28.
Di Gioia S, Bianchi P, Destro A, Grizzi F, Malesci A, Laghi L, Levrero M, Morabito A, Roncalli M. Quantitative evaluation of RASSF1A methylation in the non-lesional, regenerative and neoplastic liver. BMC Cancer. 2006;6:89.
Oh BK, Kim H, Park HJ, Shim YH, Choi J, Park C, Park YN. DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation. Int J Mol Med. 2007;20(1):65–73.
Zhang YJ, Wu HC, Shen J, Ahsan H, Tsai WY, Yang HI, Wang LY, Chen SY, Chen CJ, Santella RM. Predicting hepatocellular carcinoma by detection of aberrant promoter methylation in serum DNA. Clin Cancer Res. 2007;13(8):2378–84.
Zhang C, Li Z, Cheng Y, Jia F, Li R, Wu M, Li K, Wei L. CpG island methylator phenotype association with elevated serum alpha-fetoprotein level in hepatocellular carcinoma. Clin Cancer Res. 2007;13(3):944–52.
Chan KC, Lai PB, Mok TS, Chan HL, Ding C, Yeung SW, Lo YM. Quantitative analysis of circulating methylated DNA as a biomarker for hepatocellular carcinoma. Clin Chem. 2008;54(9):1528–36.
Chang H, Yi B, Li L, Zhang HY, Sun F, Dong SQ, Cao Y. Methylation of tumor associated genes in tissue and plasma samples from liver disease patients. Exp Mol Pathol. 2008;85(2):96–100.
Nishida N, Nagasaka T, Nishimura T, Ikai I, Boland CR, Goel A. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology. 2008;47(3):908–18.
Su H, Zhao J, Xiong Y, Xu T, Zhou F, Yuan Y, Zhang Y, Zhuang SM. Large-scale analysis of the genetic and epigenetic alterations in hepatocellular carcinoma from Southeast China. Mutat Res. 2008;641(1–2):27–35.
Lou C, Du Z, Yang B, Gao Y, Wang Y, Fang S. Aberrant DNA methylation profile of hepatocellular carcinoma and surgically resected margin. Cancer Sci. 2009;100(6):996–1004.
Hu L, Chen G, Yu H, Qiu X. Clinicopathological significance of RASSF1A reduced expression and hypermethylation in hepatocellular carcinoma. Hep Intl. 2010;4(1):423–32.
Formeister EJ, Tsuchiya M, Fujii H, Shpyleva S, Pogribny IP, Rusyn I. Comparative analysis of promoter methylation and gene expression endpoints between tumorous and non-tumorous tissues from HCV-positive patients with hepatocellular carcinoma. Mutat Res. 2010;692(1–2):26–33.
Feng Q, Stern JE, Hawes SE, Lu H, Jiang M, Kiviat NB. DNA methylation changes in normal liver tissues and hepatocellular carcinoma with different viral infection. Exp Mol Pathol. 2010;88(2):287–92.
Saelee P, Wongkham S, Chariyalertsak S, Petmitr S, Chuensumran U. RASSF1A promoter hypermethylation as a prognostic marker for hepatocellular carcinoma. Asian Pac J Cancer Prev. 2010;11(6):1677–81.
Hua D, Hu Y, Wu YY, Cheng ZH, Yu J, Du X, Huang ZH. Quantitative methylation analysis of multiple genes using methylation-sensitive restriction enzyme-based quantitative PCR for the detection of hepatocellular carcinoma. Exp Mol Pathol. 2011;91(1):455–60.
Feng Y, Xue WJ, Li P, Sha ZY, Huang H, Rui L, Li HX, Mao QS. RASSF1A hypermethylation is associated with aflatoxin B1 and polycyclic aromatic hydrocarbon exposure in hepatocellular carcinoma. Hepatogastroenterology. 2012;59(118):1883–8.
Li Z, Zhang H, Yang J, Hao T, Li S. Promoter hypermethylation of DNA damage response genes in hepatocellular carcinoma. Cell Biol Int. 2012;36(5):427–32.
Mohamed NA, Swify EM, Amin NF, Soliman MM, Tag-Eldin LM, Elsherbiny NM. Is serum level of methylated RASSF1A valuable in diagnosing hepatocellular carcinoma in patients with chronic viral hepatitis C? Arab J Gastroenterol. 2012;13(3):111–5.
Xu B, Di J, Wang Z, Han X, Li Z, Luo X, Zheng Q. Quantitative analysis of RASSF1A promoter methylation in hepatocellular carcinoma and its prognostic implications. Biochem Biophys Res Commun. 2013;438(2):324–8.
