
Abstract
Background
Noncanonical redox cofactors are emerging as important tools in cell-free biosynthesis to increase the economic viability, to enable exquisite control, and to expand the range of chemistries accessible. However, these noncanonical redox cofactors need to be biologically synthesized to achieve full integration with renewable biomanufacturing processes.
Results
In this work, we engineered Escherichia coli cells to biosynthesize the noncanonical cofactor nicotinamide mononucleotide (NMN+), which has been efficiently used in cell-free biosynthesis. First, we developed a growth-based screening platform to identify effective NMN+ biosynthetic pathways in E. coli. Second, we explored various pathway combinations and host gene disruption to achieve an intracellular level of ~ 1.5 mM NMN+, a 130-fold increase over the cell’s basal level, in the best strain, which features a previously uncharacterized nicotinamide phosphoribosyltransferase (NadV) from Ralstonia solanacearum. Last, we revealed mechanisms through which NMN+ accumulation impacts E. coli cell fitness, which sheds light on future work aiming to improve the production of this noncanonical redox cofactor.
Conclusion
These results further the understanding of effective production and integration of NMN+ into E. coli. This may enable the implementation of NMN+-directed biocatalysis without the need for exogenous cofactor supply.
Similar content being viewed by others

Introduction
In the last decade, cell free biosynthesis has emerged as a prominent tool in the production of renewable chemicals, fuels, and pharmaceuticals [1,2,3]. Cell-free systems, both purified enzyme-based and crude lysate-based, have unique advantages over whole-cell biotransformation systems. For example, environmental conditions can be varied within a wider range to favor product formation [4]; transportation issues across cell membranes are eliminated [5]; toxic compounds can be produced at much higher titers than the cell’s tolerance limit [6]. Because components of the biosynthetic pathways can be readily mix-and-matched in a combinatorial fashion, cell-free biosynthesis has also been used as a high-throughput prototyping tool to inform pathway design in whole-cell biosynthesis [7, 8].
Cofactors such as nicotinamide adenine dinucleotide (phosphate) (NAD(P)+) are essential reagents in biosynthesis. In cell-free biosynthesis, cofactors are freed from the life-sustaining roles they play in vivo. Therefore, true opportunities exist to significantly expand the toolkit of cofactors beyond what is offered by Nature to achieve desirable goals in biocatalysis. For example, cheaper noncanonical cofactors, such as 3-carbamoyl-1-phenethylpyridin-1-ium chloride (P2NA+) [9, 10], have been used in purified enzyme-based redox catalysis to increase economic viability. Noncanonical cofactors with stronger electron-accepting capability, such as 3-acetylpyridine adenine dinucleotide [11, 12], have been used to drive the thermodynamically unfavorable reactions of alcohol oxidation.
We recently developed a cell-free biosynthesis platform surrounding the noncanonical redox cofactor nicotinamide mononucleotide (NMN+) [13]. NMN+ was enzymatically cycled by pairing an engineered glucose dehydrogenase from Bacillus subtilis with a variety of enzymes to reduce activated C = C double bonds, activated C ≡ C triple bonds, nitro groups, and to supply electrons to a cytochrome P450. This system demonstrated robust temporal stability over 96 h and a total turnover number of ~ 39,000. Because of its smaller size, NMN+ has also been shown to provide a faster mass transfer rate in enzymatic biofuel cells [14].
Compared to other noncanonical cofactors which are made through chemical synthesis [15,16,17], NMN+ is particularly suited for fully renewable biomanufacturing processes because it is accessible through biosynthesis [18,19,20]. This feature is especially desirable in crude lysate-based cell-free biosynthesis and whole-cell biosynthesis, where NMN+ produced in the cells does not need to be purified or exogenously supplied, and it can be directly used for downstream biocatalysis. Importantly, since we demonstrated its successful application in E. coli whole cells to enable orthogonal electron delivery [13], NMN+ can potentially be utilized in crude lysate-based biosynthesis to control the flow of reducing power and mitigate side reactions based on the same principles [13, 21].
Although NMN+ has been biosynthesized previously in metabolically engineered E. coli [13, 19], further improving NMN+ production requires more efficient pathways and a better understanding of its metabolism in the host. While previous efforts have primarily used the nicotinamide phosphoribosyltransferases, NadV, to convert nicotinamide to NMN+, only a few NadV homologs have been tested and many other NadV-independent pathways for NMN+ biosynthesis remain unexplored. Furthermore, whether and how NMN+ accumulation impacts cell physiology remains largely unknown. In this work, we developed a growth-based screening platform to identify pathways for efficient NMN+ generation in vivo. This platform was designed by making NMN+ an essential precursor in NAD+ biosynthesis in engineered E. coli. We used this platform to demonstrate that NMN+ synthetase, NadE* from Francisella tularensis, effectively mediates an additional route for NMN+ biosynthesis in E. coli. We also bioprospected for NadV homologs based on comparative genomic data [18], and we tested their ability to produce NMN+ in combination with F. tularensis NadE*. The best NMN+ producing strain accumulated ~ 1.5 mM of intracellular NMN+ while overexpressing F. tularensis NadE* and Ralstonia solanacearum NadV simultaneously, as well as harboring a disruption in the gene encoding NMN+ amidohydrolase, PncC. Although our current highest NMN+ production titer did not cause growth inhibition, we observed inhibitory effect when very high concentrations of NMN+ was fed to the cells through a heterologous NMN+ transporter. Interestingly, we showed that this inhibitory effect can be alleviated when the transcriptional regulator of NAD+ biosynthesis, NadR, was disrupted. Together, these results provide insight for future metabolic engineering efforts aiming to further improve NMN+ biosynthesis. Compared to NAD(P)+, NMN+ has been suggested to be less expensive [22]. The development of an efficient NMN+ biosynthetic route from even cheaper precursors may further increase the economical viability of NMN+-dependent biotransformation processes.
