
Abstract
Background
Enantiopure 2-hydroxy acids are key intermediates for the synthesis of pharmaceuticals and fine chemicals. We present an enantioselective cascade biocatalysis using recombinant microbial cells for deracemization of racemic 2-hydroxy acids that allows for efficient production of enantiopure 2-hydroxy acids.
Results
The method was realized by a single recombinant Escherichia coli strain coexpressing three enzymes: (S)-2-hydroxy acid dehydrogenase, (R)-2-keto acid reductase and glucose dehydrogenase. One enantiomer [(S)-2-hydroxy acid] is firstly oxidized to the keto acid with (S)-2-hydroxy acid dehydrogenase, while the other enantiomer [(R)-2-hydroxy acid] remains unchanged. Then, the keto acid obtained reduced to the opposite enantiomer with (R)-2-keto acid reductase plus cofactor regeneration enzyme glucose dehydrogenase subsequently. The recombinant E. coli strain coexpressing the three enzymes was proven to be a promising biocatalyst for the cascade bioconversion of a structurally diverse range of racemic 2-hydroxy acids, giving the corresponding (R)-2-hydroxy acids in up to 98.5 % conversion and >99 % enantiomeric excess.
Conclusions
In summary, a cascade biocatalysis was successfully developed to prepare valuable (R)-2-hydroxy acids with an efficient three-enzyme system. The developed elegant cascade biocatalysis possesses high atom efficiency and represents a promising strategy for production of highly valued (R)-2-hydroxy acids.
Similar content being viewed by others

Background
Enantiopure 2-hydroxy acids are among the most important building blocks for synthesizing pharmaceuticals and fine chemicals [1]. For example, (R)-(-)-mandelic acid is widely used as an intermediate for the preparation of antibiotics, antiobesity drugs, and antitumor agents [2]. (R)-o-Chloromandelic acid is the most preferred chiral building block for the industrial synthesis of anti-thrombotic agent, a best-selling cardiovascular drug [3]. (R)-2-Hydroxy-4-phenylbutyric acid is an intermediate in the manufacture of angiotensin converting enzyme inhibitors [4]. (R)-3-Phenyllactic acid is used as a precursor for the synthesis of englitazone which has excellent hypoglycemic effect [5]. Due to their importance, many enantioselective routes for their synthesis have been developed and a great progress has been achieved in recent years. Traditionally, their industrial production mainly relies on the chemical approaches such as chemical kinetic resolution with chiral agent. However, it does not always satisfactorily work because of expensive agent, unsatisfied enantiomeric excess (ee) of product or low yield (e.g., <50 %).
Biocatalysis is increasingly being used to develop efficient and green processes for chiral 2-hydroxy acids synthesis [6]. Several enzymatic approaches have been reported for synthesizing chiral 2-hydroxy acids in the literature [1, 7, 8], including reduction of 2-keto acids with stereoselective 2-keto acid reductase [9], enantioselective oxidation of racemic 2-hydroxy acids with 2-hydroxy acid dehydrogenase [10] or diols with alditol oxidase [11], resolution of 2-hydroxy acids with lipase [12] or esterase [1], hydrolysis of 2-hydroxynitriles with nitrilase [13, 14], enantioselective addition of HCN to aldehydes with oxynitrilase followed by nitrilase hydrolysis [15], hydrolysis of amide with amidase [16], oxidation of L-amino acids with an L-amino acid deaminase followed by asymmetric reduction of the keto acid with an 2-hydroxyisocaproate dehydrogenase [17], and deracemization of racemic 2-hydroxy acids by cascade biocatalysis [18] (Scheme 1). Among all these approaches, deracemization of racemate by cascade biocatalysis is one of the most attractive methods because it allows to completely transform a cheap racemate into a single stereoisomeric product without byproduct. Furthermore, this enantioselective cascade biocatalysis enables multistep reactions to be performed in one pot, which circumvented yield-reducing and time-consuming isolation of intermediates [19]. Recently, several types of enantioselective cascade biocatalysis have been developed for chiral synthesis [20–25], such as the deracemization of sec-alcohols, amines and amino acids [26–30], amination of alcohols to chiral amines [31], preparation of chiral α-hydroxy ketones from epoxides [32], and biotransformation of α-hydroxy acids into chiral α-amino acids [33, 34].

