
Abstract
Background
The hypoxic tumor microenvironment accelerates the invasion and migration of colorectal cancer (CRC) cells. The aim of this study was to develop and validate a hypoxia gene signature for predicting the outcome in stage I/II CRC patients that have limited therapeutic options.
Methods
The hypoxic gene signature (HGS) was constructed using transcriptomic data of 309 CRC patients with complete clinical information from the CIT microarray dataset. A total of 1877 CRC patients with complete prognostic information in six independent datasets were divided into a training cohort and two validation cohorts. Univariate and multivariate analyses were conducted to evaluate the prognostic value of HGS.
Results
The HGS consisted of 14 genes, and demarcated the CRC patients into the high- and low-risk groups. In all three cohorts, patients in the high-risk group had significantly worse disease free survival (DFS) compared with those in the low risk group (training cohort—HR = 4.35, 95% CI 2.30–8.23, P < 0.001; TCGA cohort—HR = 2.14, 95% CI 1.09–4.21, P = 0.024; meta-validation cohort—HR = 1.91, 95% CI 1.08–3.39, P = 0.024). Compared to Oncotype DX, HGS showed superior predictive outcome in the training cohort (C-index, 0.80 vs 0.65) and the validation cohort (C-index, 0.70 vs 0.61). Pathway analysis of the high- and low-HGS groups showed significant differences in the expression of genes involved in mTROC1, G2-M, mitosis, oxidative phosphorylation, MYC and PI3K–AKT–mTOR pathways (P < 0.005).
Conclusion
Hypoxic gene signature is a satisfactory prognostic model for early stage CRC patients, and the exact biological mechanism needs to be validated further.
Similar content being viewed by others


Background
Colorectal cancer (CRC) is one of the most commonly diagnosed cancers worldwide, and ranks third in terms of morbidity and mortality [1]. About half of the CRC patients are at stages I/II, and more than a quarter of the early-stage patients (I–III) relapse after initial treatment [2]. Hypoxia is a common feature of solid tumors, and contributes to tumor progression and therapeutic recalcitrance. Intra-tumoral hypoxia is considered to be an indicator of poor prognosis [3, 4], and even regulates genes involved in the invasion and migration of CRC cells, which is consistent with recent reports indicating an association between lack of oxygen and distant metastasis [5,6,7]. Hypoxia reduces the efficacy of not only surgical resection [8], but also radiotherapy and chemotherapy [9, 10]. Only limited options are available at present for hypoxia-related targeted therapies, and there is no unequivocal evidence from clinical trials as yet regarding their efficacy, likely due to the lack of individual-based treatment [8, 11, 12]. Therefore, an accurate and non-invasive method is needed to assess tumor hypoxia. To this end, we identified a hypoxia-related gene signature (HGS) from CRC-specific transcriptomes through high-throughput expression analysis. The HGS demarcated the stage I/II CRC patients into distinct prognostic groups, and functional and pathway analyses provided new insights in the mechanism of CRC recurrence.
Materials and methods
Patients
The gene expression profiles of CRC tissue samples obtained from six public cohorts, including 309 CRC patients from the CIT/GSE39582 gene microarray dataset that served as the discovery cohort, were retrospectively analyzed. The two largest individual data sets—CIT/GSE39582 and The Cancer Genome Atlas (TCGA)—were used for training and independent validation. The meta-validation cohort consisted of the remaining four microarray data sets—GSE14333, GSE17536, GSE37892 and GSE33113—which were obtained from the Gene Expression Omnibus (GEO) database. All datasets are from the GPL570 platform ([HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array). TCGA cohort data was downloaded from Broad GDAC Firehose, and the other data sets were obtained directly in their processed format from the GEO database through Bioconductor package ‘GEOquery’. Transcripts per million (TPM) of level 3 RNA-Seq data in log2 scale was applied to calibrate the gene expression levels in TCGA cohort. The ‘combat’ algorithm of the R package ‘sva’ and the z-scores were used to correct the batch effects, in order to standardize microarray data across multiple experiments and compare them independent of the original hybridization intensities. The data of 1877 CRC patients enrolled from Sep 27 to Dec 26, 2018 was also included.