Results
Identification of NMN+ biosynthetic routes
In E. coli cells, NMN+ is only present at a nominal level, ~ 11.5 µM as previously reported [13], as the product of the DNA ligase reaction [18]. On the other hand, NMN+ accumulates to higher levels and serves as a main intermediate in NAD+ biosynthesis in other organisms [23, 24]. Here, we sought to systematically investigate the effectiveness of these heterologous NMN+ biosynthetic routes in E. coli (Fig. 1).

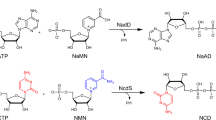
Establishing NMN+ Biosynthetic Routes in Escherichia coli. NMN+ is produced in a small amount through the DNA ligase reaction in the E. coli cell. Heterologous enzymes, shown in green, can introduce new routes to generate NMN+. E. coli endogenous genes and transport processes are shown in blue. Pathway 1 introduces an NMN+ synthase from Francisella tularensis to produce NMN+ from NaMN+. Pathway 2, NR salvage, produces NAD+ from NR. Pathway 3, NA salvage, produces NMN+ from NA. The NMN+ transporter PnuC* from Salmonella enterica, shown in orange, enabled transport of NMN+ into the cell. The endogenous pncC, was targeted for gene disruption to prevent NMN+ degradation. In the presence of NAD+, NadR inhibits transcription of the genes involved in de novo NAD+ biosynthesis, nicotinate salvage, and E. coli pnuC. NaMN+, nicotinic acid mononucleotide; NaAD+, nicotinic adenine dinucleotide; NAD+, nicotinamide adenine dinucleotide; NMN+, nicotinamide mononucleotide; NR, nicotinamide riboside; NadD, NaMN adenylyltransferase; NadR, NMN adenylyltransferase; NadE, NAD synthase; Ft NadE*, NMN synthase from Francisella tularensis; PncC, NMN amidohydrolase; NadV, nicotinamide phosphoribosyltransferase; Pnuc, nicotinamide riboside transporter; Pnuc*, a mutant PnuC enabling direct transport of NMN+ across the cell membrane
Three major NMN+ biosynthetic pathways exist in Nature (Fig. 1): Pathway 1 produces NMN+ from nicotinic acid mononucleotide (NaMN+) using NMN+ synthetase (NadE*), and it was shown to be part of the de novo NAD+ biosynthetic pathway in a small group of prokaryotes including F. tularensis [24]. Pathway 2 involves phosphorylation of nicotinamide riboside (NR) and functions to salvage NR to ultimately yield NAD+. To establish this pathway, we chose to overexpress the native NR transporter in E. coli, PnuC [25], in conjunction with two different NR kinases, Nrk1 from Saccharomyces cerevisiae [26] and NadR from Salmonella enterica [27]. Pathway 3 uses nicotinamide phosphoribosyltransferase (NadV) to convert nicotinamide (NA) to NMN+; it plays a role in NA salvage in vertebrates and some bacteria. Marinescu and coworkers demonstrated NMN+ accumulation in E. coli by heterologously expressing three NadV homologs from Haemophilus ducreyi, Shewanella oneidensis, and Mus musculus while feeding NA [19], and they showed that H. ducreyi NadV performed the best [19]. We previously showed that NadV from F. tularensis, which belongs to a different clade in the NadV phylogenetic tree to H. ducreyi NadV [18], can also effectively produce NMN+ in E. coli [13]. Here, we sought to explore more bacterial NadVs in the same family of F. tularensis NadV and compare them with H. ducreyi and F. tularensis NadV. Namely, we chose NadV homologs from R. solanacearum, Synechocystis sp., and Synechococcus elongatus [18].
All three pathways have unique advantages. While Pathway 3 requires a much cheaper substrate than Pathway 2 (NA versus NR), the latter incorporates ATP hydrolysis as a robust driving force. Since NaMN+ is an intermediate in E. coli’s de novo NAD+ biosynthesis and can be efficiently produced from central metabolites (Fig. 1), Pathway 1 has the potential to achieve complete de novo NMN+ biosynthesis from simple feed stocks such as glucose.
Evaluating the NMN+ biosynthetic pathways in vivo
We evaluated the three above-mentioned pathways in E. coli using a growth-based screening platform (Figs. 1 and 2). To link NMN+ production to cell survival, we employed E. coli strain 72c, which contains a temperature sensitive allele of nadD, an essential gene in NAD+ biosynthesis. As a result, the cells cannot grow at 42 °C [28] unless NMN+ can accumulate inside the cells and be converted to NAD+ by E. coli NadR (Figs. 1 and 2). Previous work has also established NMN+-dependent NAD+ biosynthesis to rescue NAD+ auxotrophy in E. coli [29].