Enzymatic routes for the synthesis of enantiopure (R)-2-hydroxy acids
In this work, we aim to develop an enantioselective cascade biocatalysis for deracemization of racemic 2-hydroxy acids to (R)-2-hydroxy acids via an oxidation–reduction sequence using a recombinant Escherichia coli expressing three enzymes (Scheme 2). One enantiomer [(S)-2-hydroxy acid] is firstly oxidized to the keto acid with enantioselective (S)-2-hydroxy acid dehydrogenase [(S)-2-HADH] while the other enantiomer [(R)-2-hydroxy acid] remains unchanged. The keto acid obtained is then bioreduced to the opposite enantiomer with stereoselective (R)-2-keto acid reductase [(R)-2-KAR] plus cofactor regeneration enzyme glucose dehydrogenase (GDH) subsequently. The recombinant E. coli strain coexpressing (S)-2-HADH, (R)-2-KAR and GDH was proven to be a promising biocatalyst. A wide range of 2-hydroxy acids can be deracemized to (R)-2-hydroxy acids with near-perfect stereo purity and high conversion. This method by cascade enantioselective oxidation and asymmetric reduction with a single recombinant strain represents a cheap, easy and environmental approach for synthesizing (R)-2-hydroxy acids from their racemates.

Enantioselective cascade biocatalysis for deracemization of 2-hydroxy acids to enantiopure 2-hydroxy acids via an oxidation–reduction sequence
Results and discussion
Construction of recombinant E. coli strain expressing (S)-2-HADH
We recently established a high-throughput screening method to screen stereoselective (S)-2-HADH producing strains [35]. Pseudomonas aeruginosa CCTCC M 2011394 harboring a flavine mononucleotice (FMN)-dependent (S)-2-HADH, which specifically oxidizes the (S)-isomer of 2-hydroxy acids to 2-keto acids [36], was isolated from the soil samples. Thus, (S)-2-HADH may be used as the biocatalyst for the oxidation step in the designed enantioselective cascade biocatalysis. The gene of (S)-2-HADH from P. aeruginosa CCTCC M 2011394 (GenBank accession number: KU612124) was cloned and expressed in E. coli BL21(DE3). The recombinant E. coli was cultured in LB medium at 37 °C to reach an OD600 of 0.6 and then induced by the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) at 0.1 mM. The cells were continually grown at 28 °C, 150 rpm for 12 h. The resting cells of the recombinant E. coli strain (E. coli BL21(DE3)/pET28b-HADH) were used as biocatalysts for the enantioselective oxidation of racemic 2-hydroxy acids with rac-1a as model substrate. The result showed that the activity was lower than 5.0 U/g dry cell weight (DCW) and conversion of 2-keto acid 2a was only 5.1 % after 2 h reaction (Table 1, entry 1). To achieve economic feasibility and competitiveness for the enantioselective oxidation of 2-hydroxy acids, it is necessary to find an promising (S)-2-HADH showing high activity and enantioselectivity (E). We adopted a genome mining strategy to search for more efficient (S)-2-HADHs. A pBLAST search was conducted by using (S)-2-HADH from P. aeruginosa CCTCCM 2011394 as the template in the NCBI database. Four representative (S)-2-HADHs from Burkholderia xenovorans LB400 (ABE35802.1), P. putida ATCC 12633 (AAC15503.1), P. aeruginosa NUST (AGM49308.1), P. fluorescens strain EBC191(AAW79575.1) were selected (Additional file 1: Figure S1). After being synthesized in vitro and cloned into pET28b, the four (S)-2-HADH genes were then expressed in E. coli BL21(DE3), respectively. Rac-1a was used as the model substrate to evaluate their activity and enantioselectivity. The results indicated that they all displayed oxidation activities. The resting cells of recombinant E. coli BL21(DE3) expressing the (S)-2-HADH from the B. xenovorans LB400 and P. aeruginosa NUST showed relatively higher activity (>90 U/g DCW) with excellent enantioselectivity (E > 200). After 2 h reaction, the conversions of keto acid 2a reach 49.0 and 48.9 %, respectively, which were near theoretical conversions. However, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis showed that (S)-2-HADH from B. xenovorans LB400 has been expressed in partially soluble state. In the forthcoming experiments, the (S)-2-HADH from P. aeruginosa NSUT was selected for further studies. The requirement of coenzyme in the