Construction and validation of HGS
To construct a prognostic HGS, annotated functional database MSigDB (version 6.2) [13,14,15] was used to identify a list of hypoxia-related genes with the keyword “hypoxia”, and the HGSs measured by all platforms were selected. The log-rank test was used with 1000 randomizations (80% of samples each time) to evaluate the association between each HGS and clinical outcome in the training dataset. Genes that were repeatedly significant were selected as the candidates of the hypoxia signature. To minimize the risk of over-fitting, Cox proportional hazards regression model was applied with the least absolute shrinkage and selection operator (LASSO) (glmnet, version 2.0-16). The penalty parameter was estimated by tenfold cross-validation in the training data set at 1 SE beyond the minimum partial likelihood deviance.
A time-dependent receiver operating characteristic (ROC) curve (survival ROC, version 1.0.3) at 5 years was plotted using Kaplan–Meier estimation, and used to determine the optimal HGS cutoff to separate patients in the training data set into the low-risk and high-risk groups. The HGS corresponding to the shortest distance between the ROC curve and the point representing 100% true positive rate and 0% false-positive rate was used as the cutoff value. Univariate analysis was used to evaluate the prognostic value of the HGS in stage I/II CRC patients, and in patients at all stages in the training and independent validation cohorts. In the multivariate analyses, HGS was combined with other clinical and pathological variables.
Functional annotation and analysis
To investigate the biological characteristics of the HGS, enrichment analysis was conducted for differentially expressed genes (DEGs) between the risk groups in TCGA CRC data set using R package ‘gProfileR’. Gene Set Enrichment Analysis (GSEA) was further performed using Bioconductor package ‘HTSanalyzeR’ to predict the significant pathways [16].
Statistical analysis
All statistical analysis was performed in R software (version 3.5.1; http://www.Rproject.org). Descriptive statistics were computed for all variables, and expressed as mean ± standard deviation (SD) or medians and interquartile ranges (IQR) for continuous factors, and as frequencies for categorical factors. Continuous values were compared using Student-t tests between different groups. Log-rank test was used to evaluate results of the univariate analysis of HGS and other clinico-pathological factors with disease free survival (DFS). Multivariate analysis was performed with the Cox proportional hazards regression model. The C-index was calculated by ‘survcomp’ (version 1.32.0). P values less than 0.5 were considered statistically significant.
Results
The establishment of HGS
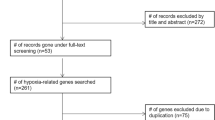
We analyzed the CIT gene microarray dataset (GSE39582) and created the discovery subset with 309 eligible CRC patients (Fig. 1). After exclusion of genes with MAD > 0.5 and less median expression, 1636 genes were retained for further analysis. Following selection of 80% of the repeatable genes via 1000 random Cox univariate regressions, we identified 106 genes that were associated with DFS, of which 14 hypoxia-related genes were selected to construct the HGS using LASSO Cox regression for stage I/II CRC patients (Fig. 2). The risk scores were calculate using the formula derived from the Cox model as follows: Risk score = − 0.013 × exp(mRNA expression level of MDM2) + 0.0733 × exp(mRNA expression level of VEGFA) + 0.112 × exp(mRNA expression level of ORAI3) + 0.043 × exp(mRNA expression level of MVD) − 0.060 × exp(mRNA expression level of TRAF3) − 0.003 × exp(mRNA expression level of CYB5R3) − 0.003 × exp(mRNA expression level of ZBTB44) − 0.045 × exp(mRNA expression level of CASP6) + 0.082 × exp(mRNA expression level of FBP1) − 0.026 × exp(mRNA expression level of CCNG1) − 0.032 × exp(mRNA expression level of FAM117B) − 0.025 × exp(mRNA expression level of PRELID2) − 0.129 × exp(mRNA expression level of RRP1B) + 0.014 × exp(mRNA expression level of GAS6). Based on time-dependent ROC curve analysis, the optimal cutoff of HGS for stratifying patients in the training set into the high and low risk groups was determined to be a satisfactory RFS cutoff at 5 years (Fig. 3b, e and h). The incidence of tumor recurrence was higher among the patients in the high-risk group compared to the low-risk group when the entire CIT dataset (n = 566) was used as a training cohort (Fig. 4a, P < 0.001).