Identification of efficient NMN+ production pathways in vivo using a growth-based screening platform. A growth-base screening platform was used to identify pathways which efficiently generated NMN+ in vivo. E. coli strain 72c, which contains a temperature sensitive allele of nadD (ts nadD), which exhibits a conditionally lethal phenotype when cultured at 42 °C because the native NAD+ biosynthesis is disrupted. Therefore, the cell must rely on intracellular NMN+ to restore NAD+ formation and growth. a Direct feeding of NMN+ into the growth medium with the overexpression of an NMN+ transporter, PnuC* from S. enterica, restored growth to levels near the wild type control, indicating the platform is effective for NMN+ production screening. b Introducing F. tularensis NadE* also restored growth with the supplementation of nicotinamide. c, d Overexpression of E. coli ‘s native nicotinamide riboside (NR) transporter PnuC, paired with S. enterica NadR (c) or S. cerevisiae Nrk1 (d) while feeding NR failed to efficiently restore growth. Screening was performed in a deep-well 96-well plate containing 1 mL of LB medium supplemented with 2 g/L d-glucose and 200 μM of NMN+ precursors, if applicable
When 200 μM NMN+ was directly fed to the cells expressing an NMN+ transporter from S. enterica PnuC* [30] (on a multiple-copy plasmid pWB302), growth was restored to levels comparable to wild type cells after 6 h (Fig. 2a). In contrast, cells carrying a control plasmid (pWB301) could not grow, suggesting the basal level of NMN+ in E. coli cells does not cause background issues in this growth-based screening, possibly because E. coli NadR has low affinity towards NMN+ [31]. These results demonstrate that the screening platform is functioning properly, and cell growth is serving as a readout for intracellular NMN+ level. We and others have previously shown that NMN+ can enter E. coli cells without expressing a heterologous transporter [13, 29]. In this work, the amount of NMN+ supplementation can be substantially reduced, suggesting that S. enterica PnuC* improves the efficiency of NMN+ transportation into E. coli cells.
Overexpression of F. tularensis NadE* (Fig. 1, Pathway 1, on the plasmid pWB303) with 200 μM nicotinamide supplementation restored growth to a similar level as directly feeding NMN+ (Fig. 2b). Nicotinamide can yield the substrate of NadE*, namely NaMN+, through E. coli ‘s native salvage pathway. Interestingly, growth restoration by F. tularensis NadE* does not depend on nicotinamide feeding (Additional file 1: Figure S1). NMN+ production using S. cerevisiae Nrk1 or S. enterica NadR (Fig. 2, Pathway 2, on the plasmids pWB304 and pWB305) with 200 μM NR supplementation was not efficient enough to restore growth (Fig. 2c, d). This may be due to the poor expression of these heterologous kinases in E. coli and their relatively high KM for NR and ATP [30]. Therefore, we did not proceed with Pathway 2.
Taken together, these results suggest that besides the well-established NadV route (Fig. 1, Pathway 3) [19], the F. tularensis NadE*-dependent pathway (Pathway 1) is also effective in E. coli for NMN+ biosynthesis.
Bioprospecting NadV homologs and optimizing NMN+ biosynthesis
After demonstrating NMN+ can be effectively generated by overexpressing F. tularensis NadE*, we examined the effects of pairing it with different NadV homologs. F. tularensis NadE* was overexpressed in a synthetic operon on a multiple-copy plasmid with each of the five NadV candidates as described above (pWB203, pDB101, pDB102, pDB103, pDB104). Cells were fed 1 mM of NA and grown for 4 h before processing and quantification of intracellular NMN+ and NAD+ levels via LC–MS analysis as previously described [13]. When using the wild type BW25113 cells as the host, ~ 12 to 51 µM of NMN+ was produced through these pathways (Fig. 3). We previously found that low levels of intracellular NMN+ could be attributed to NMN+ degradation by the NMN+ amidohydrolase, PncC [13]. Expression of the NadE*/NadV pathways in a ΔpncC strain, JW2670-1 significantly increased the intracellular NMN+ levels. When the NadV candidates were expressed without F. tularensis NadE* in JW2670-1 (on plasmids pWB303, pWB306, pWB307, pWB308, pWB309), intracellular NMN+ levels were lower for all NadVs except H. ducreyi (Fig. 3). Notably, expressing R. solanacerum and S. elongatus NadVs alone resulted in a significant growth challenge (Additional file 1: Figure S2) and the concomitant diminishing of NMN+ production (Fig. 3). However, this growth challenge could be overcome by expressing the NadV with F. tularensis NadE* (Additional file 1: Figure S2), indicating that Ft NadE* may provide a synergistic benefit to the stability, activity, or expression for some NadV candidates. Future work is needed to pinpoint the molecular mechanism behind this synergy.

Pathway combination and strain modification improved NMN+ production. NadV homologs were co-overexpressed with and without F. tularensis NadE* in wild type and ΔpncC cells. In wild type cells, introducing F. tularensis NadE* and NadV homologs only resulted in low levels of NMN+ accumulation. Disrupting the NMN+ degrading-enzyme PncC greatly increased intracellular NMN+ levels. When the NadV homologs were expressed without F. tularensis NadE*, intracellular NMN+ levels decreased for all candidates except H. ducreyi. In some cases, only expressing NadV caused a significant growth detriment (shown as n.d.). Of the NadV homologs tested, R. solanacearum NadV demonstrated the highest intracellular NMN+ production of ~ 1.5 mM in ΔpncC cells, a 130-fold increase over the cell’s basal level when expressed with F. tularensis NadE*. Cells were grown in 2xYT medium supplemented with 1 mM nicotinamide at 30 °C for 4 h. NMN+ concentration was determined by LC-MS. Ft Francisella tularensis, Hd Haemophilus ducreyi, Rs Ralstonia solanacearum, Ss Synechocystis sp., SynE Synechococcus elongatus, n.d not determined due to poor growth
Cells expressing F. tularensis NadE* and R. solanacearum NadV in the ΔpncC strain reached the highest intracellular NMN+ level of ~ 1.5 mM, a 130-fold increase over the cell’s basal NMN+ level [13], when tested under the same conditions (Fig. 3). Furthermore, the R. solanacearum NadV strain performed better than F. tularensis NadV, the NadV we used in our previous work [13], exhibiting a 2.8-fold increase in intracellular NMN+ concentration. R. solanacearum NadV also performed better than H. ducreyi NadV [19] when paired with F. tularensis NadE*.