stereoselective oxidation catalyzed by (S)-2-HADH from P. aeruginosa NSUT was investigated. The activity of (S)-2-HADH was almost lost upon flavin removal. The activity of the apoenzyme was partly reactivated by the addition of FMN. These results confirmed that the (S)-2-HADH from P. aeruginosa NUST is a flavoprotein with FMN as cofactor. The reaction that oxidizes (S)-2-hydroxy acids to 2-keto acids consists of the steps involved in substrate oxidation and FMN reduction [37]. The FMN is then reoxidized by electron transfer to the oxidant. The FMN-dependent (S)-2-HADH family can be divided into three subgroups based on the different oxidants including oxygen, flavocytochrome b2s and ubiquinone utilized in the second oxidative half-reaction [37, 38]. Operation parameters, including optimum temperature and pH of the dehydrogenation by the recombinant E. coli BL21(DE3)/pET28b-HADH were investigated. The result showed that the resting cells of recombinant E. coli BL21(DE3)/pET28b-HADH showed high activity at 35–55 °C and pH 7.5–8.5 (Additional file 1: Figure S2). The wide ranges of optimum temperature and pH are very beneficial for the cascade biocatalysis.
Construction of recombinant E. coli strain coexpressing (R)-2-KAR and GDH
Stereoselective (R)-2-KAR could reduce prochiral 2-keto acids to produce corresponding chiral 2-hydroxy acids. The gene of (R)-2-KAR cloned from Leuconostoc mesenteroides CCTCC M 2016063 (GenBank accession number: KU612125) was expressed in E. coli BL21(DE3). After cultivation, the whole cells of recombinant E. coli BL21(DE3)/pET28b-KAR were collected and disrupted by sonication. The (R)-2-KAR with N-terminal his-tag in the cell free extract was purified to homogeneity by nickel affinity chromatography. The purified (R)-2-KAR migrated as a single band and located at the position of about 32 kDa on SDS-PAGE (Additional file 1: Figure S3), which is in agreement with the molecular mass deduced from its amino acid sequence. The purified enzyme showed little activity with NADPH but full activity with NADH, indicating an NADH-dependence. For the application of reductase, the addition of expensive cofactor often makes the bioreaction less practically feasible from the viewpoint of economic aspects. A coexpression of two enzymes in one E. coli cell seems to be an efficient approach to solve this problem. Thus, we introduced a GDH from Exiguobacterium sibiricum (WP_012369122.1) for the regeneration of the oxidized cofactor (NAD+). A coexpression plasmid (pCDFDuet-KAR-GDH) containing both (R)-2-KAR and GDH genes was constructed and transformed into E. coli BL21(DE3) cells. After cultivation, the whole cells of recombinant E. coli BL21(DE3)/pCDFDuet-KAR-GDH were collected and disrupted by sonication. The SDS-PAGE of the cell free extract of the recombinant E. coli showed that the coexpressed (R)-2-KAR and GDH were clearly visible (Additional file 1: Figure S4). The (R)-2-KAR and GDH located at the position of about 32 and 28 kDa on SDS-PAGE. The effects of temperature and pH on the reduction by the recombinant E. coli BL21(DE3)/pCDFDuet-KAR-GDH were also investigated. The resting cells of the recombinant E. coli exhibit high activity at 35 °C and pH 7.5 using keto acid 2a as substrate. To test the potential of recombinant E. coli BL21(DE3)/pCDFDuet-KAR-GDH in chemical synthesis, various substrates were used for asymmetric reduction. With the assistance of GDH from E. sibiricum for cofactor regeneration, the resting cells of E. coli strain coexpressing (R)-2-KAR and GDH could reduce a wide range of prochiral 2-keto acids to corresponding (R)-2-hydroxy acids with >99 % ee. The substituents in substrates and the distance between the hydroxy group and benzene ring are the important factors to affect the catalytic ability of the biocatalyst. Among all the 2-hydroxy acids tested, substrate 2a–2m could be efficiently reduced to (R)-2-1a–1m in >86 % conversion and >99 % ee within 3.5–10 h (Table 2, entries 1–13). When the OH and OCH3 were attached to the phenyl ring of the substrates (2n–2q) and the distance between the hydroxy group and benzene ring increased (2r and 2s), the recombinant E. coli exhibited a relatively low activity (Table 2, entries 14–19).