Schematic flow chart of the study procedure

The establishment of hypoxic gene signature (HGS) using 14 hypoxia-associated genes from the LASSO COX regression

a Distribution of the HGS risk score in stage I/II CRC cohort and its correlation to recurrence in the training, TCGA and meta-validation cohorts, with risk scores as the continuous variable for individual patients. The DFS and recurrence in the different hypoxia risk groups of training cohort (b), TCGA cohort (e) and meta-validation cohort (h). Kaplan–Meier curves comparing survival of patients with low or high hypoxia risk in training cohort (c), TCGA cohort (f) and meta-validation cohort (i)

a Distribution of the HGS risk score and its correlation to recurrence in the training, TCGA cohort and meta-validation cohort, with risk scores as the continuous variable for individual patients. The DFS and recurrence in the different hypoxia risk groups of training cohort (b), TCGA cohort (e) and meta-validation cohort (h). Kaplan–Meier curves comparing survival of patients with low or high hypoxia risk in training cohort (c), TCGA cohort (f) and meta-validation cohort (i). P-values were calculated using log-rank tests and HR is short for hazard ratio
Validation of HGS
The prognostic significance of HGS was assessed with additional CRC transcription data sets that included clinical and prognostic data. Clinicopathological characteristics of three cohorts are listed in Table 1. The validation datasets consisted of TCGA datasets (n = 624) and the meta-validation cohort (n = 687), including GSE17536, GSE33113, GSE37892 and GSE14333. No significant difference was seen between the clinico-pathological features of the training and validation cohorts (Table 2). The DFS was significantly higher in the low-risk compared to the high-risk HGS group in all three cohorts (training cohort: HR = 4.35, 95% CI 2.30–8.23, P < 0.001; validation: HR = 2.14, 95% CI 1.09–4.21, P = 0.024 and meta-validation cohort: HR = 1.91, 95% CI 1.08–3.39, P = 0.024) (Fig. 3c, f and i). We compared HGS with Oncotype DX to further evaluate its prognostic value and robustness (Table 3), and found that HGS had a more optimized C-index in both training and TCGA cohorts (training cohort: 0.80 vs 0.65, TCGA cohort: 0.70 vs 0.61, Table 3). Further data mining indicated a prognostic value of HGS in all CRC cohorts (GSE39582 cohort: HR = 2.01, 95% CI 1.46–2.77, P < 0.001; TCGA cohort: HR = 1.72, 95% CI 1.14–2.62, P = 0.010; meta-validation cohort: HR = 1.94, 95% CI 1.34–2.8, P < 0.001) (Fig. 4c, f and i). Similar results were obtained in the AUC analysis (Fig. 4b, e and h).
Independent influencing factors of HGS
Univariate and multivariate analyses were used to determine whether patient age, gender, tumor stage, tumor location, pathological gene status and HGS were associated with prognosis in stage I/II CRC patients. The univariate analysis showed that HGS was significantly associated with a poor outcome in the three cohorts, (GSE39582 cohort: HR = 8.66, 95% CI 4.37–17.17, P < 0.001; TCGA cohort: HR = 2.59, 95% CI 1.08–6.25, P = 0.04; and meta-validation cohort: HR = 8.25, 95% CI 3.09–22.03, P < 0.001, Table 2). After adjusting for other factors in the multivariate analysis, it remained an independent prognostic factor (GSE39582 cohort, HR = 7.54, 95% CI 3.78–15.06, P < 0.001; TCGA cohort, HR = 2.59, 95% CI 1.08–6.25, P = 0.04; and meta-validation cohort, HR = 7.25, 95% CI 2.72–19.29, P < 0.001, Table 2).