Investigating the physiological response to NMN+ accumulation
Even though the de novo NAD+ biosynthesis pathway was unmodified, the ΔpncC cells overexpressing F. tularensis NadE* and NadV homologs had lower intracellular NAD+ levels compared to the control strain in which no proteins were overexpressed and intracellular NMN+ level was low (Fig. 3, Additional file 1: S3). This suggests that maintaining intracellular NMN+ at millimolar-range concentrations may be detrimental to cellular fitness. Although we observed no growth defects in our current best NMN+ producing strain, the potential physiological effects of NMN+ accumulation may become a bottleneck for future strain optimization.
Since the effects of NMN+ accumulation on E. coli are not well understood, we sought to stress the cells by drastically increasing the intracellular NMN+ levels in the ΔpncC strain and observe cellular growth. NMN+ concentration was titrated in the medium while expressing the exogenous NMN+ transporter S. enterica PnuC*. We found that cell growth was inhibited at high NMN+ concentrations (> 5 mM) (Fig. 4), which suggests that elevated NMN+ level may interfere with physiological processes in E. coli. Although intracellular NMN+ levels were not measured in this application, PnuC*-mediated NMN+ transport was shown to be active with as low as 200 μM of NMN+ supplementation to support growth restoration (Fig. 2a). Given the decrease in intracellular NAD+ level upon NMN+ accumulation (Additional file 1: Figure S3), we hypothesized that NMN+ may regulate NAD+ biosynthesis, and we sought to examine whether this regulation was mediated by the transcriptional regulator of NAD+ biosynthesis, NadR [32]. Interestingly, when NadR was disrupted, the growth inhibition effect of NMN+ was significantly alleviated (Fig. 4). These results suggest that NadR may indeed play a role in the physiological response to NMN+ accumulation in E. coli. NAD+ has been suggested to allosterically modulate NadR’s function [33]. Given NadR’s capability to also recognize NMN+ [31], further studies are needed to investigate whether NMN+ binding induces conformational change in the DNA-binding domain of NadR and modulates its function as a transcriptional regulator.

NMN+ accumulation affects cell physiology possibly via NadR. To determine if NMN+ accumulation impacts cell fitness, NMN+ was titrated in the growth medium of cells expressing the NMN+ transporter S. enterica PnuC*. The ΔpncC strain exhibited decreased growth at high NMN+ concentrations. Disruption of nadR significantly alleviated the growth inhibition. This suggests the NadR may mediate the physiological response to NMN+ accumulation in E. coli. Cells were grown at 30 °C for 6.5 h in a deep-well 96-well plate containing 1 mL of medium per well
Discussion
This work represents the initial steps towards filling some of the fundamental knowledge gaps that remained open in previous work on NMN+ biosynthesis in E. coli. The important work by Marinescu and coworkers [19] focused on the NadV pathway, but it left many other naturally occurring NMN+ biosynthetic routes unexplored. Our previous work [13] sought to simply recapitulate the NMN+ metabolism of F. tularensis [24] by overexpressing both NadE* and NadV from this organism, without dissecting the role of each pathway. Moreover, both efforts did not study the physiological response in E. coli to NMN+ accumulation. Beside functioning as a cofactor, NAD+ is also a universal signaling compound that allosterically controls key enzymes and transcriptional regulators in response to the fluctuating cellular redox state [31, 34,35,36,37]. Since NMN+ is an analog of NAD+, it is an open question whether NMN+, when it accumulates to a high level, can also interact with the numerous proteins that are modulated by NAD+.
The rich information provided by comparative genomic analysis can greatly aid metabolic engineering efforts. By bioprospecting NadV homologs from evolutionarily diverse organisms, we identified R. solanacearum NadV, which outperformed the two NadVs that have been previously reported as efficient NMN+-producing enzymes in E. coli [13, 19], when they are compared in the same condition at a bench scale (Fig. 3). Since F. tularensis NadE* also showed promise to produce NMN+ efficiently, a similar approach may be taken in the future to bioprospect NadE* homologs.
To gain fundamental understanding of nicotinamide cofactor biosynthesis, the mass spectrometry method reported in this work needs to be further expanded to quantify the biosynthetic intermediates of NMN+, such as NR and NaMN+. While we only produced NMN+ in oxidized form, quantification of intracellular NMNH is also important when enzymes that can cycle NMN+ are introduced. Our future work will focus on investigating the reduction potential of NMN(H) redox pair in vivo, as well as its interplay with NAD(P)/H redox pairs.
An intracellular NMN+ level of 1.5 mM is comparable to the levels of native cofactors found E. coli, and similar concentrations have been shown to enable NMN-dependent biotransformation in whole cells [13]. In the future, additional work will be performed to determine the optimal NMN+ production levels to pair with cell-free applications. Moving forward, culture medium and growth conditions can be optimized to potentially yield increased intracellular NMN+ levels. In this work, all NMN+ production was performed with laboratory standard medium and without optimization. Marinescu and coworkers demonstrated a 32.7-fold increase from 0.72 mM to 23.57 mM of intracellular NMN+ upon scale-up to a 500 mL bioreactor while optimizing pH, NA feeding concentration, and dissolved oxygen while culturing in PYA8 medium [19]. Therefore, performing a similar scale-up with our NMN+ producing strain may yield significant increases in NMN+ production.