Zhang X, Li HM, Liu Z, Zhou G, Zhang Q, Zhang T, Zhang J, Zhang C. Loss of heterozygosity and methylation of multiple tumor suppressor genes on chromosome 3 in hepatocellular carcinoma. J Gastroenterol. 2013;48(1):132–43.
Michailidi C, Soudry E, Brait M, Maldonado L, Jaffe A, Ili-Gangas C, Brebi-Mieville P, Perez J, Kim MS, Zhong X, et al. Genome-wide and gene-specific epigenomic platforms for hepatocellular carcinoma biomarker development trials. Gastroenterol Res Pract. 2014;2014:597164.
Zekri AR, Bahnasy AA, Shoeab FE, Mohamed WS, El-Dahshan DH, Ali FT, Sabry GM, Dasgupta N, Daoud SS. Methylation of multiple genes in hepatitis C virus associated hepatocellular carcinoma. J Adv Res. 2014;5(1):27–40.
Feng Y, Li P, Liu Y, Sha Z, Feng L, Wang F, Mao Q, Xue W. The association of Ala133Ser polymorphism and methylation in Ras association domain family 1A Gene with unfavorable prognosis of hepatocellular carcinoma. Hepatitis Monthly. 2015;15(10):e32145.
Huang W, Li T, Yang W, Chai X, Chen K, Wei L, Duan S, Li B, Qin Y. Analysis of DNA methylation in plasma for monitoring hepatocarcinogenesis. Genetic Test Mol Biomark. 2015;19(6):295–302.
Lin JC, Wu YC, Wu CC, Shih PY, Wang WY, Chien YC. DNA methylation markers and serum α-fetoprotein level are prognostic factors in hepatocellular carcinoma. Annals Hepatol. 2015;14(4):494–504.
Qu Z, Jiang Y, Li H, Yu DC, Ding YT. Detecting abnormal methylation of tumor suppressor genes GSTP1, P16, RIZ1, and RASSF1A in hepatocellular carcinoma and its clinical significance. Oncol Lett. 2015;10(4):2553–8.
Kanekiyo S, Iizuka N, Tsunedomi R, Tokumitsu Y, Hashimoto N, Tokuhisa Y, Maeda Y, Iida M, Sakamoto K, Tamesa T, et al. Preoperative serum methylation signature as prognostic tool after curative hepatectomy in patients with hepatocellular carcinoma. Anticancer Res. 2015;35(2):997–1007.
Villanueva A, Portela A, Sayols S, Battiston C, Hoshida Y, Méndez-González J, Imbeaud S, Letouzé E, Hernandez-Gea V, Cornella H, et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology. 2015;61(6):1945–56.
Dong X, He H, Zhang W, Yu D, Wang X, Chen Y. Combination of serum RASSF1A methylation and AFP is a promising non-invasive biomarker for HCC patient with chronic HBV infection. Diagn Pathol. 2015;10:133.
Araújo OC, Rosa AS, Fernandes A, Niel C, Villela-Nogueira CA, Pannain V, Araujo NM. RASSF1A and DOK1 Promoter Methylation Levels in Hepatocellular Carcinoma, Cirrhotic and Non-Cirrhotic Liver, and Correlation with Liver Cancer in Brazilian Patients. PLoS ONE. 2016;11(4):e0153796.
Liu ZJ, Huang Y, Wei L, He JY, Liu QY, Yu XQ, Li ZL, Zhang J, Li B, Sun CJ, et al. Combination of LINE-1 hypomethylation and RASSF1A promoter hypermethylation in serum DNA is a non-invasion prognostic biomarker for early recurrence of hepatocellular carcinoma after curative resection. Neoplasma. 2017;64(5):795–802.
Mansour LA, El Raziky M, Mohamed AA, Mahmoud EH, Hamdy S, El Sayed EH. Circulating hypermethylated RASSF1A as a molecular biomarker for diagnosis of hepatocellular carcinoma. Asian Pac J Cancer Prev. 2017;18(6):1637–43.
Wu HC, Yang HI, Wang Q, Chen CJ, Santella RM. Plasma DNA methylation marker and hepatocellular carcinoma risk prediction model for the general population. Carcinogenesis. 2017;38(10):1021–8.
Pasha HF, Mohamed RH, Radwan MI. RASSF1A and SOCS1 genes methylation status as a noninvasive marker for hepatocellular carcinoma. Cancer Biomark. 2019;24(2):241–7.
El-Bendary M, Nour D, Arafa M, Neamatallah M. Methylation of tumour suppressor genes RUNX3, RASSF1A and E-Cadherin in HCV-related liver cirrhosis and hepatocellular carcinoma. Br J Biomed Sci. 2020;77(1):35–40.
Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589–604.
Liu L, Tommasi S, Lee DH, Dammann R, Pfeifer GP. Control of microtubule stability by the RASSF1A tumor suppressor. Oncogene. 2003;22(50):8125–36.
Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet. 2000;25(3):315–9.
Agathanggelou A, Cooper WN, Latif F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 2005;65(9):3497–508.
Vos MD, Ellis CA, Bell A, Birrer MJ, Clark GJ. Ras uses the novel tumor suppressor RASSF1 as an effector to mediate apoptosis. J Biol Chem. 2000;275(46):35669–72.
Pefani DE, O’Neill E. Safeguarding genome stability: RASSF1A tumor suppressor regulates BRCA2 at stalled forks. Cell Cycle. 2015;14(11):1624–30.
Baksh S, Tommasi S, Fenton S, Yu VC, Martins LM, Pfeifer GP, Latif F, Downward J, Neel BG. The tumor suppressor RASSF1A and MAP-1 link death receptor signaling to Bax conformational change and cell death. Mol Cell. 2005;18(6):637–50.
Donninger H, Clark J, Rinaldo F, Nelson N, Barnoud T, Schmidt ML, Hobbing KR, Vos MD, Sils B, Clark GJ. The RASSF1A tumor suppressor regulates XPA-mediated DNA repair. Mol Cell Biol. 2015;35(1):277–87.
Fausti F, Di Agostino S, Sacconi A, Strano S, Blandino G. Hippo and rassf1a pathways: a growing affair. Mol Biol Int. 2012;2012:307628.
Papaspyropoulos A, Bradley L, Thapa A, Leung CY, Toskas K, Koennig D, Pefani DE, Raso C, Grou C, Hamilton G, et al. RASSF1A uncouples Wnt from Hippo signalling and promotes YAP mediated differentiation via p73. Nat Commun. 2018;9(1):424.
Ahn EY, Kim JS, Kim GJ, Park YN. RASSF1A-mediated regulation of AREG via the Hippo pathway in hepatocellular carcinoma. Mol Cancer Res. 2013;11(7):748–58.
Huang ZH, Hu Y, Hua D, Wu YY, Song MX, Cheng ZH. Quantitative analysis of multiple methylated genes in plasma for the diagnosis and prognosis of hepatocellular carcinoma. Exp Mol Pathol. 2011;91(3):702–7.
Charostad J, Astani A, Goudarzi H, Faghihloo E. DNA methyltransferases in virus-associated cancers. Rev Med Virol. 2019;29(2):e2022.
Kao WY, Yang SH, Liu WJ, Yeh MY, Lin CL, Liu CJ, Huang CJ, Lin SM, Lee SD, Chen PJ, et al. Genome-wide identification of blood DNA methylation patterns associated with early-onset hepatocellular carcinoma development in hepatitis B carriers. Mol Carcinog. 2017;56(2):425–35.
Ye C, Tao R, Cao Q, Zhu D, Wang Y, Wang J, Lu J, Chen E, Li L. Whole-genome DNA methylation and hydroxymethylation profiling for HBV-related hepatocellular carcinoma. Int J Oncol. 2016;49(2):589–602.
Okamoto Y, Shinjo K, Shimizu Y, Sano T, Yamao K, Gao W, Fujii M, Osada H, Sekido Y, Murakami S, et al. Hepatitis virus infection affects DNA methylation in mice with humanized livers. Gastroenterology. 2014;146(2):562–72.
Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61(9):3573–7.
Jang TJ, Kim DI, Shin YM, Chang HK, Yang CH. P16(INK4A) promoter hypermethylation of non-tumorous tissue adjacent to gastric cancer is correlated with glandular atrophy and chronic inflammation. Int J Cancer. 2001;93(5):629–34.
Zhao Z, Hu Y, Shen X, Lao Y, Zhang L, Qiu X, Hu J, Gong P, Cui H, Lu S, et al. HBx represses RIZ1 expression by DNA methyltransferase 1 involvement in decreased miR-152 in hepatocellular carcinoma. Oncol Rep. 2017;37(5):2811–8.
Dang S, Zhou J, Chen Y, Chen P, Ji M, Shi B, Yang Q, Hou P. Dynamic expression of ZNF382 and its tumor-suppressor role in hepatitis B virus-related hepatocellular carcinogenesis. Oncogene. 2019;38(24):4804–19.
Arzumanyan A, Friedman T, Kotei E, Ng IO, Lian Z, Feitelson MA. Epigenetic repression of E-cadherin expression by hepatitis B virus x antigen in liver cancer. Oncogene. 2012;31(5):563–72.
Lo KW, Chung GTY, To KF. Deciphering the molecular genetic basis of NPC through molecular, cytogenetic, and epigenetic approaches. Semin Cancer Biol. 2012;22(2):79–86.
