
Abstract
Regulation of amino acid’s biosynthetic pathway is of significant importance to maintain homeostasis and cell functions. Amino acids regulate their biosynthetic pathway by end-product feedback inhibition of enzymes catalyzing committed steps of a pathway. Discovery of new feedback resistant enzyme variants to enhance industrial production of amino acids is a key objective in industrial biotechnology. Deregulation of feedback inhibition has been achieved for various enzymes using in vitro and in silico mutagenesis techniques. As enzyme’s function, its substrate binding capacity, catalysis activity, regulation and stability are dependent on its structural characteristics, here, we provide detailed structural analysis of all feedback sensitive enzyme targets in amino acid biosynthetic pathways. Current review summarizes information regarding structural characteristics of various enzyme targets and effect of mutations on their structures and functions especially in terms of deregulation of feedback inhibition. Furthermore, applicability of various experimental as well as computational mutagenesis techniques to accomplish feedback resistance has also been discussed in detail to have an insight into various aspects of research work reported in this particular field of study.
Similar content being viewed by others

Introduction
Product feedback inhibition of allosteric enzymes is of paramount importance in biotechnological industries for discovery of efficient microbial strains for increased production of metabolites of interest. Discovery of product-feedback inhibition traced back to 1950’s and was first reported by Novick and Szilard for the tryptophan biosynthetic pathway and has been reported as cornerstone for regulation of cell functions and control fluxes for optimal growth in microorganisms [1, 2]. Allosteric regulation of proteins is a fundamental mechanism of cellular control e.g. regulation of the enzymes involved in biosynthesis of amino acids, nucleotides and vitamins. In case of amino acid feedback inhibition, the first enzyme in the pathway is an allosteric enzyme that binds to the end product (i.e. amino acid) which alters its active site so that it cannot mediate the enzymatic reaction needed to initiate the pathway. Ultimately, pathway is shut down as long as adequate amounts of the end product are present but inhibition is relieved and the enzyme regains its activity if the end product is used up or disappears. Owing to role of amino acids as building block of life and their wide applications in agriculture, pharmaceutical and cosmetics industries, the chemical industry is focused on various synthetic strategies for production of these biochemically distant compounds. Amino acids production industry is growing day by day at an annual rate of 7%, as reported previously [3]. Allosteric feedback inhibition of the committed step in amino acids biosynthetic pathways is thought to maintain homeostasis of end-products [4]. The consequences of dysregulating these enzymes were mainly studied in vitro [5] or in the context of biotechnological overproduction strains [6].
Extensive studies have been reported for deregulation of feedback inhibition of various amino acids biosynthetic pathways using structural information of target enzyme, in vitro and in silico mutagenesis techniques. Most allosterically regulated enzymes are oligomeric in structural composition (i.e. made up of two or more polypeptide chains) having more than one active site and allosteric sites. Although reviews over the metabolic engineering strategies of amino acid biosynthetic pathways have been reported previously [7] still structural and functional aspects of feedback inhibition is not reviewed yet. An enzyme’s function is intrinsically linked to its three dimensional structure, determining how it performs substrate binding, catalysis and regulation. The relation of enzyme structure to its functions signifies the importance of structural insights of enzyme. Keeping in view the importance of structural aspects of enzyme, here, we provide detailed insight into various amino acid biosynthetic pathways, structural details of target enzymes feedback inhibited by amino acid (each case), mutagenesis approaches (both in vitro and in silico) used to incorporate structural and conformational changes to deregulate their inhibition tendency. The structure, design and mechanism of product feedback inhibition for all enzyme targets have been discussed in detail to provide an insight into structural and functional basis of amino acid feedback inhibition. Current review will provide guidelines for designing of better feedback resistant enzymes and will facilitate biotechnologists for discovery of novel enzyme variants for increased production of amino acids at industrial scale.
Small molecule regulation of enzyme activities
Regulation of enzyme activity has profound effects on cell functions with huge practical applications. Enzyme activities can be regulated either by dissociation or binding of regulators and effectors causing conformational or structural changes that ultimately determine their catalytic activity [8, 9]. Small molecule regulation of enzyme functions has garnered much interest and plenty of new molecules have been identified with specific regulatory functions. Use of small molecule for controlling gene-product activity to precisely elucidate functions of protein target has huge significance and is termed as chemical genetics. Protein functions are regulated in two ways (1) loss of function using small molecule inhibitors, (2) gain of function using small molecule activators [10]. Cell metabolisms are controlled by various metabolic pathways and interlinked networks to generate energy alongside biomass that is transmitted to other neighboring cells. Two well established methods to regulate cell metabolism are: (a) genetic regulation, (b) small molecule inhibition or activation (For instance allosteric inhibition) [11,12,13].
Allosteric regulation controls given protein activity involved in catalysis, signal transduction, gene regulation alongside various other biological processes [14, 15]. Regulation of protein functions or dynamics due to binding of regulator at site other than enzyme’s active site is termed as “allostery”. Allosteric regulators are classified as allosteric activators and allosteric inhibitors leading to increase in protein’s activity and decrease in activity, respectively. Allosteric proteins have capacity to switch between two states i.e. active state and inactive state triggered by allosteric signal (an effector/binder) [16]. Control of protein activity by these allosteric effectors is attributed to their ability to stabilize specific conformation of target protein with distinct binding. Most protein surfaces have various potential allosteric sites except fibrous as well as structural proteins [14]. For instance, the binding of oxygen at one part of hemoglobin increases binding tendency of oxygen at other subunit represents most suitable example of allostery [17]. Although most of allosteric inhibitors have been discovered serendipitously but have more selectivity as compared to orthosteric ones [18,19,20].
Initially, the allosteric property was reported in quaternary proteins but later it has been confirmed as intrinsic characteristic of all dynamic proteins. Keeping in view the existence of these proteins as collection of active and inactive conformers, the binding of allosteric regulators causes structural changes in proteins and shifts this dynamic equilibrium either towards an active state (allosteric activator) or inactive state (allosteric inhibitor) [14, 21] as depicted in Fig. 1. Although allosteric inhibition being advantageous over competitive inhibition is more desirable but their mechanism of action is not clearly defined. Role of allosteric proteins as intermediate for signal transduction pathways is well documented where they serve as mediator, modulator or adaptor to activate partner proteins to perform their activity [22].

Mechanism of Allosteric Regulation in proteins a. Allosteric activator induces conformation changes to facilitate in substrate binding, b. Allosteric inhibitor reduces substrate binding tendency through conformation changes
Amino acid biosynthetic pathway
Amino acids are building blocks of proteins and are critical for life as they play role in synthesis of various metabolites through metabolic pathways. Amino acids biosynthesis is a complex array of alternate pathways connected as non-linear series of reactions [23]. Apart from being precursor for protein synthesis, amino acids also serve as intermediates of other biosynthetic pathways of cell like purine synthesis [24, 25]. In plants, they are catabolized through tricarboxylic acid pathway to produce energy that is utilized by cells for their growth and proliferation [26, 27].
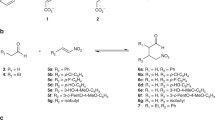
The biosynthetic pathways for the essential amino acids (i.e. acquired through dietary sources; animals cannot synthesize) are found only in microorganisms and are more complex as compared to non-essential amino acids. Owing to their common metabolic precursors, the amino acids have been classified as four families namely Aspartate family (lysine, methionine, threonine, Asparagine) [28,29,30], Pyruvate family (Alanine, leucine, isoleucine, valine) [31], Aromatic family (phenylalanine, Tyrosine, Tryptophan) [32, 33] and α-ketoglutarate family (glutamic acid, glutamine, proline, arginine) along with Histidine and serine [34].
Availability of given amino acids in living organisms are effected by either regulatory factors being capable of controlling synthesis of amino acids or either their proficient catabolism [35]. So far, over 300 amino acids have been reported out of which 20 amino acids serve as basic structural units of proteins while 10 amino acids are essential for humans and other living beings as they need to be provided through dietary sources [36, 37]. Role of amino acids in food industry, fodder, cosmetic industry, and pharmaceutical industry signifies their importance and need of high scale production to achieve market demand. Keeping in view the important role of amino acids as building block of life in human beings and animals, the chemical industry focused on various synthetic strategies for production of these biochemically distant compounds.
Different metabolic pathways involved in synthesis of amino acids are glycolysis; for branched chain amino acids (Val, Leu) alongside Ala, Gly, Cys, Ser synthesis, citric acid cycle; for Asn, Asp, Lys, Met, Thr and Ile, phosphoribosyl pyrophosphate (PRPP) pathway; for aromatic amino acid (Phe, Trp, Tyr) and histidine synthesis and shikimate pathway [38, 39]. The Shikimate biosynthetic pathway also referred as prechorismate pathway leads to synthesis of aromatic amino acids (AAA) having huge significance in terms of their role in synthesis of various secondary metabolites (i.e. serotonin and various neurotransmitters) [40, 41]. As mentioned earlier different pathways leading to synthesis of amino acids of all families are interconnected (Fig. 2) like pyruvate an important precursor through series of enzymatic reactions is converted into alanine, valine and leucine while oxaloacetate obtained from pyruvate leads to production of aspartic acid that is ultimately transformed into asparagine, methionine, threonine, lysine and isoleucine. Similarly, phosphoenolpyruvate serve as precursor for AAA while α-ketoglutarate give rise to glutamic acid, glutamine, proline and arginine. In addition, 3-phosphoglycerate follow multistep enzyme reactions to produce phosphoserine that is later converted to serine, cysteine and glycine. The details of amino acid biosynthesis in diatoms has been reviewed previously by Mariusz A. Bromke [42]. Current review is focused in structural aspects of enzymes involved in feedback inhibition and regulation of biosynthetic pathway of amino acids.

Amino acids biosynthetic pathways
Despite the role of amino acids for accurate functioning of cells, homeostasis is maintained through metabolic regulation of biosynthetic pathways achieved either through (i) controlled synthesis of enzyme or (ii) feedback inhibition of enzyme by end product (i.e. amino acid). This end product inhibition of enzymes is counteracted either by sufficient availability of substrate or by heavy metal cation to facilitate balanced concentration of given metabolite/intermediate/amino acid for proper functioning of cell [16, 39, 43, 44].
Feedback inhibition of amino acids biosynthetic pathway
Discovery of product feedback inhibition dated back to 1950, has been attributed as cornerstone of metabolic regulation that has been reported to sufficiently control metabolic fluxes to facilitate proficient growth [1, 2]. For a given biosynthetic pathway, the inhibition of first committed step by end product is termed as feedback inhibition while inhibition at sites other than active sites is termed as allosteric inhibition with huge biological significance. Binding of allosteric regulator to enzyme causes conformational changes in enzyme structure and perturb its activity [35]. Allosteric inhibition of amino acids biosynthetic pathways is well documented and 16 amino acids have ability to feedback inhibit their own biosynthetic pathway by targeting allosteric sites of enzyme catalyzing first committed step. Deregulation of feedback inhibition is of utmost importance to maintain cell homeostasis and has been mostly reported in vitro studies to improve production of various amino acids with huge industrial impact [4, 45]. The details of amino acids alongside enzyme target with feedback inhibition has been summarized in Table 1.