In addition, host selection may play a significant role in efficient NMN+ biosynthesis. While most industrial model hosts including E. coli and S. cerevisiae utilize a nicotinic acid adenine dinucleotide (NaAD)-mediated route for de novo NAD+ biosynthesis, a small group of prokaryotes use NMN+ as the primary precursor to NAD+ [18, 24]. Since NMN+ adopts a distinct role and is naturally maintained at a higher level in these organisms [23, 24], the physiological responses to intracellular NMN+ accumulation may be different. Thus, organisms which utilize NMN+-mediated NAD+ biosynthesis may be interesting targets for metabolic engineering.
Ultimately, efficient and cost-effective production and purification of NMN+ is key for the long-term viability of NMN+-based cell-free biotransformation. Once upstream pathways for the renewable production of NMN+ are further established, NMN+ will need to be extracted and purified before use in cell-free systems. Cells can be isolated through centrifugation, washed, and lysed through homogenization to isolate NMN+ from cellular debris. Alternatively, cells can also be permeabilized to release NMN+ across the cell membrane, allowing for fewer steps of isolating NMN+ from cell mass. Finally, a major advantage of producing NMN+ in vivo is the direct compatibility with crude lysate-based cell-free and whole-cell biosynthesis. By using cells that are capable of both producing intracellular NMN+ and expressing enzymes of interest, crude lysates or whole cells can be directly used for NMN+-dependent biosynthesis without the exogenous supply of redox cofactors.
Conclusions
In this work, we explored routes to efficiently produce NMN+ in E. coli. After surveying the routes for NMN+ production in vivo, bioprospecting NadVs enabled the production of 1.5 mM of NMN+ using the NadV from R. solanacearum. Under the conditions tested, R. solanacearum outperformed the previous best NadV’s shown to accumulate NMN+ efficiently [13, 19]. In addition, this work began to elucidate the physiological effects of NMN+ accumulation in E. coli. However, further investigation is necessary to maintain productivity as NMN+ levels are further increased. Ultimately, advancing noncanonical redox cofactor biosynthesis in microorganisms may enable the application of self-sustained, fully renewable cell-free and whole-cell biocatalysis.
Methods
Plasmid and strain construction
All molecular cloning was performed in E. coli XL1-Blue cells (Stratagene). A summary of strains and plasmids used in this study can be found in Table 1. Plasmids were assembled by Gibson Isothermal DNA Assembly [38]. Polymerase chain reaction (PCR) fragments were generated using PrimeSTAR Max DNA Polymerase (TaKaRa). The method for plasmid construction is described below.
The yqhD gene was isolated from E. coli BL21 chromosomal DNA by PCR. The resulting PCR fragment was gel purified and assembled into a ColE1 ori, AmpR vector backbone by Gibson isothermal DNA assembly method. We used the yqhD-harboring plasmid (pWB301) as a control vector in the growth rescue experiments, because it expresses a similar sized-protein to the NMN+-producing enzymes using the same promoter, and hence may cause similar growth burden. The gene product of yqhD has unrelated function to NMN+ biosynthesis.
E. coli pnuC, S. enterica nadR, S. cerevisiae NRK1, Synechocystis sp nadV, and S. elongatus nadV were isolated by PCR from their respective chromosomal DNA. F. tularensis nadE*, R. solanacearum nadV, and H. ducreyi nadV genes were amplified from E. coli codon optimized synthesized DNA templates and assembled as described above.
S. enterica PnuC* is generated by site-directed mutagenesis based on the wild type S. enterica pnuC gene [30]. The S. enterica pnuC gene was isolated by PCR from chromosomal DNA and assembled as discussed above. To perform the KA insertion, which has been shown to enable NMN+ transport [30], a sequence of AAAGCA was inserted directly after the 321 base pair, as shown in red text in the Supplemental Information. This resulted in the insertion of a lysine and an alanine residue at the 108 and 109 residue positions. The insertion was introduced by PCR. The subsequent mutant PCR fragments were assembled at discussed above. To generate multi-gene plasmids, the genes were inserted sequentially with a ribosome binding site preceding each gene in the synthetic operon.
Gene sequences are listed in the Additional file.
Growth-based screening platform
Plasmids (selected from pWB301-305) were transformed into E. coli strains BW25113 and 72c [28] using the Mix & Go E. coli Transformation Kit (Zymo Research).
Overnight cultures were grown in LB medium supplemented with 2 g/L d-glucose, 0.1 mM IPTG, appropriate antibiotics in test tubes at 30 °C while shaking at 250 r.p.m. for 16 h. For the growth assay, cells were cultured in 1 mL of LB medium supplemented with 2 g/L d-glucose, 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG), appropriate antibiotics, and 200 µM of feeding compound, if applicable, in a 2 mL deep-well plate, with square wells, sealed with air permeable film. The medium was inoculated with 1% v/v overnight cultures. Cultures were grown at 42 °C while shaking at 250 r.p.m.. Cell growth was monitored by measuring optical density at 600 nm. When applicable, media contained 100 mg/L ampicillin to maintain the plasmids.