Deracemization of 2-hydroxy acids with the mixtures of recombinant E. coli BL21(DE3)/pET28b-HADH and E. coli BL21(DE3)/pCDFDuet-KAR-GDH
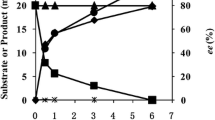
For developing a process for deracemization of racemic 2-hydroxy acids, we coupled the asymmetric oxidation with the opposite stereoselective reduction. Recombinant E. coli BL21(DE3)/pET28b-HADH and E. coli BL21(DE3)/pCDFDuet-KAR-GDH were cultivated, separately, to achieve the resting cells. The cells of the mixed two strains were designed as the catalytic system. The enantioselective cascade biocatalysis for deracemization of 1a–1s was carried out by a one-pot strategy. The conversion, ee of products and reaction time were detected (Table 3). The result showed that the (R)-isomers of substrates (1a–1m) were obtained in high conversions (>90 %) with >99 % ee. In the case of 1n–1s (Table 3, entries 14–19), (R)-2-hydroxy acids were obtained with relative low conversion (34.6–76.4 %) within 6 h. For the 1p–1q (Table 3, entry 16–17), (R)-2-hydroxy acids were obtained with high enantiomeric excess (>99 %), which indicated that the (S)-isomer of 2-hydroxy acids were completely oxidized. The lower conversion might be due to the accumulation of 2-keto acids. Figure 1 shows a typical deracemization progress for the preparation of (R)-1a from 1a with the mixtures of recombinant E. coli BL21(DE3)/pET28b-HADH and E. coli BL21(DE3)/pCDFDuet-KAR-GDH. Oxidation was the faster step in the overall reaction because 2a was accumulated in the cascade reaction. After 150 min reaction, the conversion to (R)-1a from racemate was 97 %.

Time course of deracemization of rac-1a with the mixtures of recombinant E. coli strain. The freshly prepared cells of E. coli BL21(DE3)/pET28b-HADH and E. coli BL21(DE3)/pCDFDuet-KAR-GDH were mixed in 10 mL of phosphate buffer (100 mM, pH 7.5) to a cell density of 4 and 8 g DCW/L, respectively
Construction of recombinant E. coli coexpressing (S)-2-HADH, (R)-2-KAR and GDH for deracemization of 2-hydroxy acids by cascade biocatalysis
In order to avoid the respective cultivation of the two recombinant strains and reduce the cell concentration in the cascade biocatalysis, we attempted to use a single recombinant strain coexpressing (S)-2-HADH, (R)-2-KAR and GDH for the cascade biocatalysis. The recombinant E. coli strain expressing all the necessary enzymes was constructed. The pET28b-HADH and pCDFDuet-KAR-GDH with different antibiotic selection were introduced to E. coli BL21(DE3). The recombinant strain was selected with LB agar plates containing streptomycin and kanamycin. A three-enzyme coexpression strain E. coli BL21(DE3)/pET28b-HADH/pCDFDuet-KAR-GDH was screened. The SDS-PAGE of the cell free extract of the recombinant E. coli showed that the coexpressed (S)-2-HADH, (R)-2-KAR and GDH were clearly visible (Additional file 1: Figure S5). The constructed three-enzyme system was used for one-pot cascade biocatalysis. The recombinant E. coli BL21(DE3)/pET28b-HADH/pCDFDuet-KAR-GDH was cultured to achieve the resting cells. The cascade oxidation–reduction reaction catalyzed by the resting cells of the recombinant E. coli were performed at 35 °C and pH 7.5 (Table 4). The results showed that most of those substrates (1a–1m) can be obtained in up to 98.5 % conversion and >99 % ee in a shorter reaction time as compared to the mixtures of two recombinant E. coli. The reason for this may be that the multienzyme in one recombinant strain avoid the transfer of substrates in different cells. Figure 2 shows a typical time course for production of (R)-1a from racemic 1a with a single recombinant E. coli BL21(DE3)/pET28b-HADH/pCDFDuet-KAR-GDH expressing all the necessary enzymes. After 100 min reaction, (S)-1a was almost completely converted to (R)-1a. The results confirmed that the three-enzyme coexpressing system was more efficient.