Pathways analysis of HGS predicted risk group
Gene ontology and KEGG pathway enrichment analysis of the DEGs and the GSEA showed a significant enrichment of metabolic pathways such as mTROC1 (P = 0.0001), G2-M (P = 0.0001), mitosis (P = 0.0001), oxidative phosphorylation (P = 0.0001), MYC (P = 0.0001), and PI3K–AKT–mTOR (P = 0.0039) (Fig. 5).

a Functional annotation of the HGS. Enrichment analysis of the DEGs between risk groups. b GSEA showed that mTROC1, G2-M, mitosis, oxidative phosphorylation, MYC and PI3K–AKT–mTOR were downregulated in high hypoxia risk patients
Discussion
The current therapeutic modality for early stage CRC is surgical resection. Nevertheless, the recurrence rate of stage I/II CRC patients after surgery is still higher than 20% [17]. Despite identifying numerous genes that affect the recurrence and metastasis of CRC [18, 19], no prognostic gene signature has been validated so far. Effective prognostic biomarkers are therefore urgently needed to predict the DFS rate and risk of relapse after treatment in early-stage CRC patients. In this study, we developed a novel predictive hypoxia-related 14-gene signature for CRC, and validated it in multiple cohorts. The results suggest that the HGS can successfully predict the DFS of CRC patients after treatment.
Oxygen provides energy for cell growth and division, and is a key signaling molecule. The hypoxia inducible factors (HIFs) respond to changes in oxygen levels and cellular energy status, and trigger a transcriptional program [20] that mediates malignant transformation and progression. Not surprisingly, lack of oxygen and overexpression of HIF is associated with poor prognosis in cancer patients [21, 22]. Furthermore, tumor cells induce pro-angiogenic factors to vascularize the tumor in order to survive and proliferate under hypoxic condition, which are regulated along with the hypoxia-related genes [23]. In fact, HIF inhibitors also improve the efficacy of anti-angiogenesis drugs during cancer treatment [21, 22, 24]. Consistent with these previous studies, we found that hypoxia-related genes worsened CRC prognosis by affecting genes involved in the cell cycle, indicating that hypoxia-related drug targets can potentially improve CRC prognosis.
Several studies have shown an association between tumor hypoxia and poor therapeutic outcome in cancer patients. Oxygen deficiency reduces the efficacy of surgical resection and increases metastatic potential of tumors [25, 26]. The current endogenous markers of hypoxia cannot accurately monitor intra-tumor oxygen levels, which limits the efficacy of hypoxia-targeting drugs [8, 27]. The HGS stratified the stage I/II CRC patients into high- and low-risk groups that differed significantly in terms of DFS during a 5-year follow-up. The C index results of the 14-gene hypoxia signature showed its clinical superiority to Oncotype DX. This novel prognostic tool can thus identify CRC patients with highly hypoxic tumors that at risk of treatment failure, and enable clinicians to make informed decisions regarding treatment regimens. It may also help in calculating the possibility of tumor recurrence after surgery.
Several research groups have developed hypoxia-targeted therapy against solid tumors to improve patient survival, although clinical trials have not yielded satisfactory results [27,28,29]. Therefore, there is an urgent need for better therapeutic targets to improve the prognosis of CRC patients. We observed a significant enrichment of cell cycle/metabolism-related genes and functions, such as mTROC1, E2F, G2-M, mitosis, oxidative phosphorylation, MYC and PI3K–AKT–mTOR (P < 0.005), in the high-risk, low DFS group. Previous studies have found a correlation between these targets and CRC development, although they did not link these targets to tumor hypoxia [30,31,32,33,34,35]. Further studies are needed to clarify the effects of hypoxia on cell cycle in order to identify more targets and improve the prognosis of early stage CRC patients.