Few amino acids have the tendency to feedback inhibit multiple enzyme targets and their deregulation signifies their role to improve industrial production by identifying new and better strains. For instance, arginine and proline synthesized as final product from oxaloacetate pathway feedback inhibit N-acetyl-L-glutamate kinase (NAGK) and N-acetyl-L-glutamate synthase (in case of arginine) and glutamate kinase in case of proline [64]. Similarly, lysine has capacity to feedback inhibit multiple enzyme targets namely diaminopimilate decarboxylase, dihydrodipicolinate synthase, homocitrate synthase and aspartokinase III [65, 66]. In few cases, same enzyme is targeted by multiple amino acids as feedback inhibitors like aspartokinase is feedback inhibited by both lysine and threonine [67]. Amino acid biosynthetic pathway and effect of feedback inhibition alongside use of various approaches for deregulation of inhibition has remained focus of researchers especially in industrial biotechnology. Various reports over deregulation of feedback inhibition of individual biosynthetic pathways have been previously reported by our research center. For instance, Geng et al., reported successful alleviation of feedback inhibition of DHDPSE.coli through mutations at inhibitor binding site and identified E. coli MG1655 strain with improved L-lysine production yield by 46%. They utilized structural characteristics of L-lysine-sensitive DHDPS E.coli and L-lysine-insensitive DHDPSC.glutamicum and reported new enzyme variants through point mutations at specified sites with improved lysine production [68]. Similarly, Zhen et al., utilized combination of SCA and MD approach to accomplish successful deregulation of the allosteric inhibition of aspartokinase i.e. an industrial enzyme for increased amino acid production [69]. Recently, we have used the computational mutagenesis method to identify new mutant structures with potential deregulated feedback inhibition by tryptophan for anthranilate synthase from S. marcescens (Sadia et al., under revisions).
Here, we summarized structural characteristics of various enzyme targets and effect of mutations on their structures and functions especially in terms of deregulation of feedback inhibition. Applicability of various experimental as well as computational techniques (i.e. site directed mutagenesis, site specific mutagenesis etc.) to accomplish feedback resistance has also been discussed in details to have an insight into various aspects of research work reported in this particular field of study.
Structural characteristics of enzymes targeted by amino acid feedback inhibition
The amino acids can be broadly grouped as four categories or families following different pathways for their synthesis. The details of amino acids capable of inhibiting committed step of their own biosynthesis, their categories, pathways as per their common precursors and their feedback sensitive enzyme targets are shown schematically in Fig. 3.

Schematic Representation of Amino acid’s Feedback Inhibition
Detailed structural features of various enzymes belonging to different families of amino acids have been provided in next section.
Glutamate family of amino acids
Structural insights into enzyme targeted by feedback inhibition of arginine
L-arginine has huge significance at industrial level especially, cosmetic industry, pharmaceutical and food industry. Microbial fermentation is employed for synthesis of arginine at industrial scale [70, 71, 72, 73]. Arginine biosynthetic route of microorganisms as well as plants comprises of eight steps where first five steps lead to production of ornithine i.e. precursor for arginine [74]. Biosynthesis of arginine in case of E.coli follows linear route where hydrolysis of N-acetylornithine directly lead to production of ornithine and first enzyme N-acetylglutamate synthase (NAGS) is feedback inhibited by arginine and catalyzes rate determining step of route [47]. Few microorganisms like yeasts, algae and few bacteria like P. aeruginosa follow a different route where acetyl group from acetylornithine is recycled to glutamate and in this case the N-acetylglutamate kinase (NAGK) is feedback inhibited by arginine [75]. Another pathway has also been reported involving novel family of transcarbamylases for biosynthesis of arginine [76]. The N-acetylglutamate kinase (NAGK) may serve as arginine-inhibitable or arginine-insensitive systems in different organisms.
N-acetyl-l-glutamate synthase (NAGS)
First step of arginine biosynthetic pathway in E. coli and other microorganisms following linear route is catalyzed by N-acetyl-l-glutamate synthase (NAGS) that is inhibited by arginine [77]. Amino acid kinase (AAK) domain of both acetylglutamate kinase (NAGK) and acetylglutamate synthase (NAGS) are similar hexameric systems formed by amino acid kinase dimers connected through N-terminal α helices [78, 79]. Comparison of characteristic features of 3D-crystal structures of NAGS from different microorganisms like M. tuberculosis and N. gonorrhea provided insights into conformational arrangements needed for inhibition and regulation of enzyme activity and function. Both NAGSM. tuberculosis (PDB ID: 6ADD, 5YO2 [80] and NAGSN. Gonorrhea (PDB ID: 2R98 [81] crystal structures exists as dimer stacked together to form hexamer arrangement that is necessary for feedback inhibition by arginine. Although both NAGS comprises of distant V-cleft alongside glutamine and AcCoA binding sites still some differences were observed marked by dotted circle/box in Fig. 4a, b. The acceptor binding mode in case of NAGSM. tuberculosis is different due to much deeper binding pocket formed by fragment (i.e. 17 residues longer than NAGSN. Gonorrhea) connecting α4 and β6. The arrangement of amino acids involved at inter-junction of two monomer units to form dimer structure are depicted in Fig. 4c [46]. Binding of L-arginine causes major changes in quaternary structure of NAGS by inducing contraction in hexamer leading to increase in width of internal cavity as revealed by differences observed in binding of arginine and N-acetylglutamine (NLQ) (Fig. 4d, e).

Comparison of 3D-crystal structures of NAGS from different microorganisms a. NAGS from M. tuberculosis, b. Superimposed structure of NAGS from M. tuberculosis and N. gonorrhoeae, c. Amino acids arrangement at interjunction of two monomer units, d. Binding of N-acetylglutamine (NLQ) and e arginine inside NAGS, f. Sequence alignment for NAGS sequences retrieved from different organisms; Reproduced from Refs. [46, 80, 81]
Sequence alignment for NAGS sequences retrieved from different organisms revealed fully conserved and partially conserved amino acids around L-arginine-binding site suggesting role of key amino acids for binding of arginine. Mutations at these sites most probably deregulate feedback inhibition caused by arginine and will aid in discovery of feedback resistant strains with improved production of L-arginine. Li et al. reported comparison of two forms of N-acetyl-l-glutamate synthase (NAGS) belonging to N. gonorrhoeae i.e. l-arginine bound T-form (inactive) and R-form (active) with bound CoA and L-glutamate [47, 81, 82]. These NAGS are comprised of single polypeptide chain having two domains i.e. N-terminal AAK (L-arginine binding site) and C-terminal NAT domain (substrate binding site).
In case of Yeast like Saccharomyces cerevisiae, the NAGS and NAGK play their role as complex and are targeted by feedback inhibition of arginine [81, 83, 84]. In addition, previous studies showed that bifunctional NAGS/NAGK complex reported from Maricaulis maris and Xanthomonas campestris have capability to oligomerize as tetramer where both NAT and AAK domains are similar to NAGS of N. gonorrhoeae but differences are observed for linker between two domains as well as their relative orientations [85]. Furthermore, it was revealed that NAGS belonging to both human and mouse are tetrameric in form just like bacterial NAGS/NAGK complex [86].
N-acetyl-l-glutamate kinase (NAGK)
N-acetyl-l-glutamate kinase (NAGK) also termed as argB is rate limiting enzyme of cyclic route for l-arginine biosynthetic pathway as in case of Corynebacteria where acetyl group of N-acetyl-ornithine is recycled to generate l-glutamate [72, 73, 87]. Yet, metabolic control should occur on the production of acetylglutamate, regardless of its origin. Therefore, feedback inhibition on both the synthase and the kinase is believed to be general for organisms using cyclic ornithine synthesis. [88, 89]. In contrast, NAG synthetase would appear to be a less suitable target, because in those organisms that recycle the acetyl group in the route of ornithine synthesis it only plays a purely anaplerotic role [90].
The key structural features of NAGK (3D-crystal structure; Protein data bank) from various microorganisms have been summarized in Tables 2, 3 to provide a glimpse of conformational arrangements governing feedback inhibition of arginine. The differences among arginine-sensitive and arginine-insensitive NAGK will provide clue about the discovery of new mutant structures with better production of arginine.
Arginine insensitive NAGK belonging to E. coli are homodimer while those arginine sensitive type of NAGK are hexamer comprising of three homodimeric units structurally similar to NAGK of E. coli. Arginine feedback inhibition is not necessarily attributed to hexameric form but hexamer system play major role for proper functioning and stability of NAGK and enhances arginine sensitivity [94].
Aspartate family of amino acids
Aspartate family includes lysine, threonine and methionine that regulate their own synthesis by end product feedback inhibition. Lysine and threonine synthesis is achieved both by DAP biosynthetic pathway or AAA biosynthetic pathway and enzymes catalyzing committed step of these routes are feedback inhibited by end product amino acids. The key enzyme targeted by lysine feedback inhibition are dihydrodipicolinate synthase (DHDPS), aspartokinase (AK) and while threonine feedback inhibit Homoserine kinase (HSK). Similarly, Homoserine O-acetyltransferase (HTA) is feedback inhibited by methionine.
Aspartokinase III The aspartate kinase the first enzyme of aspartate pathway is feedback inhibited by l-lysine in C. glutamicum. Three forms of aspartokinase (AK I, AK II and AK III) have been reported in different bacterial species. In case of B. subtilis and E. coli, the aspartokinase II and aspartokinase III are feedback inhibited by l-lysine and l-threonine [95, 96].
The regulation of lysine biosynthesis varies among organisms. Only aspartokinase is regulated for the biosynthesis of lysine in C. glutamicum. It is inhibited by l-lysine plus l-threonine; either l-lysine or l-threonine alone does not inhibit aspartokinase. In E. coli, many enzymes of lysine biosynthesis are regulated by the end-products of the pathway. There are three isozymes of aspartokinase in E. coli: aspartokinase I is repressed by l-threonine plus l-isoleucine and inhibited by l-threonine, aspartokinase II is repressed by l-methionine, and aspartokinase III is repressed and inhibited by l-lysine. Aspartokinase I and II are bifunctional enzymes and show both aspartokinase and homoserine dehydrogenase activities [97,98,99].
Comparing crystal structure (retrieved from protein data bank) of aspartokinase from different microorganisms namely C. glutamicum and E.coli [54, 100, 101] revealed presence of ACT domains in monomer, dimer or tetramer structures. This ACT domain is characteristic feature of various kinases, 3PGDH, AK-I and AK-III where ligand binds i.e. serine, threonine and lysine binds similar region as depicted in Fig. 5.

Comparison of ACT domains of a. Ser-binding site of 3PGDH, b. Thr-binding site of CgAKβ, c. Lys-binding site of AtAKI d. Overall structure of α2β2-type aspartokinase (AK) from C. glutamicum, e. Threonine-binding site of AK of C. glutamicum, f. Lysine-binding site of AK of C. glutamicum; Reproduced from Refs. [100, 101]
Comparison of AKC. glutamicum and AKE.coli identified presence of two state conformation (T-state and R-state) in case of E. coli where each monomer unit is comprised of two ACT domain at C-terminus while each monomer unit of tetramer structure of C. glutamicum has single ACT domain [101, 102]. The AK from C. glutamicum is feedback inhibited by both lysine and threonine in a concerted way while AK of T. thermophilus is feedback inhibited by threonine only [103,104,105]. Ayako et al., reported crystal structure of AK from C. glutamicum as: inhibitory complex bound to lysine and threonine, (ii). Active complex bound to threonine. Comparison of these two forms of AK revealed that inhibitory form is stabilized by conformational changes caused by binding of inhibitors. Structure of AK is heterotetramer comprised of two α subunits and two β subunits where dimerization of homo-oligomeric AK involves interaction among regulatory domains. The crystal structure with bound lysine and threonine exists as in active T-form [101]. AK structure bound to both lysine and threonine follow a concerted mechanism for inhibition involving binding of threonine first following lysine binding lead to closed conformation Fig. 5.
Enzyme targeted by feedback inhibition of histidine
ATP-phosphoribosyl transferase (ATP-PRT)
Histidine biosynthetic pathway found in bacteria, plants and fungi comprises of ten steps and has been considered as well recognized target for discovery of various antimicrobial drugs. The enzyme ATP-phosphoribosyl transferase (ATP-PRT) catalyzing first committed step of pathway is feedback inhibited by end product histidine. The reaction catalyzed by ATP-PRT involves synthesis of N′-5′-phosphoribosyl-ATP (PR-ATP) by condensation of ATP with 5-phosphoribosyl-a-1-pyrophosphate (PRPP) [106]. The enzyme ATP-PRT play major role to regulate microbial growth as histidine biosynthetic pathway is connected with various other metabolic pathway [51, 107]. Histidine allosterically inhibit ATP-PRT enzyme at site other than active pocket termed as allosteric site while AMP, GTP and ADP serve as competitive inhibitors [50].