Time course of deracemization of rac-1a with a single recombinant E. coli strain. The freshly prepared cells of E. coli BL21(DE3)/pET28b-HADH/pCDFDuet-KAR-GDH were resuspended in 10 mL phosphate buffer (100 mM, pH 7.5) to a cell concentration of 8 g DCW/L
Conclusions
In summary, a cascade biocatalysis was successfully developed to prepare valuable (R)-2-hydroxy acids with an efficient three-enzyme system, which was constructed by coexpressing (S)-2-HADH, (R)-2-KAR and GDH. The recombinant E. coli strain coexpressing the three enzymes was proven to be a promising biocatalyst for the cascade bioconversion of a structurally diverse range of racemic 2-hydroxy acids, giving the corresponding (R)-2-hydroxy acids in up to 98.5 % conversion and >99 % ee. The developed elegant cascade biocatalysis possesses high atom efficiency and represents a promising strategy for production of highly valued (R)-2-hydroxy acids.
Methods
Materials
1a–1s, (R)-1a, (S)-1a and benzoyl formic acid were provided from J&K Chemical Co., Ltd. (Shanghai, China). All other chemicals used were of analytical grade and commercially available. DNA purification kits, restriction endonucleases, T4 DNA ligase, Pfu DNA polymerase, and Taq DNA polymerase were purchased from Shenergy Biocolor BioScience and Technology Company (Shanghai, China). The pGEM-T (Promega, Madison, WI, USA) was used as cloning vector. The pETDuet-1 and pET28b (Novagen, Darmstadt, Germany) were used as expression vector.
Construction of recombinant E. coli BL21(DE3)/pET28b-HADH
The gene encoding (S)-2-HADH from P. aeruginosa CCTCC M 2011394 was amplified via PCR with a series of primers (F-1 GGTGAAACACAACCGCGA; R-1 AGGGCATCCAATCTGGGC, F-2 CTATGGTTCCAGCTCTATGTG; R-2 AGTTGGCGACCGCCGTG, F-3 CATGCCGCAACTGGCCAA; R-3 TGCGGTCGATATCCGCTTT). (S)-2-HADH gene was amplified via PCR with primers (F- GGAATTCCATATGATGATCATTTCCGCTTCCACC, R- CCCAAGCTTTCAGGCGCCCAGTTCGCGGACCA) designed according to the sequence similarity. The PCR product was digested with NdeI and HindIII and ligated into pET28b. The recombinant plasmids were transformed into E. coli BL21(DE3) for expression. A pBLAST search was conducted by using (S)-2-HADH from P. aeruginosa CCTCCM 2011394 as the template in the NCBI database. The (S)-2-HADH from B. xenovorans LB400 (ABE35802.1), P. putida (AAC15503.1), P. aeruginosa NUST (AGM49308.1), P. fluorescens strain EBC191 (AAW79575.1) were selected. Nucleotide sequences of (S)-2-HADHs from these strains were synthesized using the polymerase chain reaction assembly method [39]. The coding genes were ligated into pET28b and expressed in E. coli BL21(DE3). For the selection of E. coli BL21(DE3) transformants, 50 μg/mL kanamycin was added to the Luria–Bertani (LB) medium (5 g yeast extract, 10 g tryptone, and 10 g NaCl in 1 L of distilled water). The requirement of coenzyme in the stereoselective oxidation catalyzed by (S)-2-HADH from P. aeruginosa NSUT was investigated according to the method as described previously [40].
Construction of recombinant E. coli BL21(DE3)/pCDFDuet-KAR-GDH
The gene of (R)-2-KAR from L. mesenteroides CCTCC M 2016063 was amplified via PCR with primers (F-AGGCCATGGGTAAAATCGCAATTGCCG, R-AATCTCGAGGATCTCGAAGTTCTCTTGC). The gene of GDH from E. sibiricum (WP_012369122.1) was synthesized in vitro. The genes of (R)-2-KAR and GDH were cloned and inserted into the coexpression vector pCDFDuet. The pCDFDuet-KAR-GDH was transformed into E. coli BL21(DE3) for expression. For the selection of E. coli BL21(DE3) transformants (E. coli BL21(DE3)/pCDFDuet-KAR-GDH), 50 μg/mL streptomycin was added to the LB medium.