In conclusion, we identified a prognostic hypoxia-associated gene signature using genome-wide analysis to predict DFS in patients with stage I/II CRC. These hypoxia-associated DEGs are potential therapeutic targets against CRC. However, our study is beset with the limitations associated with all retrospective studies, in addition to systematic errors resulting from analyzing samples from disparate databases. Therefore, further clinical and pharmacological tests are needed to validate our results.
Conclusions
We developed a novel HGS to stratify stage I and II CRC patients into high- and low-risk groups with greater accuracy compared to the currently used clinicopathological risk factors. A “risk prediction model” was also constructed using the HGS, the scores of which can be readily applied to independent prospective cohorts. HGS is a highly promising prognostic tool for personalized treatment regimens and clinical management of stage I/II CRC patients.
Availability of data and materials
TCGA cohort data was downloaded from Broad GDAC Firehose (http://gdac.broadinstitute.org/). The datasets generated and analyzed during the current study are available in the GSE39582 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE39582), TCGA (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga), GSE14333 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE14333), GSE17536 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE17536), GSE37892 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37892) and GSE33113 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33113).
Abbreviations
- CRC:
-
colorectal cancer
- HGS:
-
hypoxic gene signature
- TCGA:
-
The Cancer Genome Atlas
- GEO:
-
Gene Expression Omnibus
- TPM:
-
Transcripts per million
- ROC:
-
receiver operating characteristic
- DEGs:
-
differentially expressed genes
- GSEA:
-
Gene Set Enrichment Analysis
- SD:
-
standard deviation
- IQR:
-
interquartile ranges
- DFS:
-
disease free survival
- HIFs:
-
hypoxia inducible factors
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
van der Stok EP, Spaander MCW, Grunhagen DJ, Verhoef C, Kuipers EJ. Surveillance after curative treatment for colorectal cancer. Nat Rev Clin Oncol. 2017;14(5):297–315.
Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58(7):1408–16.
Vaupel P, Mayer A, Hockel M. Tumor hypoxia and malignant progression. Methods Enzymol. 2004;381:335–54.
Yang L, Zhang W, Wang Y, et al. Hypoxia-induced miR-214 expression promotes tumour cell proliferation and migration by enhancing the Warburg effect in gastric carcinoma cells. Cancer Lett. 2018;414:44–56.
Li H, Rokavec M, Jiang L, Horst D, Hermeking H. Antagonistic effects of p53 and HIF1A on microRNA-34a regulation of PPP1R11 and STAT3 and hypoxia-induced epithelial to mesenchymal transition in colorectal cancer cells. Gastroenterology. 2017;153(2):505–20.
Kim CW, Oh ET, Kim JM, et al. Hypoxia-induced microRNA-590-5p promotes colorectal cancer progression by modulating matrix metalloproteinase activity. Cancer Lett. 2018;416:31–41.
Walsh JC, Lebedev A, Aten E, Madsen K, Marciano L, Kolb HC. The clinical importance of assessing tumor hypoxia: relationship of tumor hypoxia to prognosis and therapeutic opportunities. Antioxid Redox Signal. 2014;21(10):1516–54.
Li F, Mei H, Gao Y, et al. Co-delivery of oxygen and erlotinib by aptamer-modified liposomal complexes to reverse hypoxia-induced drug resistance in lung cancer. Biomaterials. 2017;145:56–71.
Rechavi G, Brok-Simoni F, Ben-Bassat I, Ramot B. Spurious haemoglobinopathy. Lancet. 1986;1(8488):1035.
Betensky RA, Louis DN, Cairncross JG. Influence of unrecognized molecular heterogeneity on randomized clinical trials. J Clin Oncol. 2002;20(10):2495–9.
Semenza GL. Angiogenesis in ischemic and neoplastic disorders. Annu Rev Med. 2003;54:17–28.
Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–50.
Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–40.
Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–25.
Wang X, Terfve C, Rose JC, Markowetz F. HTSanalyzeR: an R/Bioconductor package for integrated network analysis of high-throughput screens. Bioinformatics. 2011;27(6):879–80.
Markle B, May EJ, Majumdar AP. Do nutraceutics play a role in the prevention and treatment of colorectal cancer? Cancer Metastasis Rev. 2010;29(3):395–404.