Crystal structural analysis of tertiary and quaternary ATP-PRT revealed two different forms. The most widely found homo-hexameric form, encountered in bacteria, fungi, and plants, while second form i.e. the hetero-octameric type, limited to few bacterial strains. ATP-PRT belonging to M. tuberculosis is homo-hexameric class, also found in S. enterica and E. coli [51, 108]. Crystallographic data revealed that L-histidine binding converts active dimer of MtATP-PRT into an inactive hexamer [50]. Furthermore, the L-histidine-bound hexameric form causes major conformational changes, having twist of domain III compared to domain I and II. Crystallographic studies also revealed that L-histidine binds at a site approximately 30 Å away from the active site, suggesting allosteric nature of inhibition. Finally, it was observed that L-histidine interacts with allosteric site involving carboxyl group of Asp218, the hydroxyl moiety of Thr238, and the backbone amide oxygen of Ala273 [50]. The mycobacterial ATP-PRT enzyme comprises of ten helices and 15 β-strands arranged as three domains. Domain I contains a central β-sheet consisting of four parallel β-strands and two anti-parallel strands while domain II also has four parallel β-strands and one anti-parallel β-strand surrounded by two α-helices on each side. Catalytic site (substrate binding site) of ATP-PRT lies at intersection of domain I and II involving mostly interaction with amino acids of domain II while allosteric inhibitor histidine binds in domain III. The catalytic domain of ATP-PRT of both mycobacteria and glutamine binding E. coli are similar to each other [50, 109].
The ATP-PRT enzyme of proteobacteria has shorter form without domain III where HisG forms complex with HisZ. Crystal structure of both apo and AMP:His complex is in hexamer form comprised of trimer of dimers where histidine bound hexameric complex is more compact than apo form as shown in Fig. 6. In case of dimer, the interactions mainly involve domain I and II of catalytic domain while hexamer interface involve histidine binding domain (domain III). Histidine binding stabilizes hexamer form (inactive) and also influences catalytic site for substrate binding due to steric hindrance [50].

a. Cartoon representation of the ATP-PRTase monomer (M. tb and C. jejuni) where ligand AMP and His are shown in ball-and-stick, b. Cartoon representation of ATP-PRT trimer structure ATP (open domain III domain) and His bound (More close domain III domain), c. Hexamer structure of ATP-PRT (C. jejuni) with bound His, and AMP; Reproduced from Ref. [49] and [50]
Histidine binding alongside AMP causes conformational changes in ATP-PRT structure where active site of enzyme is closed and amino acid residues involved in binding of PRPP also disrupt that hindered catalytic activity [50].
Aromatic amino acids family of amino acids
Enzyme targeted by feedback inhibition of tryptophan
Aromatic amino acids namely tryptophan, phenylalanine and tyrosine follow Shikimate pathway for their biosynthesis. Despite their role as building block for protein, they also serve as precursor for various pharmaceutical products. For instance, Tryptophan serve as precursor for various metabolites with significant pharmacological importance especially, vinblastine and vincristine two well-known anticancer drugs while Tyrosine is precursor for neurotransmitter dopamine, p-hydroxystyrene and p-hydroxycinnamic acid, needed for manufacturing of novel materials, pharmaceuticals and nutraceuticals [110,111,112,113,114,115]. Chorismate is a branch point biosynthesis of phenylalanine, tyrosine and tryptophan alongside other metabolites. The shikimic acid pathway of bacteria is regulated and homeostasis is maintained through end product feedback inhibition for enzyme of branch point [116]. The Anthranilate synthase (AS) catalyzes first committed step of pathway and is feedback inhibited by Tryptophan. On the other hand, the l-phenylalanine and l-tyrosine are formed from chorismic acid via prephenate, which undergoes either decarboxylation/dehydration or decarboxylation/dehydrogenation, followed by a transamination to generate given amino acids. The enzymes namely prephenate dehydrogenase and prephenate dehydratase catalyzes first step of tyrosine pathway and phenylalanine pathway, respectively and are feedback inhibited by their end product.
Anthranilate synthase (AS)
Tryptophan belong to aromatic amino acids family and its biosynthesis proceeds via a common pathway to chorismate [117]. The first step of tryptophan biosynthetic pathway is catalyzed by anthranilate synthase (AS; EC 4.1.3.27) that is feedback inhibited by final product i.e. tryptophan. Anthranilate synthase comprises of two polypeptide chains that form a complex TrpE2:TrpG2 and catalyzes the first reaction branching from the shikimate pathway toward the biosynthesis of tryptophan occurring in bacteria, fungi and plants [118]. AS is composed of two non-identical subunits where TrpE subunit binds chorismate (CA) and is the site of formation of anthranilate from CA and NH3 while TrpG belongs to glutamine amidotransferase family [119]. The reactions catalyzed by TrpE and TrpG involves: (i) Glutamine-dependent AS reaction requires both subunits while (ii) Ammonia-dependent AS reaction requires either α-subunit or both subunits [120]. Glen et al. reported the crystal structure of anthranilate synthase (AS) from Serratia marcescens in the presence of its substrates and inhibitor l-tryptophan. The trpE of S. marcescens comprises of domain I (composed of 1–57, 178–308, 468–520 amino acid residues) and domain II (composed of antiparallel β-sheet from 9 to 12 strands (β4, β4a, β4b) alongside two α-helices (α1a,α1b). The junction of two domains contains various amino acids causing tryptophan feedback inhibition while another neighboring crevice attached to trpG contains amino acid residues involved in substrate binding [63].
Comparison of crystal structures of AS from various organisms revealed different arrangement of TrpG and TrpE in heterotetrameric complex. The structural composition of AS from S. marcesens is comparable to AS from S. solfataricus while AS of S. typhimurium represents different structure as depicted in Fig. 7.

Comparison of Anthranilate synthase structure from S. solfataricus, S. marcesens and S. typhimurium; Reproduced from Ref. [63]
Structural characteristics of TrpG and TrpE from S. marcescens is comparable to S. solfataricus where Gly328, Thr329, His398 and Gly485 from chorismate binding domain play vital role in binding of chorismate while Ser40, Pro291, Met293, Val453, Tyr455 are involved in binding of tryptophan. Similarly, glutamine, pyruvate and benzoate binding site are also shown as different regions in Fig. 6a, b. Anthranilate synthesis need both sub units (α- and β) of anthranilate synthase involving reaction of chorismate with glutamine while only α-subunit is needed for reaction of chorismate and ammonia (at high concentrations of ammonium) [121, 122].
The second enzyme of tryptophan biosynthetic pathway anthranilate phosphoribosyl transferase (AnPRT) is also feedback inhibited by L-tryptophan. The AnPRT (TrpD) catalyzes reaction of PRPP and anthranilate to N-(5′-phosphoribosyl)-anthranilate (PRA) and PPi. Crystal structure revealed AnPRT as homodimer having N-terminal domain comprising of six α-helices and C-terminal domain formed by eight α-helices surrounding seven stranded β-sheet [123]. The active site is present at interface of C- and N-terminal domains as revealed by crystal structure of S. solfataricus, P. carotovorum, T. thermophiles, X. campestris, M. tuberculosis [124,125,126,127,128].
The enzyme anthranilate phosphoribosyl transferase (AnPRT) belonging to enteric bacteria exists as complex with anthranilate synthase (AS). Bifunctional TrpD: TrpE complex is responsible for the first step in tryptophan biosynthesis i.e. the transfer of an amino group from glutamine to chorismate and the formation of anthranilate. The Feedback inhibition of anthranilate phosphoribosyl transferase (AnPRT) is observed in its complex form with AS and sensitivity to tryptophan feedback inhibition is lost once complex is distorted [129].
Pyruvate family of amino acids
Enzyme targeted by feedback inhibition of leucine
The branched chain amino acids follow pyruvate pathway for their synthesis and use pyruvate or 2-ketobutyrate as precursor formed through acetohydroxy acid synthase (AHAS). The pyruvate pathway branches at isopropylmalate (IPM) where 2-ketoisovalerate and acetyl CoA is converted to α-isopropylmalate catalyzed by α-isopropylmalate synthase (α-IPMS) regulated by leucine feedback inhibition [130]. The IPM pathway has been reported as key route for synthesis of leucine for bacteria, fungi and plants [131,132,133].
α-isopropylmalate synthase (α-IPMS)
The α-isopropylmalate synthase catalyzing first committed step of leucine biosynthetic pathway branched from pyruvate route is feedback inhibited by end product leucine. The reaction catalyzed by IPMS involves Claisen condensation of α-ketoisovalerate and AcCoA to produce α-isopropylmalate [134]. Luiz et al., reported crystal structure of α-IPMS from M. tuberculosis, a monomeric structure comprising of two domains i.e. N-terminal (catalytic site) and C-terminal (regulatory site) connected through two subdomains depicted in Fig. 8. The catalytic domain exits as (α/β)8 TIM barrel where linker domain I comprises of α10 and two short β-strands, while subdomain II composed of α11-α13. Similarly, regulatory domain located at C-terminal is comprised of two βββα units. They also identified that C-terminal domain markedly effect activity of enzyme [135, 136].

Crystal structure of α-IPMS from M. tuberculosis; Reproduced from Ref. [136]
Structural analysis of α-IPMS revealed binding of Zn2+ and α-ketoisovalerate into active site while leucine exhibits reversible feedback inhibition by binding inside regulatory domain. Feedback inhibition of α-IPMS by leucine follow time dependent slow onset inhibition where complex formed by leucine binding inside regulatory domain isomerizes to tightly bound complex. The linker domain facilitate in transmission of inhibitory signals towards catalytic site [134]. Cavalieri et al., reported L-leucine insensitive α-IPMS from S. cerevisiae mutations at leucine binding site [137] while another mutant structure reported for α-IPMSMtbTyr410Phe mutant also showed insensitivity to leucine binding [138].
Deregulation of amino acids feedback inhibition
Applicability of basic principles for deregulation of histidine feedback inhibition (Case study I)
It is a generalized fact that structural characteristics of given enzyme system has marked effect on deregulation of feedback inhibition of amino acids. Enzymes existing as monomer, dimer or oligomer complexes comprised of different domains, chains, and sites. Mutations at any of these sites negatively effects binding of inhibitors and ultimately inhibition tendency alongside catalytic activity of that enzyme complex is disrupted. Here, we discussed applicability of various mechanisms and strategies employed to accomplish deregulation of feedback inhibition for histidine biosynthetic pathway (i.e. taken as example).
Histidine has huge significance as per its role in various pharmaceutical products and also serve as precursor for production of various bioactive compounds. Owing to its role in various industries like pharmaceutical and cosmetic industry, the aim of biotechnologists is to enhance its production at industrial scale by implementing various available techniques and approaches. Bioinformatician, as well as experimentalists are contributing their efforts to accomplish deregulation of enzyme′s feedback inhibition. Commonly used strategies are (1). Reducing interaction and binding of feedback inhibitor inside pocket, (2). Blocking entry of inhibitor into binding domain, (3). Mutations at other parts/sites of enzyme to eliminate metastability of protein due to inhibitor binding, (4). Effect of mutations at interface of dimer or multimer enzyme, (5). Competitive feedback inhibition by end product (i.e. amino acid). The structural characteristics of ATP-phosphoribosyl transferase (ATP-PRT) enzyme (feedback inhibited by end product histidine) revealed two forms i.e. homo-hexamer or hetero-octamer. Detailed discussion over use of various techniques and strategies for deregulation of feedback inhibition in HisG are provided in next section.