Construction of recombinant E. coli BL21(DE3)/pET28b-HADH/pCDFDuet-KAR-GDH
pET28b-HADH and pCDFDuet-KAR-GDH with different antibiotic selection were transformed into E. coli BL21(DE3). The recombinant strain (E. coli BL21(DE3)/pET28b-HADH/pCDFDuet-KAR-GDH) was selected with LB agar plates containing 50 μg/mL streptomycin and 50 μg/mL kanamycin.
Microorganisms and culture conditions
P. aeruginosa CCTCCM 2011394 was cultured at 30 °C in rich medium containing 10 g glucose, 10 g yeast extract, 2.5 g K2HPO4·3H2O, 2.5 g KH2PO4, 0.2 g MgSO4·7 H2O, 0.03 g FeSO4·7 H2O and 1.0 g NaCl in 1 L of distilled water (pH 7.0). L. mesenteroides CCTCC M 2016063 was cultured at 30 °C in complete medium containing 10 g glucose, 10 g yeast extract, 10 g tryptone, 5 g NaCl and 5 g beef extract in 1 L of distilled water (pH 7.0). E. coli BL21(DE3) (Novagen, Darmstadt, Germany) were used for gene expression. The E. coli strains were grown at 37 °C in LB medium. The recombinant E. coli strains were grown at 37 °C in LB medium containing appropriate antibiotics (50 μg/mL streptomycin, or 50 μg/mL kanamycin, or both) to reach an OD600 of 0.6 and then induced by adding IPTG at 0.1 mM. The strains were cultured continually at 28 °C, 150 rpm for 12 h. The whole cells were collected by centrifugation at 9000×g under 4 °C for 10 min, washed twice with 100 mM phosphate buffer (pH 7.5) for activity test and biotransformation.
Cascade deracemization of 2-hydroxy acids with mixture of the resting cells of recombinant E. coli BL21(DE3)/pET28b-HADH and E. coli BL21(DE3)/pCDFDuet-KAR-GDH
The freshly prepared whole cells of E. coli BL21(DE3)/pET28b-HADH and E. coli BL21(DE3)/pCDFDuet-KAR-GDH were suspended in 10 mL of phosphate buffer (100 mM, pH 7.5) to a cell concentration of 4 and 8 g DCW/L, respectively. Racemic 2-hydroxy acids 1a–1s was added to the mixture at a final concentration of 20 mM. The mixtures were shaken at 35 °C and 150 rpm in 50-mL flasks. Samples were taken at regular intervals and the reactions were terminated through centrifugation (12,000×g, 4 °C, 5 min). The conversion and ee of products were determined by chiral high-performance liquid chromatographic (HPLC) method.
Cascade deracemization of 2-hydroxy acids with the resting cells of recombinant E. coli BL21(DE3)/pET28b-HADH/pCDFDuet-KAR-GDH
The freshly prepared cells of E. coli BL21(DE3)/pET28b-HADH/pCDFDuet-KAR-GDH were resuspended to a cell concentration of 8 g DCW/L in 10 mL phosphate buffer (100 mM, pH 7.5) containing 20 mM 1a–1s. The mixtures were shaken at 35 °C and 150 rpm in 50-mL flasks. Samples were taken at regular intervals and the reactions were terminated through centrifugation (12,000×g, 4 °C, 5 min). The conversion and ee values were determined by chiral HPLC method.
Analytical methods
The determination of (R)-1a–1s, (S)-1a–1s and 2a–2s was performed by chiral HPLC equipped with a chiral column (ChirobioticTM R 250 × 4.6 mm, particle size 5 μm, Sigma, USA). The flow rate was set at 1.0 mL/min. The mobile phase was composed of 0.5 % NH4OH-CH3OH (10:90, v/v). The eluate was monitored at 215 nm. Enantioselectivity (E) was calculated from conversion and ee as described previously [41].
Abbreviations
- (S)-2-HADH:
-
(S)-2-hydroxy acid dehydrogenase
- (R)-2-KAR:
-
(R)-2-keto acid reductase
- GDH:
-
glucose dehydrogenase
- FMN:
-
flavine mononucleotice
- DCW:
-
dry cell weight
- SDS-PAGE:
-
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- IPTG:
-
isopropyl β-d-1-thiogalactopyranoside
References
Gröger H. Enzymatic routes to enantiomerically pure aromatic α-hydroxy carboxylic acids: a further example for the diversity of biocatalysis. Adv Synth Catal. 2001;343:547–58.