Lo KW, Kwong J, Hui ABY, Chan SYY, To KF, Chan SC, Chow LSN, Teo PML, Johnson PJ, Huang DP. High frequency of promoter hypermethylation of RASSF1A in nasopharyngeal carcinoma. Cancer Res. 2001;61(10):3877–81.
Shivakumar L, Minna J, Sakamaki T, Pestell R, White MA. The RASSF1A tumor suppressor blocks cell cycle progression and inhibits cyclin D1 accumulation. Mol Cell Biol. 2002;22(12):4309–18.
Lassaletta L, Patrón M, González T, Martinez-Glez V, Rey JA, Gavilan J. RASSF1A methylation and cyclin D1 expression in vestibular schwannomas. Acta Neuropathol. 2007;114(4):431–3.
Lambert MP, Paliwal A, Vaissière T, Chemin I, Zoulim F, Tommasino M, Hainaut P, Sylla B, Scoazec JY, Tost J, et al. Aberrant DNA methylation distinguishes hepatocellular carcinoma associated with HBV and HCV infection and alcohol intake. J Hepatol. 2011;54(4):705–15.
van Vlodrop IJ, Niessen HE, Derks S, Baldewijns MM, van Criekinge W, Herman JG, van Engeland M. Analysis of promoter CpG island hypermethylation in cancer: location, location, location! Clin Cancer Res. 2011;17(13):4225–31.
Yao S, Hu Q, Kerns S, Yan L, Onitilo AA, Misleh J, Young K, Lei L, Bautista J, Mohamed M, et al. Impact of chemotherapy for breast cancer on leukocyte DNA methylation landscape and cognitive function: a prospective study. Clin Epigenet. 2019;11(1):45.
Harat M, Blok M, Harat A, Soszyńska K. The impact of adjuvant radiotherapy on molecular prognostic markers in gliomas. OncoTargets Ther. 2019;12:2215–24.
Heimbach JK, Kulik LM, Finn RS, Sirlin CB, Abecassis MM, Roberts LR, Zhu AX, Murad MH, Marrero JA. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology. 2018;67(1):358–80.
EASL Clinical Practice Guidelines. Management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236.
Altman DG, McShane LM, Sauerbrei W, Taube SE. Reporting recommendations for Tumor Marker Prognostic Studies (REMARK): explanation and elaboration. PLoS Med. 2012;9(5):e1001216.
Dammann RH, Richter AM, Jiménez AP, Woods M, Küster M, Witharana C. Impact of natural compounds on DNA methylation levels of the tumor suppressor gene RASSF1A in cancer. Int J Mol Sci. 2017;18(10):2160.
Morrissey C, Martinez A, Zatyka M, Agathanggelou A, Honorio S, Astuti D, Morgan NV, Moch H, Richards FM, Kishida T, et al. Epigenetic inactivation of the RASSF1A 3p213 tumor suppressor gene in both clear cell and papillary renal cell carcinoma. Cancer Res. 2001;61(19):7277–81.
Flotho C, Claus R, Batz C, Schneider M, Sandrock I, Ihde S, Plass C, Niemeyer CM, Lübbert M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia. 2009;23(6):1019–28.
Balch C, Yan P, Craft T, Young S, Skalnik DG, Huang TH, Nephew KP. Antimitogenic and chemosensitizing effects of the methylation inhibitor zebularine in ovarian cancer. Mol Cancer Ther. 2005;4(10):1505–14.
Acknowledgements
We would like to thank Editage (https://www.editage.com) for English language editing.
Funding
This study supported by grants funded by Tsinghua University-Peking Union Medical College Hospital Cooperation Project (Grant No. PTQH201904552), Peking Union Medical College Graduate Student Innovation Fund (Grant No. 2019-1002-45), National Natural Science Foundation of China, No. 81972698, Chinese Academy of Medical Sciences (CAMS) Initiative for Innovative Medicine (No. 2017-I2M-4-002) and Chen Xiao-Ping Foundation for the Development of Science and Technology of Hubei Province (No. CXPJJH11900001-2019215).
Author information
Authors and Affiliations
Contributions
GX, XZ and JX designed the study, performed the literature search and screening, performed the data analyses and wrote the manuscript. YX and BJ designed the study, retrieved the literature and data, analyzed the retrieved data and participated in the writing of manuscript. LS, HY, FX and SD assisted in the designing of the study, performed the literature search and screening, assisted in the data analyses and participated in the writing of manuscript. YM and HX designed the study and supervised the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Additional tables and figures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xu, G., Zhou, X., Xing, J. et al. Identification of RASSF1A promoter hypermethylation as a biomarker for hepatocellular carcinoma. Cancer Cell Int 20, 547 (2020). https://doi.org/10.1186/s12935-020-01638-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-020-01638-5