Reducing binding of feedback inhibitor inside pocket/regulatory domain Allosteric inhibitor binding sites have been previously engineered by various research groups to accomplish reduced binding of inhibitor inside pocket. Reducing interaction tendency of inhibitor by mutations of key amino acids of binding domain is of paramount importance for deregulation of feedback inhibition. Cho et al., reported crystal structure of ATP-Phosphoribosyl transferase (HisG) and identified key amino acids involved in binding of allosteric inhibitor histidine [50]. Zhang et al. employed this approach of reducing binding interaction with pocket and reported site directed mutagenesis of key amino acids (Asn215, Leu231, Thr235, and Ala270) involved in feedback inhibition identified through sequence identity comparison of HisG from M. tuberculosis and C. glutamicum. The N215K/L231F/T235A mutant of HisG resulted in higher yield of histidine [170]. Similarly, G233H/T235Q mutants of Gly233 amino acid residue reported by Schendzielorz and co-workers enhanced production of histidine [171]. Similar approach was utilized against DHDPS from C. glutamicum where four amino acids of regulatory domain directly interacting with l-lysine were mutated by Feng et al. [89]. They reported total deregulation of feedback inhibition for His56Lys and Glu84Thr suggesting essentiality of these two amino acids for l-lysine binding and inhibition. As mentioned earlier, the histidine binding into allosteric site (Domain III) of ATP-PRT results in more compact and inactive form of enzyme. Identifying key amino acids involved in binding of L-histidine like Asp218, Thr238, and Ala273 [146] and sequence identity comparison among ATP-PRT of different sources will facilitate in achieving mutations at these sites to accomplish deregulation. Computational as well as experimental mutagenesis approaches can be employed. Similarly, comparison of feedback sensitive and feedback insensitive forms of ATP-PRT enzyme belonging to different sources through sequence identity calculation also facilitate to discover new enzyme variants of interest. For instance, Yoshimi et al., reported three mutants of aspartokinase III i.e. Thr344Met, Ser345Leu, and Gly323Asp with considerable lysine-insensitivity [172] while Met318Ile and Thr352Ile of E. coli AKIII (lysine-insensitive AKIII) were reported by Falco et al. [173].
Blocking entry of inhibitor into binding domain Modifying the key amino acid residues positioned at the entrance of feedback inhibitor binding site facilitate to block entry of inhibitor Fig. 9a, b. Steric hindrance, change in hydrophobicity or hydrophilicity and charge modification are key contributor to achieve deregulation of feedback inhibition in this case. This could be an effective strategy to achieve deregulation of ATP-PRT enzyme as previously reported for deregulation in feedback inhibition for NAGK from C. crenatum by mutations at Glu19 position located at entrance of arginine binding site. Where the feedback inhibition of l-arginine was most deregulated in the Glu19Tyr, Glu19Trp and Glu19Phe mutants [93]. Now, in case of ATP-PRT enzyme histidine bound structure exists as trimer or hexamer form where histidine binding site is located at junction of two monomeric units and blocking entry of histidine in this site can be achieved by mutations of amino acids at entrance point.

Strategies for deregulation of feedback inhibition in HisG
Eliminate metastability of protein due to inhibitor binding As histidine binding introduces compactness in trimeric, or multimeric structure of ATP-PRT enzyme. Mutations or engineering of sites other than feedback inhibitor binding site may cause conformational changes alongside disruption of enzyme stability that ultimately destroy catalytic activity of enzyme. Owing to effect of histidine binding with key amino acids of binding domain, the modifications on other sites or positions of enzyme cause conformational changes and allosterically effect catalytic activity as well as inhibitor binding tendency of ATP-PRT. Plenty of approaches i.e. both experimental and computational are in common practice to introduce mutations in enzyme structure for discovery of new enzyme variants. This particular approach has been successfully employed by various research groups for aspartokinase III to identify enzyme mutant structures with deregulated inhibition for instance Gln298Gly and Ala279Thr (using point mutation approach) [174,175,176].
Effect of mutations at interface of dimer or multimer enzyme Histidine need ATP-PRT enzyme in trimer or hexamer form to show its feedback inhibition as Histidine binding domain lie at junction of two monomeric units. Mutations at interface of dimer structure may disrupt inter-domain communication and transfer of signals leading to loss of catalytic activity and feedback inhibition as shown in Fig. 9c, d. Most of enzymes targeted by feedback inhibitor exists as dimer, tetramer or hexamer structure and mutations at junction of these monomeric units may affect feedback inhibition, on the other hand catalytic activity of enzyme is also effected and coping this situation is of paramount importance. This approach have been successfully employed to achieve deregulation of α-IPMS (mutant Y410F) with complete loss of sensitivity towards feedback inhibition by leucine [138].
As histidine has its own binding domain/regulatory domain and its interactions with pocket affects catalytic activity through allosteric effect. On the other hand, few amino acids like L-threonine showed feedback inhibition as competitive inhibitor and disrupting their binding inside active pocket will ultimately destroy the catalytic activity. Multiple strategies could be employed to retain wild-type activity while decreasing affinity for the competitive inhibitor. Like engineer new interactions that support substrate binding but weaken inhibitor binding, effecting hydrophobicity, creating steric bulk or decreasing strength of van dar Waals interactions.
Plenty of mutagenesis approaches including both experimental and computational techniques are in common practice to achieve deregulation in targets of interest. Commonly used in vitro mutagenesis techniques are Directed evolution technique [177, 178], Site directed mutagenesis (SDM) [179], Site saturation mutagenesis (SSM) [180, 181], Random mutagenesis [182], Chemical mutagenesis, Error-prone PCR (ep-PCR) [183, 184], Rolling circle amplification-PCR (RCA-PCR) [185,186,187,188]. The summary of mutant structures of enzymes with deregulated feedback inhibition by amino acids have been provided in Table 4.
No significant studies have been reported for mutational studies over Anthranilate synthase (AS), Anthranilate phophoribosyl transferase (AS-PRT), Pyrroline carboxylase reductase, Glutamate 5-kinase (GK), and Homoserine-O-acetyltransferase (HSAT) enzyme targets regulated by feedback inhibition of their own end product amino acids.
In addition, the development of bioinformatics algorithms enabled computational approaches to provide more precise guidance for enzyme engineering and make it more efficient and less laborious. The success of rational design depends on in-depth knowledge about sequence and structure features of target proteins. Various computational mutagenesis approaches have been reported to get in depth sights of protein functions, structure, stability and thermodynamic characteristics (Table 5). The folding and interaction among various amino acid residues of globular proteins depend on proteins sequences while mutating these amino acids will disrupt these folding patterns hence different protein conformations are obtained with varied thermodynamic characteristics. For instance, computational saturation mutagenesis (CoSM) uses molecular dynamic equilibration, sidechain flips and energy minimization to improve side conformations in mutants enable prediction of stability changes with better accuracy and correlation with the experimentally deciphered stability changes. Similarly, elastic network contact model (ENCoM) based on normal mode analysis (NMA) rely on amino-acid’s nature and help to calculate vibrational entropy changes upon mutations [203, 204]. Unfolding mutation screen (UMS) another well-known computational mutagenesis technique employed to evaluate effect of given mutations on structure and functions of protein using unfolding propensity and display of data as interactive heat maps. The UMS approach is advantageous over previously used techniques as it does not need prior knowledge of protein structure and function [205,206,207].
Literature revealed that none of above mentioned in silico mutagenesis techniques have been employed for mutations prediction to deregulate feedback inhibition of enzymes. Using combination of various in silico approaches to design new enzyme variants with deregulated feedback inhibition is of worth importance.
Future perspectives
Current review emphasized need of new mutagenesis techniques to accomplish more efficient enzyme variants aimed for increased production of amino acids. The detailed description of enzyme structures here affirmed that (a). Engineering of regulatory domain is key contributor to accomplish deregulation of feedback inhibition, (b). Blocking entry of inhibitor into binding domain by modifying the key amino acid residues positioned at the entrance of feedback inhibitor binding site, steric hindrance and charge modification is of paramount importance for deregulation, (c). In case of dimer, tetramer or hexameric structures, mutations at interface or junction of two monomeric units may disrupt signal transfer and ultimately destroy enzyme activity.
Biotechnologists, microbiologists and other research groups need to develop new strategies and approaches with better efficacy. Rational mutagenesis technique like site directed mutagenesis (SDM) and site saturation mutagenesis (SSM) have many advantages over random mutagenesis approach. Amalgamation of in vitro and in silico would be more effective approach and focus should be on more reliable and efficient software designs.
Concluding remarks
Detailed literature search over amino acid biosynthetic pathways, structural details of target enzymes feedback inhibited by amino acid, mutagenesis approaches (both in vitro and in silico) used to incorporate structural and conformational changes to deregulate their inhibition tendency have been summarized. The structure, design and mechanism of product feedback inhibition for all enzyme targets have been discussed in detail to provide an insight into structural and functional basis of amino acid feedback inhibition. Bacterial amino acids biosynthetic pathways are interconnected as different amino acids may be produced using same common precursor as starting point thus has been categorized into various groups. Majority of amino acid’s biosynthetic pathways (16 out of 20 amino acids) are regulated by allosteric feedback inhibition. Structural analysis of enzyme targets that are feedback inhibited by their own end product revealed that they exists mostly as dimer, tetramer or hexamer structure comprising of multiple subunits having more than one binding and allosteric sites. Although some enzyme targets also exist as monomer structures but allosteric inhibition is mostly observed in dimer or oligomeric systems. Traditional methods used for discovery of new bacterial strains like random mutagenesis have been replaced by rational mutagenesis approach and plenty of new computer assisted methods are in use now. Identifying key sites and domains in given enzyme structure keeping in view their role in feedback inhibition prior to mutations is of worth importance that will facilitate new enzyme discovery efficiently. Current review will provide guidelines for designing of better feedback resistant enzymes and will facilitate biotechnologists for discovery of novel enzyme variants for increased production of amino acids at industrial scale.
Availability of data and materials
The materials and datasets supporting the review are provided within the article.
Abbreviations
- TCA:
-
Tricarboxylic acid
- PRPP:
-
Phosphoribosyl pyrophosphate
- SDM:
-
Site directed mutagenesis
- SSM:
-
Site saturation mutagenesis
- AAA:
-
Aromatic amino acids
- NAGK:
-
N-Acetyl-l-glutamate kinase
- NAGS:
-
N-Acetylglutamate synthase
- AAK:
-
Amino acid kinase
- ATP-PRT:
-
ATP-phosphoribosyl transferase
- DAP:
-
Diaminopimelic acid pathway
- AK:
-
Aspartokinase
- DHDPS:
-
Dihydrodipicolinate synthase
- AS:
-
Anthranilate synthase
- CA:
-
Chorismate
- AnPRT:
-
Anthranilate phosphoribosyl transferase
- HSK:
-
Homoserine kinase
- SAT:
-
Serine acetyltransferase
- OASS:
-
O-acetylserine sulfhydrylase
- PSP:
-
Phosphoserine phosphatase
- GK:
-
Glutamate kinase
- ep-PCR:
-
Error-prone PCR
- RCA-PCR:
-
Rolling circle amplification-PCR
- NMA:
-
Normal mode analysis
- UMS:
-
Unfolding mutation screen
- CMP:
-
Comprehensive mutational profile
References
Umbarger HE. Evidence for a negative-feedback mechanism in the biosynthesis of isoleucine. Science. 1956;123:848–848.
Yates RA, Pardee AB. Control of pyrimidine biosynthesis in Escherichia coli by a feed-back mechanism. J Biol Chem. 1956;221:757–70.
Leuchtenberger W, Huthmacher K, Drauz K. Biotechnological production of amino acids and derivatives: current status and prospects. Appl Microbiol Biotechnol. 2005;69:1–8.