Tang KW, Yi JM, Huang KL, Zhang GL. Biphasic recognition chiral extraction: a novel method for separation of mandelic acid enantiomers. Chirality. 2009;21:390–5.
Ema T, Ide S, Okita N, Sakai T. Highly efficient chemoenzymatic synthesis of methyl (R)-o-chloromandelate, a key intermediate for clopidogrel, via asymmetric reduction with recombinant Escherichia coli. Adv Synth Catal. 2008;350:2039–44.
Bai YL, Yang ST. Biotransformation of R-2-hydroxy-4-phenylbutyric acid by d-lactate dehydrogenase and Candida boidinii cells containing formate dehydrogenase coimmobilized in a fibrous bed bioreactor. Biotechnol Bioeng. 2005;92:137–46.
Urban FJ, Moore BS. Synthesis of optically-active 2-benzyldihydrobenzopyrans for the hypoglycemic agent englitazone. J Heterocycl Chem. 1992;29:431–8.
Chen X, Wu QQ, Zhu DM. Enzymatic synthesis of chiral 2-hydroxy carboxylic acids. Process Biochem. 2015;50:759–70.
Gruber CC, Lavandera I, Faber K, Kroutil W. From a racemate to a single enantiomer: deracemization by stereoinversion. Adv Synth Catal. 2006;348:1789–805.
Ricca E, Brucher B, Schrittwieser JH. Multi-Enzymatic cascade reactions: overview and perspectives. Adv Synth Catal. 2011;353:2239–62.
Guo JL, Mu XQ, Zheng CG, Xu Y. A highly stable whole-cell biocatalyst for the enantioselective synthesis of optically active alpha-hydroxy acids. J Chem Technol Biotechnol. 2009;84:1787–92.
Gao C, Qiu JH, Li JC, Ma CQ, Tang HZ, Xu P. Enantioselective oxidation of racemic lactic acid to d-lactic acid and pyruvic acid by Pseudomonas stutzeri SDM. Bioresour Technol. 2009;100:1878–80.
van Hellemond EW, Vermote L, Koolen W, Sonke T, Zandvoort E, Heuts D, Janssen DB, Fraaije MW. Exploring the biocatalytic scope of alditol oxidase from streptomyces coelicolor. Adv Synth Catal. 2009;351:1523–30.
Campbell RF, Fitzpatrick K, Inghardt T, Karlsson O, Nilsson K, Reilly JE, Yet L. Enzymatic resolution of substituted mandelic acids. Tetrahedron Lett. 2003;44:5477–81.
Kumar S, Mohan U, Kamble AL, Pawar S, Banerjee UC. Cross-linked enzyme aggregates of recombinant Pseudomonas putida nitrilase for enantioselective nitrile hydrolysis. Bioresour Technol. 2010;101:6856–8.
Yamamoto K, Oishi K, Fujimatsu I, Komatsu KI. Production of R-(-)-mandelic acid from mandelonitrile by Alcaligenes faecalis ATCC 8750. Appl Environ Microbiol. 1991;57:3028–32.
Sosedov O, Matzer K, Burger S, Kiziak C, Baum S, Altenbuchner J, Chmura A, van Rantwijk F, Stolz A. Construction of recombinant Escherichia coli catalysts which simultaneously express an (S)-oxynitrilase and different nitrilase variants for the synthesis of (S)-mandelic acid and (S)-mandelic amide from benzaldehyde and cyanide. Adv Synth Catal. 2009;351:1531–8.
Wang YS, Cheng F, Zheng RC, Wang YJ, Zheng YG. Characterization of an enantioselective amidase with potential application to asymmetric hydrolysis of (R, S)-2, 2-dimethylcyclopropane carboxamide. World J Microbiol Biotechnol. 2011;27:2885–92.
Gourinchas G, Busto E, Killinger M, Richter N, Wiltschi B, Kroutil W. A synthetic biology approach for the transformation of L-α-amino acids to the corresponding enantiopure (R)- or (S)-α-hydroxy acids. Chem Commun. 2015;51:2828–31.