Intracellular NMN+ generation and quantification
NMN+ generation and quantification were performed as previously reported [13]. Briefly, plasmids (selected from pWB203, pDB102-104, and pLZ301) were transformed into E. coli strains BW25113 and JW2670-1 (ΔpncC) as described previously. Overnight cultures were grown in 2xYT medium containing 0.1 mM IPTG, 2 g/L d-glucose, and appropriate antibiotics for 12 h at 30 °C at 250 rotations per minute (r.p.m.). To cultivate cells for nucleotide analysis, cells were grown in a 50 mL conical tube containing 10 mL of 2xYT media supplemented with 0.5 mM IPTG, 1 mM nicotinamide, and appropriate antibiotics. Cultures were inoculated with 1% v/v overnight culture. Tubes were incubated at 30 °C with shaking at 250 r.p.m. for 4 h. All media contained 100 mg/L ampicillin to maintain the plasmids.
Cells were processed as previously reported [13]. Briefly, 1 mL of cell culture was pelleted, washed with 1 mL of deionized water, and lysed by resuspension in 1 mL of 95 °C 1% formic acid containing 1 µM of 1-methylnicotinamide as an internal standard. Lysates were quenched in an ice bath before pelleting cell debris. Supernatants were run on a Waters ACQUITY Ultra Performance Liquid Chromatograph with a Waters ACQUITY UPLC CSH C18 column (1.7 µM × 2.1 mm x 50 mm). Mobile phases used for separation were (A) water with 2% acetonitrile and 0.2% acetic acid and (B) acetonitrile with 0.2% acetic acid. MS/MS detection was performed by a Waters Micromass Quattro Premier XE Mass Spectrometer. The detailed UPLC and MS parameters were reported previously [13]. Values from LC–MS/MS were correlated back to intracellular concentration using the number of cells per unit OD600nm in 1 mL of culture = 1× 109 and the intracellular volume of an E. coli cell as 1 ×10−15 L per cell [39].
Cell growth with high concentrations of NMN+
A plasmid containing the NMN+ transporter S. enterica PnuC* (pWB302) was transformed into E. coli strains JW2670-1 (ΔpncC) and MX101 (ΔpncC ΔnadR). Overnight cultures were grown in 2xYT medium supplemented with 1 g/L d-glucose, 0.1 mM IPTG, and 100 mg/L ampicillin at 30 °C while shaking at 250 r.p.m. for ~ 14 h. 1 mL of overnight culture was pelleted in a 1.5 mL microcentrifuge tube and washed twice with 1 mL of 1 × M9 salts. The washed cells were resuspended in 1 mL of 1x M9 salts and used for inoculations. For the growth assay, cells were cultured in 1 mL of M9 minimal medium (1x M9 salts, 1 g/L glucose, 1 mM MgSO4, 0.1 mM CaCl2, 40 mg/L FeSO4, 1x A5 trace metals with cobalt) containing 0.5 mM IPTG in a 2 mL deep-well plate, with square wells, sealed with air permeable film. The medium was inoculated with 1% v/v of washed overnight cultures. Cultures were grown at 30 °C while shaking at 250 r.p.m.. Cell growth was monitored by measuring optical density at 600 nm. Media contained 100 mg/L ampicillin to maintain the plasmid.
Availability of data and materials
The datasets used and/or analyzed during this study are available from the corresponding author on reasonable request.
Abbreviations
- NAD+ :
-
Nicotinamide adenine dinucleotide
- NADP+ :
-
Nicotinamide adenine dinucleotide phosphate
- P2NA+ :
-
3-carbomoyl-1-phenethylpyridin-1-ium chloride
- NMN+ :
-
Nicotinamide mononucleotide
- NadV:
-
Nicotinamide phosphoribosyltransferases
- NadE*:
-
Nicotinamide mononucleotide synthase
- PncC:
-
Nicotinamide mononucleotide
- NaMN+ :
-
Nicotinic acid mononucleotide
- NR:
-
Nicotinamide riboside
- PnuC:
-
Nicotinamide riboside transporter
- PnuC*:
-
Mutant nicotinamide riboside transporter
- Nrk1:
-
Nicotinamide riboside kinase from Saccharomyces cerevisiae
- NadR:
-
Nicotinamide riboside kinase (Salmonella enterica)
- NA:
-
Nicotinamide
- LC–MS:
-
Liquid chromatography-mass spectrometry
- NaAD:
-
Nicotinic acid adenine dinucleotide
- PCR:
-
Polymerase chain reaction
- IPTG:
-
Isopropyl-β-d-thiogalactopyranoside
- r.p.m.:
-
Rotations per minute
References
Bowie JU, Sherkhanov S, Korman TP, Valliere MA, Opgenorth PH, Liu H. Synthetic Biochemistry: The Bio-inspired Cell-Free Approach to Commodity Chemical Production. Trends Biotechnol. 2020. in press.
Dudley QM, Karim AS, Jewett MC. Cell-free metabolic engineering: biomanufacturing beyond the cell. Biotechnol J. 2015;10(1):69–82.
Wilding KM, Schinn S, Long EA, Bundy BC. The emerging impact of cell-free chemical biosynthesis. Curr. Opin. Biotech. 2018;53:115–21.
Honda K, Kimura K, Ninh PH, Taniguchi H, Okano K, Ohtake H. In vitro bioconversion of chitin to pyruvate with thermophilic enzymes. J Biosci Bioeng. 2017;124(3):297–301.
Wang W, Liu M, You C, Li Z, Zhang YHP. ATP-Free biosynthesis of a high-energy phosphate metabolite fructose 1,6-diphosphate by in vitro metabolic engineering. Metab Eng. 2017;42:168–74.
Kay JE, Jewett MC. A cell-free system for production of 2,3-butanediol is robust to growth-toxic compounds. Metab Eng. Commun. 2020;10:e00114.
Karim AS, Jewett MC. A cell-free framework for rapid biosynthetic pathway prototyping and enzyme discovery. Metab Eng. 2016;36:116–26.