Reznik E, Christodoulou D, Goldford JE, Briars E, Sauer U, Segrè D, Noor E. Genome-scale architecture of small molecule regulatory networks and the fundamental trade-off between regulation and enzymatic activity. Cell Rep. 2017;20:2666–77.
Schomburg I, Chang A, Placzek S, Söhngen C, Rother M, Lang M, Munaretto C, Ulas S, Stelzer M, Grote A. BRENDA in 2013: integrated reactions, kinetic data, enzyme function data, improved disease classification: new options and contents in BRENDA. Nucleic Acids Res. 2012;41:D764–72.
Hirasawa T, Shimizu H. Recent advances in amino acid production by microbial cells. Curr Opin Biotechnol. 2016;42:133–46.
Huccetogullari D, Luo ZW, Lee SY. Metabolic engineering of microorganisms for production of aromatic compounds. Microb Cell Fact. 2019;18:1–29.
Kim Y, Phillips JA, Liu H, Kang H, Tan W. Using photons to manipulate enzyme inhibition by an azobenzene-modified nucleic acid probe. Proc Natl Acad Sci. 2009;106:6489–94.
Zhou C, Yang Z, Liu D. Reversible regulation of protein binding affinity by a DNA machine. J Am Chem Soc. 2012;134:1416–8.
Bishop AC, Chen VL. Brought to life: targeted activation of enzyme function with small molecules. J Chem Biol. 2009;2:1–9.
Gutteridge A, Kanehisa M, Goto S. Regulation of metabolic networks by small molecule metabolites. BMC Bioinform. 2007;8:88.
Reichard P. Ribonucleotide reductases: the evolution of allosteric regulation. Arch Biochem Biophys. 2002;397:149–55.
Yeang C-H, Vingron M. A joint model of regulatory and metabolic networks. BMC Bioinform. 2006;7:332.
Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4:474–82.
Kuriyan J, Eisenberg D. The origin of protein interactions and allostery in colocalization. Nature. 2007;450:983–90.
Tzeng S-R, Kalodimos CG. Protein dynamics and allostery: an NMR view. Curr Opin Struct Biol. 2011;21:62–7.
Shen A. Allosteric regulation of protease activity by small molecules. Mol BioSyst. 2010;6:1431–43.
Groebe DR. In search of negative allosteric modulators of biological targets. Drug Discovery Today. 2009;14:41–9.
Hardy JA, Wells JA. Searching for new allosteric sites in enzymes. Curr Opin Struct Biol. 2004;14:706–15.
Lee GM, Craik CS. Trapping moving targets with small molecules. Science. 2009;324:213–5.
Del Sol A, Tsai C-J, Ma B, Nussinov R. The origin of allosteric functional modulation: multiple pre-existing pathways. Structure. 2009;17:1042–50.
Dennis EA, Bradshaw RA. Transduction mechanisms in cellular signaling: cell signaling collection. Cambridge: Academic Press; 2011.
Hernández-Montes G, Díaz-Mejía JJ, Pérez-Rueda E, Segovia L. The hidden universal distribution of amino acid biosynthetic networks: a genomic perspective on their origins and evolution. Genome Biol. 2008;9:1–15.
Rose AJ. Amino Acid nutrition and metabolism in health and disease. Multidiscipl Digit Publ Instit. 2019;11:2623.
Rose W. The sequence of events leading to the establishment of the amino acid needs of man. Am J Public Health Nations Health. 1968;58:2020–7.
Kirma M, Araújo WL, Fernie AR, Galili G. The multifaceted role of aspartate-family amino acids in plant metabolism. J Exp Bot. 2012;63:4995–5001.
Less H, Galili G. Coordinations between gene modules control the operation of plant amino acid metabolic networks. BMC Syst Biol. 2009;3:14.
Paulus H. Biosynthesis of the aspartate family of amino acids In Bacillus subtilis and other gram-positive bacteria american society of microbiology. Berlin: Springer; 1993.
Wittmann C, Becker J. The L-lysine story: from metabolic pathways to industrial production In amino acid biosynthesis pathways regulation and metabolic engineering. Berlin: Springer; 2007.
Viola RE. The central enzymes of the aspartate family of amino acid biosynthesis. Acc Chem Res. 2001;34:339–49.
Amorim Franco TM, Blanchard JS. Bacterial branched-chain amino acid biosynthesis: structures, mechanisms, and drugability. Biochemistry. 2017;56:5849–65.
Parthasarathy A, Cross PJ, Dobson RC, Adams LE, Savka MA, Hudson AO. A three-ring circus: metabolism of the three proteogenic aromatic amino acids and their role in the health of plants and animals. Front Mol Biosci. 2018;5:29.
Tzin V, Galili G. The biosynthetic pathways for shikimate and aromatic amino acids in Arabidopsis thaliana. Arabid book/American Soc Plant Biol. 2010;8:e0132.
Wu N, Yang M, Gaur U, Xu H, Yao Y, Li D. Alpha-ketoglutarate: physiological functions and applications. Biomol Ther. 2016;24:1.
Galili G, Amir R, Fernie AR. The regulation of essential amino acid synthesis and accumulation in plants. Annu Rev Plant Biol. 2016;67:153–78.
Massey K, Blakeslee C, Pitkow H. A review of physiological and metabolic effects of essential amino acids. Amino Acids. 1998;14:271–300.
Xu J-Z, Yang H-K, Zhang W-G. NADPH metabolism: a survey of its theoretical characteristics and manipulation strategies in amino acid biosynthesis. Crit Rev Biotechnol. 2018;38:1061–76.
Maeda H, Dudareva N. The shikimate pathway and aromatic amino acid biosynthesis in plants. Annu Rev Plant Biol. 2012;63:73–105.
Pratelli R, Pilot G. Regulation of amino acid metabolic enzymes and transporters in plants. J Exp Bot. 2014;65:5535–56.
Fernstrom JD, Fernstrom MH. Tyrosine, phenylalanine, and catecholamine synthesis and function in the brain. J Nutr. 2007;137:1539S-1547S.
Fischer K, Kammerer B, Gutensohn M, Arbinger B, Weber A, Häusler RE, Flügge U-I. A new class of plastidic phosphate translocators: a putative link between primary and secondary metabolism by the phosphoenolpyruvate/phosphate antiporter. Plant Cell. 1997;9:453–62.
Bromke MA. Amino acid biosynthesis pathways in diatoms. Metabolites. 2013;3:294–311.
Stryer L. Biochemistry. New York: WH Freeman and Company; 1995.
Sturani E, Datta P, Hughes M, Gest H. Regulation of enzyme activity by specific reversal of feedback inhibition. Science. 1963;141:1053–4.
Reaves ML, Young BD, Hosios AM, Xu Y-F, Rabinowitz JD. Pyrimidine homeostasis is accomplished by directed overflow metabolism. Nature. 2013;500:237–41.
Yang X, Wu L, Ran Y, Xu A, Zhang B, Yang X, Zhang R, Rao Z, Li J. 2017. Crystal structure of L-glutamate N-acetyltransferase ArgA from Mycobacterium tuberculosis. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics. 1865: 1800–1807.
Ramón-Maiques S, Fernández-Murga ML, Gil-Ortiz F, Vagin A, Fita I, Rubio V. Structural bases of feed-back control of arginine biosynthesis, revealed by the structures of two hexameric N-acetylglutamate kinases, from Thermotoga maritima and Pseudomonas aeruginosa. J Mol Biol. 2006;356:695–713.
Pye VE, Tingey AP, Robson RL, Moody PC. The structure and mechanism of serine acetyltransferase from Escherichia coli. J Biol Chem. 2004;279:40729–36.
Mittelstädt G, Moggré GJ, Panjikar S, Nazmi AR, Parker EJ. Campylobacter jejuni adenosine triphosphate phosphoribosyltransferase is an active hexamer that is allosterically controlled by the twisting of a regulatory tail. Protein Sci. 2016;25:1492–506.
Cho Y, Sharma V, Sacchettini JC. Crystal structure of ATP Phosphoribosyltransferase from Mycobacterium tuberculosis. J Biol Chem. 2003;278:8333–9.
Lohkamp B, McDermott G, Campbell SA, Coggins JR, Lapthorn AJ. The structure of Escherichia coli ATP-phosphoribosyltransferase: identification of substrate binding sites and mode of AMP inhibition. J Mol Biol. 2004;336:131–44.
Gallagher DT, Gilliland GL, Xiao G, Zondlo J, Fisher KE, Chinchilla D, Eisenstein E. Structure and control of pyridoxal phosphate dependent allosteric threonine deaminase. Structure. 1998;6:465–75.
Lonhienne T, Low YS, Garcia MD, Croll T, Gao Y, Wang Q, Brillault L, Williams CM, Fraser JA, McGeary RP. Structures of fungal and plant acetohydroxyacid synthases. Nature. 2020;586:317–21.
Kotaka M, Ren J, Lockyer M, Hawkins AR, Stammers DK. Structures of R-and T-state Escherichia coli aspartokinase III: mechanisms of the allosteric transition and inhibition by lysine. J Biol Chem. 2006;281:31544–52.
Yang Q, Yu K, Yan L, Li Y, Chen C, Li X. Structural view of the regulatory subunit of aspartate kinase from Mycobacterium tuberculosis. Protein Cell. 2011;2:745–54.
Hadfield A, Kryger G, Ouyang J, Petsko GA, Ringe D, Viola R. Structure of aspartate-β-semialdehyde dehydrogenase from Escherichia coli, a key enzyme in the aspartate family of amino acid biosynthesis. J Mol Biol. 1999;289:991–1002.
Mirwaldt C, Korndorfer I, Huber R. The crystal structure of dihydrodipicolinate synthase from Escherichia coli at 2 5 Å resolution. J Mol Biol. 1995;246:227–39.
Clausen T, Huber R, Laber B, Pohlenz H-D, Messerschmidt A. Crystal structure of the pyridoxal-5′-phosphate dependent cystathionine β-lyase from Escherichia coliat 183 Å. J Molecular Biol. 1996;262:202–24.
Vanhooke JL, Thoden JB, Brunhuber NM, Blanchard JS, Holden HM. Phenylalanine dehydrogenase from Rhodococcus sp M4: high-resolution X-ray analyses of inhibitory ternary complexes reveal key features in the oxidative deamination mechanism. Biochemistry. 1999;38:2326–39.
Marco-Marín C, Gil-Ortiz F, Pérez-Arellano I, Cervera J, Fita I, Rubio V. A novel two-domain architecture within the amino acid kinase enzyme family revealed by the crystal structure of Escherichia coli glutamate 5-kinase. J Mol Biol. 2007;367:1431–46.
Dey S, Hu Z, Xu XL, Sacchettini JC, Grant GA. The effect of hinge mutations on effector binding and domain rotation in Escherichia coli D-3-phosphoglycerate dehydrogenase. J Biol Chem. 2007;282:18418–26.
Sekula B, Ruszkowski M, Dauter Z. Structural analysis of phosphoserine aminotransferase (Isoform 1) from Arabidopsis thaliana–the enzyme involved in the phosphorylated pathway of serine biosynthesis. Front Plant Sci. 2018;9:876.
Spraggon G, Kim C, Nguyen-Huu X, Yee M-C, Yanofsky C, Mills SE. The structures of anthranilate synthase of Serratia marcescens crystallized in the presence of (i) its substrates, chorismate and glutamine, and a product, glutamate, and (ii) its end-product inhibitor, L-tryptophan. Proc Natl Acad Sci. 2001;98:6021–6.
Pérez-Arellano I, Carmona-Álvarez F, Martínez AI, Rodríguez-Díaz J, Cervera J. Pyrroline-5-carboxylate synthase and proline biosynthesis: from osmotolerance to rare metabolic disease. Protein Sci. 2010;19:372–82.