Al-Temaimi RA, Tan TZ, Marafie MJ, Thiery JP, Quirke P, Al-Mulla F. Identification of 42 genes linked to stage II colorectal cancer metastatic relapse. Int J Mol Sci. 2016;17(5):598.
De Rosa M, Pace U, Rega D, et al. Genetics, diagnosis and management of colorectal cancer. Oncol Rep. 2015;34(3):1087–96.
Moniz S, Biddlestone J, Rocha S. Grow(2): the HIF system, energy homeostasis and the cell cycle. Histol Histopathol. 2014;29(5):589–600.
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–32.
Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. 2004;9(Suppl 5):10–7.
Pahlman S, Mohlin S. Hypoxia and hypoxia-inducible factors in neuroblastoma. Cell Tissue Res. 2018;372(2):269–75.
Rapisarda A, Uranchimeg B, Scudiero DA, et al. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002;62(15):4316–24.
Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996;56(19):4509–15.
Sullivan R, Graham CH. Hypoxia-driven selection of the metastatic phenotype. Cancer Metastasis Rev. 2007;26(2):319–31.
Bennewith KL, Dedhar S. Targeting hypoxic tumour cells to overcome metastasis. BMC Cancer. 2011;11:504.
Overgaard J. Clinical evaluation of nitroimidazoles as modifiers of hypoxia in solid tumors. Oncol Res. 1994;6(10–11):509–18.
Overgaard J. Hypoxic modification of radiotherapy in squamous cell carcinoma of the head and neck—a systematic review and meta-analysis. Radiother Oncol. 2011;100(1):22–32.
Kim T, Jeon YJ, Cui R, et al. Role of MYC-regulated long noncoding RNAs in cell cycle regulation and tumorigenesis. J Natl Cancer Inst. 2015. https://doi.org/10.1093/jnci/dju505.
Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer. 2010;102(12):1746–52.
Oliveira A, Beyer G, Chugh R, et al. Triptolide abrogates growth of colon cancer and induces cell cycle arrest by inhibiting transcriptional activation of E2F. Lab Invest. 2015;95(6):648–59.
Satoh K, Yachida S, Sugimoto M, et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc Natl Acad Sci USA. 2017;114(37):E7697–706.
Sisinni L, Maddalena F, Condelli V, et al. TRAP1 controls cell cycle G2-M transition through the regulation of CDK1 and MAD2 expression/ubiquitination. J Pathol. 2017;243(1):123–34.
Wang Y, Kuang H, Xue J, Liao L, Yin F, Zhou X. LncRNA AB073614 regulates proliferation and metastasis of colorectal cancer cells via the PI3K/AKT signaling pathway. Biomed Pharmacother. 2017;93:1230–7.
Acknowledgements
Not applicable.
Funding
This work was supported by National Key Clinical Discipline, the Fundamental Research Funds for the young teacher training program of sun yat-sen university (18ykpy02) and “5010 Clinical Research Programme” of Sun Yat-sen University (2010012).
Author information
Authors and Affiliations
Contributions
YZ, YT, YR, XL and FG contributed to study concept and design, acquisition, analysis, interpretation of data and drafting of the manuscript. YT, JX, ZY, YC, JK, CL, XC, XL, FG, XW and PL contributed to data collections and manuscript review. YZ, XL, FG, XW and PL contributed to study concept and design, analysis and interpretation of data and critical revision of the manuscript for important intellectual content. XW and PL supervised the study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This is a retrospective trial from public datasets with minimal risk and we petition for waiver of ethics consent.
Consent for publication
We have obtained consents to publish this paper from all the participants of this study.
Competing of interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zou, Yf., Rong, Ym., Tan, Yx. et al. A signature of hypoxia-related factors reveals functional dysregulation and robustly predicts clinical outcomes in stage I/II colorectal cancer patients. Cancer Cell Int 19, 243 (2019). https://doi.org/10.1186/s12935-019-0964-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-019-0964-1