Kelwick R, Ricci L, Chee SM, Bell D, Webb AJ, Freemont PS. Cell-free prototyping strategies for enhancing the sustainable production of polyhydoxyalkanoates bioplastics. Synth Bio. 2018;3(1):1–12.
Nowak C, Pick A, Csepei L, Sieber V. Characterization of biomimetic cofactors according to stability, redox potentials, and enzymatic conversion by NADH oxidase from Lactobacillus pentosus. ChemBioChem. 2017;18(19):1944–9.
Nowak C, Pick A, Lommes P, Sieber V. Enzymatic reduction of nicotinamide biomimetic cofactors using an engineered glucose dehydrogenase: providing a regeneration system for artificial cofactors. ACS Catal. 2017;7(8):5202–8.
Kaplan NO, Ciotti MM, Stolzenbach FE. Reaction of pyridine nucleotide analogues with dehydrogenases. J Biol Chem. 1956;221:833–44.
Kaufman RA. Use of Nicotinamide Adenine Dinucleotide (NAD) Analogs to Measure Ethanol. European Patent Specification Patent No. EP 1 242 440 B1, filed December 27, 2000, and published September 16, 2015.
Black WB, Zhang L, Mak WS, Maxel S, Cui Y, King E, Fong B, Sanchez Martinez A, Siegel JB, Li H. Engineering a nicotinamide mononucleotide redox cofactor system for biocatalysis. Nat Chem Biol. 2020;16(1):87–94.
Campbell E, Meredith M, Minteer SD, Banta S. Enzymatic biofuel cells utilizing a biomimetic cofactor. Chem Commun. 2012;48:1898–900.
Ji DB, Wang L, Liu W, Hou S, Zhao KZ. Synthesis of NAD analogs to develop bioorthogonal redox system. Sci China Chem. 2013;56(3):296–300.
Paul CE, Arends IWCE, Hollmann F. Is simpler better? synthetic nicotinamide cofactor analogues for redox chemistry. ACS Catal. 2014;4(3):788–97.
Makarov MV, Migaud ME. Syntheses and chemical properties of β-nicotinamide riboside and its analogues and derivatives. Beilstein J Org Chem. 2019;15:401–30.
Gazzaniga F, Stebbins R, Chang SZ, McPeek MA, Brenner C. Microbial NAD metabolism: lessons from comparative genomics. Microbiol Mol Bio Rev. 2009;73(3):529–41.
Marinescu GC, Popescu R, Stoian G, Dinischiotu A. β-nicotinamide mononucleotide (NMN) production in Escherichia coli. Sci Rep. 2018;8:12278.
Sinclair DA, EAR PH. Biological Production of NAD Precursors and Analogs. WO Patent WO 2015/069860 AI, filed November 6, 2014, and issued, May 14, 2015.
Paul CE, Gargiulo S, Opperman DJ, Lavandera I, Gotor-Fernández V, Gotor V, Taglieber A, Arends IWCE, Hollmann F. Mimicking nature: synthetic nicotinamide cofactors for C = C bioreduction using enoate reductases. Org Lett. 2013;15(1):180–3.
Rollin JA, Tam TK, Zhang YHP. New biotechnology paradigm: cell-free biosystems for biomanufacturing. Green Chem. 2013;15:1708–19.
Evans C, Bogan KL, Song P, Burant CF, Kennedy RT, Brenner C. NAD+ metabolite levels as a function of vitamins and calorie restriction: evidence for different mechanisms of longevity. BMC Chem Biol. 2010;10(2):1–10.
Sorci L, Martynowski D, Rodionov DA, Eyobo Y, Zogaj X, Klose KE, Nikolaev EV, Magni G, Zhang H, Osterman AL. Nicotinamide mononucleotide synthetase is the key enzyme for an alternative route of NAD biosynthesis in Francisella tularensis. PNAS. 2009;106(9):3083–8.
Sauer E, Merdanovic M, Mortimer AP, Bringmann G, Reidl J. PnuC and the utilization of the nicotinamide riboside analog 3-aminopyridine in haemophilus influenzae. Antimicrob Agents Chemother. 2004;48(12):4532–41.
Belenky P, Christensen KC, Gazzaniga F, Pletnev AA, Brenner C. Nicotinamide riboside and nicotinic acid riboside salvage in fungi and mammals. J Biol Chem. 2009;284(1):158–64.
Kurnasov OV, Polanuyer BM, Ananta S, Sloutsky R, Tam A, Gerdes S, Osterman A. Ribosylnicotinamide kinase domain of NadR protein: identification and implications in NAD biosynthesis. J Bacteriol. 2002;184(24):6906–17.
Stancek M, Isaksson LA, Rydén-Aulin M. fusB is an allele of nadD, encoding nicotinate mononucleotide andeylyltransferase in Escherichia coli. Microbiology. 2003;149(9):2427–33.
Wang X, Zhou YJ, Wang L, Liu W, Liu Y, Peng C, Zhao ZK. Engineering Escherichia coli nicotinic acid mononucleotide adenylyltransferase for fully active amidated NAD biosynthesis. Appl Environ Microbiol. 2017;83(13):e00692-17.
Grose JH, Bergthorsson U, Xu Y, Sterneckert J, Khodaverdian B, Roth JR. Assimilation of nicotinamide mononucleotide requires periplasmic AphA phosphatase in Salmonella enterica. J Bacteriol. 2005;187(13):4521–30.