Galili G. Regulation of lysine and threonine synthesis. Plant Cell. 1995;7:899.
Rosner A. Control of lysine biosynthesis in Bacillus subtilis: inhibition of diaminopimelate decarboxylase by lysine. J Bacteriol. 1975;121:20–8.
Chen Z, Bommareddy RR, Frank D, Rappert S, Zeng A-P. Deregulation of feedback inhibition of phosphoenolpyruvate carboxylase for improved lysine production in Corynebacterium glutamicum. Appl Environ Microbiol. 2014;80:1388–93.
Geng F, Chen Z, Zheng P, Sun J, Zeng A-P. Exploring the allosteric mechanism of dihydrodipicolinate synthase by reverse engineering of the allosteric inhibitor binding sites and its application for lysine production. Appl Microbiol Biotechnol. 2013;97:1963–71.
Chen Z, Rappert S, Sun J, Zeng A-P. Integrating molecular dynamics and co-evolutionary analysis for reliable target prediction and deregulation of the allosteric inhibition of aspartokinase for amino acid production. J Biotechnol. 2011;154:248–54.
Alvares TS, Meirelles CM, Bhambhani YN, Paschoalin VM, Gomes PS. L-Arginine as a potential ergogenic aidin healthy subjects. Sports Med. 2011;41:233–48.
Park SH, Kim HU, Kim TY, Park JS, Kim S-S, Lee SY. Metabolic engineering of Corynebacterium glutamicum for L-arginine production. Nat Commun. 2014;5:1–9.
Shin JH, Lee SY. Metabolic engineering of microorganisms for the production of L-arginine and its derivatives. Microb Cell Fact. 2014;13:166.
Xu M, Rao Z, Dou W, Yang J, Jin J, Xu Z. Site-directed mutagenesis and feedback-resistant N-acetyl-L-glutamate kinase (NAGK) increase Corynebacterium crenatum L-arginine production. Amino Acids. 2012;43:255–66.
Fernández-Murga ML, Gil-Ortiz F, Llácer JL, Rubio V. Arginine biosynthesis in Thermotoga maritima: characterization of the arginine-sensitive N-acetyl-L-glutamate kinase. J Bacteriol. 2004;186:6142–9.
Slocum RD. Genes, enzymes and regulation of arginine biosynthesis in plants. Plant Physiol Biochem. 2005;43:729–45.
Glansdorff N, Xu Y. Microbial arginine biosynthesis: pathway, regulation and industrial production in amino acid biosynthesis pathways, regulation and metabolic engineering. Berlin: Springer; 2006.
Tatibana M, Shigesada K, Mori M. Acetylglutamate synthetase. New York: John Wiley and Sons; 1976.
Marvil DK, Leisinger T. N-acetylglutamate synthase of Escherichia coli: purification, characterization, and molecular properties. J Biol Chem. 1977;252:3295–303.
Sancho-Vaello E, Fernández-Murga ML, Rubio V. Mechanism of arginine regulation of acetylglutamate synthase, the first enzyme of arginine synthesis. FEBS Lett. 2009;583:202–6.
Das U, Singh E, Dharavath S, Subhramanyam UKT, Pal RK, Vijayan R, Menon S, Kumar S, Gourinath S, Srinivasan A. Structural insights into the substrate binding mechanism of novel ArgA from Mycobacterium tuberculosis. Int J Biol Macromol. 2019;125:970–8.
Shi D, Sagar V, Jin Z, Yu X, Caldovic L, Morizono H, Allewell NM, Tuchman M. The crystal structure of N-acetyl-L-glutamate synthase from Neisseria gonorrhoeae provides insights into mechanisms of catalysis and regulation. J Biol Chem. 2008;283:7176–84.
Min L, Jin Z, Caldovic L, Morizono H, Allewell NM, Tuchman M, Shi D. Mechanism of allosteric inhibition of N-acetyl-L-glutamate synthase by L-arginine. J Biol Chem. 2009;284:4873–80.
Pauwels K, Abadjieva A, Hilven P, Stankiewicz A, Crabeel M. The N-acetylglutamate synthase/N-acetylglutamate kinase metabolon of Saccharomyces cerevisiae allows co-ordinated feedback regulation of the first two steps in arginine biosynthesis. Eur J Biochem. 2003;270:1014–24.
Sancho-Vaello E, Fernández-Murga ML, Rubio V. Functional dissection of N-acetylglutamate synthase (ArgA) of Pseudomonas aeruginosa and restoration of its ancestral N-acetylglutamate kinase activity. J Bacteriol. 2012;194:2791–801.
Shi D, Li Y, Cabrera-Luque J, Jin Z, Yu X, Zhao G, Haskins N, Allewell NM, Tuchman M. A novel N-acetylglutamate synthase architecture revealed by the crystal structure of the bifunctional enzyme from Maricaulis maris. PLoS ONE. 2011;6: e28825.
Zhao G, Jin Z, Allewell NM, Tuchman M, Shi D. Crystal structure of the N-acetyltransferase domain of human N-acetyl-L-glutamate synthase in complex with N-acetyl-L-glutamate provides insights into its catalytic and regulatory mechanisms. PLoS ONE. 2013;8: e70369.
Hoare D, Hoare S. Feedback regulation of arginine biosynthesis in blue-green algae and photosynthetic bacteria. J Bacteriol. 1966;92:375–9.
Haas D. LEISINGER T: N-Acetylglutamate 5-phosphotransferase of Pseudomonas aeruginosa: purification and ligand-directed association-dissociation. Eur J Biochem. 1975;52(365):375.
Gil F, Ramon-Maiques S, Marina A, Fita I, Rubio V. N-Acetyl-L-glutamate kinase from Escherichia coli: cloning of the gene, purification and crystallization of the recombinant enzyme and preliminary X-ray analysis of the free and ligand-bound forms. Acta Crystallogr D Biol Crystallogr. 1999;55:1350–2.
Cunin R, Glansdorff N, Pierard A, Stalon V. Biosynthesis and metabolism of arginine in bacteria. Microbiol Rev. 1986;50:314.
de Cima S, Gil-Ortiz F, Crabeel M, Fita I, Rubio V. Insight on an arginine synthesis metabolon from the tetrameric structure of yeast acetylglutamate kinase. PLoS ONE. 2012;7: e34734.
Ramón-Maiques S, Marina A, Gil-Ortiz F, Fita I, Rubio V. Structure of acetylglutamate kinase, a key enzyme for arginine biosynthesis and a prototype for the amino acid kinase enzyme family, during catalysis. Structure. 2002;10:329–42.
Zhang J, Xu M, Ge X, Zhang X, Yang T, Xu Z, Rao Z. Reengineering of the feedback-inhibition enzyme N-acetyl-l-glutamate kinase to enhance l-arginine production in Corynebacterium crenatum. J Ind Microbiol Biotechnol. 2017;44:271–83.
Fernández-Murga ML, Rubio V. Basis of arginine sensitivity of microbial N-acetyl-L-glutamate kinases: mutagenesis and protein engineering study with the Pseudomonas aeruginosa and Escherichia coli enzymes. J Bacteriol. 2008;190:3018–25.
Korosh TC, Markley AL, Clark RL, McGinley LL, McMahon KD, Pfleger BF. Engineering photosynthetic production of L-lysine. Metab Eng. 2017;44:273–83.
Gunji Y, Tsujimoto N, Shimaoka M, Ogawa-Miyata Y, Sugimoto S, Yasueda H. Characterization of the L-lysine biosynthetic pathway in the obligate methylotroph Methylophilus methylotrophus. Biosci Biotechnol Biochem. 2004;68(1449):1460.
Cahyanto MN, Kawasaki H, Nagashio M, Fujiyama K, Seki T. Regulation of aspartokinase, aspartate semialdehyde dehydrogenase, dihydrodipicolinate synthase and dihydrodipicolinate reductase in Lactobacillus plantarum. Microbiology. 2006;152:105–12.
Ogawa-Miyata Y, Kojima H, Sano K. Mutation analysis of the feedback inhibition site of aspartokinase III of Escherichia coli K-12 and its use in L-threonine production. Biosci Biotechnol Biochem. 2001;65:1149–54.
Ekechukwu CR, Burns TA, Melton T. Selection and Characterization of aspartokinase feedback-insensitive mutants of Azotobacter vinelandii. Appl Environ Microbiol. 1995;61:3189–91.
Yoshida A, Tomita T, Kurihara T, Fushinobu S, Kuzuyama T, Nishiyama M. Structural insight into concerted inhibition of α2β2-type aspartate kinase from Corynebacterium glutamicum. J Mol Biol. 2007;368:521–36.
Yoshida A, Tomita T, Kuzuyama T, Nishiyama M. Mechanism of concerted inhibition of α2β2-type hetero-oligomeric aspartate kinase from Corynebacterium glutamicum. J Biol Chem. 2010;285:27477–86.
Archer J, Solow-Cordero D, Sinskey A. A C-terminal deletion in Corynebacterium glutamicum homoserine dehydrogenase abolishes allosteric inhibition by L-threonine. Gene. 1991;107:53–9.
Kobashi N, Nishiyama M, Tanokura M. Aspartate kinase-independent lysine synthesis in an extremely thermophilic bacterium, Thermus thermophilus: lysine is synthesized via α-aminoadipic acid not via diaminopimelic acid. J Bacteriol. 1999;181:1713–8.
Umbarger HE. Amino acid biosynthesis and its regulation. Ann Rev Biochem. 1978;47:533.
Horie A, Tomita T, Saiki A, Kono H, Taka H, Mineki R, Fujimura T, Nishiyama C, Kuzuyama T, Nishiyama M. Discovery of proteinaceous N-modification in lysine biosynthesis of Thermus thermophilus. Nat Chem Biol. 2009;5:673–9.
Winkler M, Ramos-Montan EM. Biosynthesis of histidine. EcoSal Plus. 2009;3(10):1128.
Brenner M, Ames BN. The histidine operon and its regulation In metabolic regulation. Amsterdam: Elsevier; 1971.
Sn P, JoP P, Gr L-M, Kelly G. Mechanism of feedback allosteric inhibition of ATP phosphoribosyltransferase. Biochemistry. 2012;51(8027):8038.
Sun YJ, Rose J, Wang BC, Hsiao CD. The structure of glutamine-binding protein complexed with glutamine at 1.94 Å resolution: comparisons with other amino acid binding proteins. J Mol Biol. 1998;278:219–29.
Neuss N, T: Spectrum of biological activities of indole alkaloids. Indole and biogenetically related alkaloids/edited by JD Phillipson and MH Zenk 1980.
Niyogi KK, Fink GR. Two anthranilate synthase genes in Arabidopsis: defense-related regulation of the tryptophan pathway. Plant Cell. 1992;4:721–33.
Shiio I, Miyajima R, Nakagawa M. Regulation of aromstic amino acid biosynthesis in Brevibacterium flavum: I. regulation of anthranilaste synthetase. J Biochem. 1972;72:1447–55.
Crawford IP, Yanofsky C. Comparative studies on the regulation of tryptophan synthesi. Crit Rev Biochem. 1980;8:175–89.
Bongaerts J, Krämer M, Müller U, Raeven L, Wubbolts M. Metabolic engineering for microbial production of aromatic amino acids and derived compounds. Metab Eng. 2001;3:289–300.
Lütke-Eversloh T, Santos CNS, Stephanopoulos G. Perspectives of biotechnological production of L-tyrosine and its applications. Appl Microbiol Biotechnol. 2007;77:751–62.
Pohnert G, Zhang S, Husain A, Wilson DB, Ganem B. Regulation of phenylalanine biosynthesis. studies on the mechanism of phenylalanine binding and feedback inhibition in the Escherichia coli P-protein. Biochemistry. 1999;38:12212–7.
Miozzari G, Niederberger P, Hütter R. Tryptophan biosynthesis in Saccharomyces cerevisiae: control of the flux through the pathway. J Bacteriol. 1978;134:48–59.
Zalkin H, Hwang LH. Anthranilate Synthetase from Serratia marcescens on the properties and relationship to the enzyme from Salmonella typhimurium. J Biol Chem. 1971;246:6899–907.
Knöchel T, Ivens A, Hester G, Gonzalez A, Bauerle R, Wilmanns M, Kirschner K, Jansonius JN. The crystal structure of anthranilate synthase from Sulfolobus solfataricus: functional implications. Proc Natl Acad Sci. 1999;96:9479–84.
Poulsen C, Bongaert RJ, Verpoorte R. Purification and characterization of anthranilate synthase from Catharanthus roseus. Eur J Biochem. 1993;212:431–40.
Bohlmann J, Lins T, Martin W, Eilert U. Anthranilate synthase from Ruta graveolens (Duplicated AS [alpha] genes encode tryptophan-sensitive and tryptophan-insensitive isoenzymes specific to amino acid and alkaloid biosynthesis). Plant Physiol. 1996;111:507–14.
Tozawa Y, Hasegawa H, Terakawa T, Wakasa K. Characterization of rice anthranilate synthase α-subunit genesOASA1 and OASA2. tryptophan accumulation in transgenic rice expressing a feedback-insensitive mutant of OASA1. Plant Physiol. 2001;126:1493–506.
Ivens A, Mayans O, Szadkowski H, Wilmanns M, Kirschner K. Purification, characterization and crystallization of thermostable anthranilate phosphoribosyltransferase from Sulfolobus solfataricus. Eur J Biochem. 2001;268:2246–52.
Lee CE, Goodfellow C, Javid-Majd F, Baker EN, Lott JS. The crystal structure of TrpD, a metabolic enzyme essential for lung colonization by Mycobacterium tuberculosis, in complex with its substrate phosphoribosylpyrophosphate. J Mol Biol. 2006;355:784–97.
Kim C, Xuong N-H, Edwards S, Yee M-C, Spraggon G, Mills SE. The crystal structure of anthranilate phosphoribosyltransferase from the enterobacterium Pectobacterium carotovorum. FEBS Lett. 2002;523:239–46.
Marino M, Deuss M, Svergun DI, Konarev PV, Sterner R, Mayans O. Structural and mutational analysis of substrate complexation by anthranilate phosphoribosyltransferase from Sulfolobus solfataricus. J Biol Chem. 2006;281:21410–21.
Cookson TV, Evans GL, Castell A, Baker EN, Lott JS, Parker EJ. Structures of Mycobacterium tuberculosis anthranilate phosphoribosyltransferase variants reveal the conformational changes that facilitate delivery of the substrate to the active site. Biochemistry. 2015;54:6082–92.
Castell A, Short FL, Evans GL, Cookson TV, Bulloch EM, Joseph DD, Lee CE, Parker EJ, Baker EN, Lott JS. The substrate capture mechanism of Mycobacterium tuberculosis anthranilate phosphoribosyltransferase provides a mode for inhibition. Biochemistry. 2013;52:1776–87.
Sugimoto S. Regulation of tryptophan biosynthesis by feedback inhibition of the second-step enzyme, anthranilate phosphoribosyl-transferase, in Brevibacterium flavum. Agric Biol Chem. 1983;47:2295–305.
Stieglitz B, Calvo J. Distribution of the isopropylmalate pathway to leucine among diverse bacteria. J Bacteriol. 1974;118:935–41.
Kohlhaw G, Leary T, Umbarger HE. α-Isopropylmalate Synthase from Salmonella typhimurium PURIFICATION AND PROPERTIES. J Biol Chem. 1969;244:2218–25.
De Kraker J-W, Luck K, Textor S, Tokuhisa JG, Gershenzon J. Two Arabidopsis genes (IPMS1 and IPMS2) encode isopropylmalate synthase, the branchpoint step in the biosynthesis of leucine. Plant Physiol. 2007;143:970–86.
Webster R, Gross S. The α-isopropylmalate synthetase of Neurospora. I. The kinetics and end product control of α-isopropylmalate synthetase function. Biochemistry. 1965;4:2309–18.
De Carvalho LP, Argyrou A, Blanchard JS. Slow-onset Feedback inhibition: inhibition of Mycobacterium t uberculosis α-Isopropylmalate synthase by l-leucine. J Am Chem Soc. 2005;127:10004–5.
De Carvalho LPS, Blanchard JS. Kinetic and chemical mechanism of α-isopropylmalate synthase from Mycobacterium tuberculosis. Biochemistry. 2006;45:8988–99.
Huisman FH, Koon N, Bulloch EM, Baker HM, Baker EN, Squire CJ, Parker EJ. Removal of the C-terminal regulatory domain of α-isopropylmalate synthase disrupts functional substrate binding. Biochemistry. 2012;51:2289–97.
Cavalieri D, Casalone E, Bendoni B, Fia G, Polsinelli M, Barberio C. Trifluoroleucine resistance and regulation of α-isopropyl malate synthase in Saccharomyces cerevisiae. Mol Gen Genet MGG. 1999;261:152–60.
De Carvalho LPS, Frantom PA, Argyrou A, Blanchard JS. Kinetic evidence for interdomain communication in the allosteric regulation of α-isopropylmalate synthase from Mycobacterium tuberculosis. Biochemistry. 2009;48:1996–2004.
da Costa TPS, Muscroft-Taylor AC, Dobson RC, Devenish SR, Jameson GB, Gerrard JA. How essential is the ‘essential’active-site lysine in dihydrodipicolinate synthase? Biochimie. 2010;92:837–45.
Dobson RC, Griffin MD, Roberts SJ, Gerrard JA. Dihydrodipicolinate synthase (DHDPS) from Escherichia coli displays partial mixed inhibition with respect to its first substrate, pyruvate. Biochimie. 2004;86:311–5.
Dogovski C, Atkinson SC, Dommaraju SR, Hor L, Dobson R, Hutton C, Gerrard JA, Perugini M. Lysine biosynthesis in bacteria: an unchartered pathway for novel antibiotic design. Encycl Life Support syst. 2009;11:116–36.
Hutton CA, Perugini MA, Gerrard JA. Inhibition of lysine biosynthesis: an evolving antibiotic strategy. Mol BioSyst. 2007;3:458–65.
Rice EA, Bannon GA, Glenn KC, Jeong SS, Sturman EJ, Rydel TJ. Characterization and crystal structure of lysine insensitive Corynebacterium glutamicum dihydrodipicolinate synthase (cDHDPS) protein. Arch Biochem Biophys. 2008;480:111–21.
Blickling S, Beisel H-G, Bozic D, Knäblein J, Laber B, Huber R. Structure of dihydrodipicolinate synthase of Nicotiana sylvestris reveals novel quaternary structure. J Mol Biol. 1997;274:608–21.
Vauterin M, Frankard V, Jacobs M. Functional rescue of a bacterial dapA auxotroph with a plant cDNA library selects for mutant clones encoding a feedback-insensitive dihydrodipicolinate synthase. Plant J. 2000;21:239–48.
Devenish SR, Huisman FH, Parker EJ, Hadfield AT, Gerrard JA. 2009. Cloning and characterisation of dihydrodipicolinate synthase from the pathogen Neisseria meningitidis. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics. 1794 8 1168–1174.
Kaur N, Gautam A, Kumar S, Singh A, Singh N, Sharma S, Sharma R, Tewari R, Singh TP. Biochemical studies and crystal structure determination of dihydrodipicolinate synthase from Pseudomonas aeruginosa. Int J Biol Macromol. 2011;48:779–87.
da Costa TPS, Desbois S, Dogovski C, Gorman MA, Ketaren NE, Paxman JJ, Siddiqui T, Zammit LM, Abbott BM, Robins-Browne RM. Structural determinants defining the allosteric inhibition of an essential antibiotic target. Structure. 2016;24:1282–91.
Joerger AC, Mayer S, Fersht AR. Mimicking natural evolution in vitro: an N-acetylneuraminate lyase mutant with an increased dihydrodipicolinate synthase activity. Proc Natl Acad Sci. 2003;100:5694–9.
Brush A, Paulus H. The enzymic formation of O-acetylhomoserine in Bacillus subtilis and its regulation by methionine and S-adenosylmethione. Biochem Biophys Res Commun. 1971;45:735–41.
Duclos B, Cortay J, Bleicher F, Ron E, Richaud C, Saint Girons I, Cozzone A. Nucleotide sequence of the metA gene encoding homoserine trans-succinylase in Escherichia coli. Nucleic Acids Res. 1989;17:2856.
Shiio I, Ozaki H. Feedback inhibition by methionine and S-adenosylmethionine, and desensitization of homoserine O-acetyltransferase in Brevibacterium flavum. J Biochem. 1981;89:1493–500.
Mirza IA, Nazi I, Korczynska M, Wright GD, Berghuis AM. Crystal structure of homoserine transacetylase from Haemophilus influenzae reveals a new family of α/β-hydrolases. Biochemistry. 2005;44:15768–73.
Thangavelu B, Pavlovsky AG, Viola R. Structure of homoserine O-acetyltransferase from Staphylococcus aureus: the first Gram-positive ortholog structure. Acta Crystallographica Sect F Struct Biol Commun. 2014;70:1340–5.
Chiu H-J, Abdubek P, Astakhova T, Axelrod HL, Carlton D, Clayton T, Das D, Deller MC, Duan L, Feuerhelm J. The structure of Haemophilus influenzae prephenate dehydrogenase suggests unique features of bifunctional TyrA enzymes. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:1317–25.
Hudson GS, Wong V, Davidson BE. Chorismate mutase/prephenate dehydrogenase from Escherichia coli K12: purification, characterization, and identification of a reactive cysteine. Biochemistry. 1984;23:6240–9.
Kredich NM, Tomkins GM. The enzymic synthesis of L-cysteine in Escherichia coli and Salmonella typhimurium. J Biol Chem. 1966;241:4955–65.
Takagi H, Ohtsu I. L-Cysteine metabolism and fermentation in microorganisms amino acid fermentation. Berlin: Springer; 2016.
Olsen LR, Huang B, Vetting MW, Roderick SL. Structure of serine acetyltransferase in complexes with CoA and its cysteine feedback inhibitor. Biochemistry. 2004;43:6013–9.
Grant GA. A new family of 2-hydroxyacid dehydrogenases. Biochem Biophys Res Commun. 1989;165:1371–4.
Bell JK, Grant GA, Banaszak LJ. Multiconformational states in phosphoglycerate dehydrogenase. Biochemistry. 2004;43:3450–8.
Dey S, Hu Z, Xu XL, Sacchettini JC, Grant GA. D-3-Phosphoglycerate dehydrogenase from Mycobacterium tuberculosis is a link between the Escherichia coli and mammalian enzymes. J Biol Chem. 2005;280:14884–91.
Grant GA. Contrasting catalytic and allosteric mechanisms for phosphoglycerate dehydrogenases. Arch Biochem Biophys. 2012;519:175–85.
Ho C-L, Noji M, Saito M, Saito K. Regulation of serine biosynthesis in Arabidopsis: crucial role of plastidic 3-phosphoglycerate dehydrogenase in non-photosynthetic tissues. J Biol Chem. 1999;274:397–402.
Shimizu Y, Sakuraba H, Doi K, Ohshima T. Molecular and functional characterization of D-3-phosphoglycerate dehydrogenase in the serine biosynthetic pathway of the hyperthermophilic archaeon Sulfolobus tokodaii. Archiv Biochem Biophys. 2008;470:120–8.
Thompson JR, Bell JK, Bratt J, Grant GA, Banaszak LJ. V max regulation through domain and subunit changes. the active form of phosphoglycerate dehydrogenase. Biochemistry. 2005;44:5763–73.