Raffaelli NL, Lorenzi T, Mariani PL, Emanuelli M, Amici A, Ruggieri S, Magni G. The Escherichia coli NadR regulator is endowed with nicotinamide mononucleotide adenylyltransferase activity. J Bacteriol. 1999;181(17):5509–11.
Tritz GJ, Chandler JL. Recognition of a gene involved in the regulation of nicotinamide adenine dinucleotide biosynthesis. J Bacteriol. 1973;114(1):128–36.
Zhu NO, Olivera BM, Roth JR. Activity of the nicotinamide mononucleotide transport system is regulated in Salmonella typhimurium. J Bacteriol. 1991;173(3):1311–20.
Euro L, Belevich G, Bloch DA, Verkhovsky MI, Wikström M, Verkhovskaya M. The Role of the Invariant Glutamate 95 in the Catalytic Site of Complex I from Escherichia coli. Biochim. Biophys. Acta, Bioenerg. 2009;1787(1):68-73.
Leonardo MR, Dailly Y, Clark DP. Role of NAD in regulating the adhE gene of Escherichia coli. J Bacteriol. 1996;178(20):6013–8.
Rodionov DA, De Ingeniis J, Mancini C, Cimadamore F, Zhang H, Osterman AL, Raffaelli N. Transcriptional regulation of NAD metabolism in bacteria: nrtR family of Nudix-related regulators. Nucleic Acids Res. 2008;36(6):2047–59.
Shone CC, Fromm HJ. Steady-state and pre-steady-state kinetics of coenzyme a linked aldehyde dehydrogenase from Escherichia coli. Biochemistry. 1981;20(26):7494–501.
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343–5.
Findik BT, Randall LL. Determination of the intracellular concnetraiton of the export chaperone SecB in Escherichia coli. PLoS ONE. 2017;12(8):e0183231.
Acknowledgements
We thank the University of California, Irvine Mass Spectrometry Facility and Dr. Felix Grun for help with LC–MS.
Funding
H.L. acknowledges support from University of California, Irvine, the National Science Foundation (NSF) (award no. 1847705), and the National Institutes of Health (NIH) (award no. DP2 GM137427). W.B.B. acknowledges support from Graduate Assistance in Areas of National Need fellowship funded by the U.S. Department of Education. D.A. acknowledges support from the Federal Work Study Program funded by the U.S. Department of Education. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the NSF.
Author information
Authors and Affiliations
Contributions
HL conceived the research. WBB, LZ, EK designed and conducted growth-based screening. WBB, DB, DA performed study of prospective NadV homologs. WBB designed and WBB, DA, DB, LZ, EK, performed the intracellular NMN+ and NAD+ level analysis. WBB and DA investigated physiological response to intracellular NMN+. All authors analyzed the data and interpreted results. HL, WBB, DA wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Francisella tularensis NadE*-based Growth Restoration is Not Nicotinamide Feeding Dependent. A growth restoration platform was used to screen pathways for the efficient generation of nicotinamide mononucleotide (NMN+). The Escherichia coli strain 72c [1], note this reference number is for additional files only] contains a temperature sensitive allele of nadD (ts nadD). As a result, this strain cannot grow at 42 °C. By overexpressing Francisella tularensis nadE*, cells are able to produce NMN+, which can then be converted to NAD+, and thus restoring growth. We observed no dependence of growth restoration with feeding 200 µM nicotinamide (NA). This indicates either efficient NMN+ generation can be achieved through channeling the intermediate nicotinic acid mononucleotide (NaMN+) from E. coli ‘s native de novo NAD+ biosynthesis pathway, or LB medium used in this experiment already contains sufficient precursors for this pathway. Screening was performed in a deep-well 96-well plate containing 1 mL of LB medium supplemented with 2 g/L d-glucose and 200 μM of NA if applicable. Detailed conditions are described in the Methods section. Figure S2.Francisella tularensis NadE* Expression Alleviates R. solanacearum NadV Growth Challenge. BW25113 ΔpncC cells expressing R. solanacearum NadV demonstrate a growth challenge. However, when paired with F. tularensis NadE*, the growth challenge is alleviated. Interestingly, this challenge is not seen when H. ducreyi NadV is expressed. Therefore, F. tularensis NadE* may play a synergistic role in stability, activity, or expression of some NadV candidates. Cells were cultured identically to the intracellular NMN+ generation cultures described in the Methods section. Cell growth was monitored by measuring optical density at 600 nm. Abbreviations indicate source of genes: Ft, Francisella tularensis, Hd, Haemophilus ducreyi; Rs, Ralstonia solanacearum. Figure S3. Intracellular NAD+ Decreases in NMN+ Accumulating Strains From Fig. 3, co-overexpression of NMN+ generating Francisella tularensis NadE* and NadVs increases intracellular NMN+ when the NMN+ degrading PncC is disrupted. However, as shown here, NAD+ levels decreased in cells expressing F. tularensis NadE* and NadV compared to cells without overexpression. This potentially indicates NMN+ plays a regulatory role in NAD+ biosynthesis. Cells were cultured in 2xYT medium supplemented with 1 mM nicotinamide at 30 °C for 4 h. Intracellular NAD+ concentrations were determined by UPLC-MS/MS. Detailed conditions and analytical techniques are described in the Methods section.DNA Sequences of Genes Used in This Study: List of DNA sequences of the genes used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Black, W.B., Aspacio, D., Bever, D. et al. Metabolic engineering of Escherichia coli for optimized biosynthesis of nicotinamide mononucleotide, a noncanonical redox cofactor. Microb Cell Fact 19, 150 (2020). https://doi.org/10.1186/s12934-020-01415-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-020-01415-z