Xu XL, Grant GA. Regulation of Mycobacterium tuberculosis D-3-phosphoglycerate dehydrogenase by phosphate-modulated quaternary structure dynamics and a potential role for polyphosphate in enzyme regulation. Biochemistry. 2014;53:4239–49.
Hong Z, Lakkineni K, Zhang Z, Verma DPS. Removal of feedback inhibition of Δ1-pyrroline-5-carboxylate synthetase results in increased proline accumulation and protection of plants from osmotic stress. Plant Physiol. 2000;122:1129–36.
Seddon AP, Zhao KY, Meister A. Activation of glutamate by γ-glutamate kinase: formation of γ-cis-cycloglutamyl phosphate, an analog of γ-glutamyl phosphate. J Biol Chem. 1989;264:11326–35.
Zhang Y, Shang X, Deng A, Chai X, Lai S, Zhang G, Wen T. Genetic and biochemical characterization of Corynebacterium glutamicum ATP phosphoribosyltransferase and its three mutants resistant to feedback inhibition by histidine. Biochimie. 2012;94:829–38.
Schendzielorz G, Dippong M, Grünberger A, Kohlheyer D, Yoshida A, Binder S, Nishiyama C, Nishiyama M, Bott M, Eggeling L. Taking control over control: use of product sensing in single cells to remove flux control at key enzymes in biosynthesis pathways. ACS Synth Biol. 2014;3:21–9.
Kikuchi Y, Kojima H, Tanaka T. Mutational analysis of the feedback sites of lysine-sensitive aspartokinase of Escherichia coli. FEMS Microbiol Lett. 1999;173:211–5.
Falco S, Guida T, Locke M, Mauvais J, Sanders C, Ward R, Webber P. Transgenic canola and soybean seeds with increased lysine. Bio/Technology. 1995;13:577–82.
Chen Z, Meyer W, Rappert S, Sun J, Zeng A-P. Coevolutionary analysis enabled rational deregulation of allosteric enzyme inhibition in Corynebacterium glutamicum for lysine production. Appl Environ Microbiol. 2011;77:4352–60.
Dong X, Zhao Y, Zhao J, Wang X. Characterization of aspartate kinase and homoserine dehydrogenase from Corynebacterium glutamicum IWJ001 and systematic investigation of L-isoleucine biosynthesis. J Ind Microbiol Biotechnol. 2016;43:873–85.
Sindelar G, Wendisch VF. Improving lysine production by Corynebacterium glutamicum through DNA microarray-based identification of novel target genes. Appl Microbiol Biotechnol. 2007;76:677–89.
Singhania RR, Patel AK, Thomas L, Goswami M, Giri BS, Pandey A. Industrial enzymes In Industrial biorefineries & white biotechnology. Amsterdam: Elsevier; 2015.
Zeymer C, Hilvert D. Directed evolution of protein catalysts. Annu Rev Biochem. 2018;87:131–57.
B-z Zhang Zhang X, An X, Ran D, Zhou Y, Lu J, Tong Y. An easy-to-use site-directed mutagenesis method with a designed restriction site for convenient and reliable mutant screening. J Zhejiang Univ Sci B. 2009;10:479–82.
Chronopoulou EG, Labrou NE. Site-saturation mutagenesis: a powerful tool for structure-based design of combinatorial mutation libraries. Current Protocols Protein Sci. 2011;63(26):21–6.
Steffens DL, Williams JG. Efficient site-directed saturation mutagenesis using degenerate oligonucleotides. JBT. 2007;18:147.
Labrou NE. Random mutagenesis methods for in vitro directed enzyme evolution. Curr Protein Pept Sci. 2010;11:91–100.
McCullum EO, Williams BA, Zhang J, Chaput JC. Random mutagenesis by error-prone PCR In In vitro mutagenesis protocols. Berlin: Springer; 2010.
Ye J, Wen F, Xu Y, Zhao N, Long L, Sun H, Yang J, Cooley J, Pharr GT, Webby R. Error-prone pcr-based mutagenesis strategy for rapidly generating high-yield influenza vaccine candidates. Virology. 2015;482:234–43.
Ding X, Snyder AK, Shaw R, Farmerie WG, Song W-Y. Direct retransformation of yeast with plasmid DNA isolated from single yeast colonies using rolling circle amplification. Biotechniques. 2003;35:774–9.
Fire A, Xu S-Q. Rolling replication of short DNA circles. Proc Natl Acad Sci. 1995;92:4641–5.
Fujii R, Kitaoka M, Hayashi K. One-step random mutagenesis by error-prone rolling circle amplification. Nucleic Acids Res. 2004;32:e145–e145.
Liu D, Daubendiek SL, Zillman MA, Ryan K, Kool ET. Rolling circle DNA synthesis: small circular oligonucleotides as efficient templates for DNA polymerases. J Am Chem Soc. 1996;118:1587–94.
Xu M, Rao Z, Dou W, Jin J, Xu Z. Site-directed mutagenesis studies on the L-arginine-binding sites of feedback inhibition in N-acetyl-L-glutamate kinase (NAGK) from Corynebacterium glutamicum. Curr Microbiol. 2012;64:164–72.
Zhao Q, Luo Y, Dou W, Zhang X, Zhang X, Zhang W, Xu M, Geng Y, Rao Z, Xu Z. Controlling the transcription levels of argGH redistributed L-arginine metabolic flux in N-acetylglutamate kinase and ArgR-deregulated Corynebacterium crenatum. J Ind Microbiol Biotechnol. 2016;43:55–66.
Peverelli MG, da Costa TPS, Kirby N, Perugini MA. Dimerization of bacterial diaminopimelate decarboxylase is essential for catalysis. J Biol Chem. 2016;291:9785–95.
Kalinowski J, Cremer J, Bachmann B, Eggeling L, Sahm H, Pühler A. Genetic and biochemical analysis of the aspartokinase from Corynebacterium glutamicum. Mol Microbiol. 1991;5:1197–204.
Omori K, Komatsubara S. Role of serine 352 in the allosteric response of Serratia marcescens aspartokinase I-homoserine dehydrogenase I analyzed by using site-directed mutagenesis. J Bacteriol. 1993;175:959–65.
Petit C, Kim Y, Lee S-K, Brown J, Larsen E, Ronning DR, Suh J-W, Kang C-M. Reduction of feedback inhibition in homoserine kinase (ThrB) of Corynebacterium glutamicum enhances L-threonine biosynthesis. ACS Omega. 2018;3:1178–86.
Huo X, Viola RE. Substrate specificity and identification of functional groups of homoserine kinase from Escherichia coli. Biochemistry. 1996;35:16180–5.
Kumar S, Mazumder M, Dharavath S, Gourinath S. Single residue mutation in active site of serine acetyltransferase isoform 3 from Entamoeba histolytica assists in partial regaining of feedback inhibition by cysteine. PLoS ONE. 2013;8: e55932.
Denk D. BÖCK A: L-cysteine biosynthesis in Escherichia coli: nucleotide sequence and expression of the serine acetyltransferase (cysE) gene from the wild-type and a cysteine-excreting mutant. Microbiology. 1987;133:515–25.
Nakamori S, Kobayashi S, Kobayashi C, Takagi H. Overproduction of l-Cysteine andl-Cystine by Escherichia coli strains with a genetically altered serine acetyltransferase. Appl Environ Microbiol. 1998;64(1607):1611.
Casey AK, Hicks MA, Johnson JL, Babbitt PC, Frantom PA. Mechanistic and bioinformatic investigation of a conserved active site helix in α-isopropylmalate synthase from Mycobacterium tuberculosis, a member of the DRE-TIM metallolyase superfamily. Biochemistry. 2014;53:2915–25.
Schmidheini T, Sperisen P, Paravicini G, Hütter R, Braus G. A single point mutation results in a constitutively activated and feedback-resistant chorismate mutase of Saccharomyces cerevisiae. J Bacteriol. 1989;171:1245–53.
Schnappauf G, Sträter N, Lipscomb WN, Braus GH. A glutamate residue in the catalytic center of the yeast chorismate mutase restricts enzyme activity to acidic conditions. Proc Natl Acad Sci. 1997;94:8491–6.
Lütke-Eversloh T, Stephanopoulos G. Feedback inhibition of chorismate mutase/prephenate dehydrogenase (TyrA) of Escherichia coli: generation and characterization of tyrosine-insensitive mutants. Appl Environ Microbiol. 2005;71:7224–8.
Frappier V, Chartier M, Najmanovich RJ. ENCoM server: exploring protein conformational space and the effect of mutations on protein function and stability. Nucleic Acids Res. 2015;43:W395–400.
Vedithi SC, Rodrigues CH, Portelli S, Skwark MJ, Das M, Ascher DB, Blundell TL, Malhotra S. Computational saturation mutagenesis to predict structural consequences of systematic mutations in the beta subunit of RNA polymerase in Mycobacterium leprae. Comput Struct Biotechnol J. 2020;18:271–86.
McCafferty C, Sergeev YV. Unfolding mutation screen for inherited eye disease. Invest Ophthalmol Vis Sci. 2016;57:3162–3162.
McCafferty CL, Sergeev YV. Dataset of eye disease-related proteins analyzed using the unfolding mutation screen. Sci Data. 2016;3:1–10.
McCafferty CL, Sergeev YV. Global computational mutagenesis provides a critical stability framework in protein structures. PLoS ONE. 2017;12: e0189064.
Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–7.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Current Protocol Human Gene. 2013;76:21–7.
Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, De Castro E, Duvaud S, Flegel V, Fortier A, Gasteiger E. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012;40:W597–603.
Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–35.
Chen C-W, Chang K-P, Ho C-W, Chang H-P, Chu Y-W. KStable: A computational method for predicting protein thermal stability changes by k-star with regular-mRMR feature selection. Entropy. 2018;20:988.
Folkman L, Stantic B, Sattar A, Zhou Y. EASE-MM: sequence-based prediction of mutation-induced stability changes with feature-based multiple models. J Mol Biol. 2016;428:1394–405.
Guarnera E, Tan ZW, Zheng Z, Berezovsky IN. AlloSigMA: allosteric signaling and mutation analysis server. Bioinformatics. 2017;33:3996–8.
Schymkowitz JW, Rousseau F, Martins IC, Ferkinghoff-Borg J, Stricher F, Serrano L. Prediction of water and metal binding sites and their affinities by using the Fold-X force field. Proc Natl Acad Sci. 2005;102:10147–52.
Tan ZW, Tee W-V, Guarnera E, Booth L, Berezovsky IN. AlloMAPS: allosteric mutation analysis and polymorphism of signaling database. Nucleic Acids Res. 2019;47:D265–70.
Pahari S, Li G, Murthy AK, Liang S, Fragoza R, Yu H, Alexov E. SAAMBE-3D: Predicting effect of mutations on protein–protein interactions. Int J Mol Sci. 2020;21:2563.
Acknowledgements
Authors are thankful to National Key Research and Development Program of China and Science and Technology Partnership Program, Ministry of Science of China for providing support for current study.
Funding
This work was funded by the National Key Research and Development Program of China (2018YFA0900300); the International Partnership Program of Chinese Academy of Sciences (153D31KYSB20170121); Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-PTJS-001, TSBICIP-KJGG-005); Science and Technology Partnership Program, Ministry of Science of China (KY202001017).
Author information
Authors and Affiliations
Contributions
SN conceived, designed and wrote the original manuscript. PL and UF performed the bibliographic research, review and editing of the manuscript. HM was involved in funding acquisition, conceptualization, project administration, supervision of current study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Authors give their full consent for publication of article.
Competing interests
The authors declared that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Naz, S., Liu, P., Farooq, U. et al. Insight into de-regulation of amino acid feedback inhibition: a focus on structure analysis method. Microb Cell Fact 22, 161 (2023). https://doi.org/10.1186/s12934-023-02178-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-023-02178-z