Xue YP, Zheng YG, Zhang YQ, Sun JL, Liu ZQ, Shen YC. One-pot, single-step deracemization of 2-hydroxyacids by tandem biocatalytic oxidation and reduction. Chem Commun. 2013;49:10706–8.
Schrittwieser JH, Sattler J, Resch V, Mutti FG, Kroutil W. Recent biocatalytic oxidation–reduction cascades. Curr Opin Biotechnol. 2011;15:249–56.
Chen X. Sequential one-pot multienzyme (OPME) systems for the synthesis of carbohydrates and glycoconjugates. Glycobiology. 2015;25:1291.
Classen T, Korpak M, Schölzel M, Pietruszka J. Stereoselective enzyme cascades: an efficient synthesis of chiral γ-butyrolactones. ACS Catal. 2014;4:1321–31.
Liu J, Li Z. Cascade biotransformations via enantioselective reduction, oxidation, and hydrolysis: preparation of (R)-δ-lactones from 2-alkylidenecyclopentanones. ACS Catal. 2013;3:908–11.
Sattler JH, Fuchs M, Tauber K, Mutti FG, Faber PK, Pfeffer J, Haas T, Kroutil PW. Redox self-sufficient biocatalyst network for the amination of primary alcohols. Angew Chem Int Ed. 2012;124:9290–3.
Xue R, Woodley JM. Process technology for multi-enzymatic reaction systems. Bioresour Technol. 2012;115:183–95.
Lopez-Gallego F, Schmidt-Dannert C. Multi-enzymatic synthesis. Curr Opin Chem Biol. 2010;14:174–83.
Voss CV, Gruber CC, Kroutil W. Deracemization of secondary alcohols through a concurrent tandem biocatalytic oxidation and reduction. Angew Chem Int Ed. 2008;47:741–5.
Li YL, Xu JH, Xu Y. Deracemization of aryl secondary alcohols via enantioselective oxidation and stereoselective reduction with tandem whole-cell biocatalysts. J Mol Catal B Enzym. 2010;64:48–52.
Songür R, Atici EM, Mehmetoğlu Ü. Deracemization of 1-phenyl ethanol via tandem biocatalytic oxidation and reduction. Curr Opin Biotechnol. 2011;22(Supplement 1):S56–7.
Kato DI, Miyamoto K, Ohta H. Preparation of optically active 4-chlorophenylalanine from its racemate by deracemization technique using transformant Escherichia coli cells. Biocatal Biotransform. 2005;23:375–9.
Caligiuri A, D’Arrigo P, Gefflaut T, Molla G, Pollegioni L, Rosini E, Rossi C, Servi S. Multistep enzyme catalysed deracemisation of 2-naphthyl alanine. Biocatal Biotransform. 2006;24:409–13.
Mutti FG, Knaus T, Scrutton NS, Breuer M, Turner NJ. Conversion of alcohols to enantiopure amines through dual-enzyme hydrogen-borrowing cascades. Science. 2015;349:1525–9.
Zhang J, Wu S, Wu J, Li Z. Enantioselective cascade biocatalysis via epoxide hydrolysis and alcohol oxidation: one-pot synthesis of (R)-α-hydroxy ketones from meso- or racemic epoxides. ACS Catal. 2015;5:51–8.
Fan CW, Xu GC, Ma BD, Bai YP, Zhang J, Xu JH. A novel d-mandelate dehydrogenase used in three-enzyme cascade reaction for highly efficient synthesis of non-natural chiral amino acids. J Biotechnol. 2015;195:67–71.
Resch V, Fabian WMF, Kroutil W. Deracemisation of mandelic acid to optically pure non-natural l-phenylglycine via a redox-neutral biocatalytic cascade. Adv Synth Catal. 2010;352:993–7.
Xue YP, Wang W, Wang YJ, Liu ZQ, Zheng YG, Shen YC. Isolation of enantioselective alpha-hydroxyacid dehydrogenases based on a high-throughput screening method. Bioprocess Biosyst Eng. 2012;35:1515–22.
Xue YP, Tian FF, Ruan LT, Liu ZQ, Zheng YG, Shen YC. Concurrent obtaining of aromatic (R)-2-hydroxyacids and aromatic 2-ketoacids by asymmetric oxidation with a newly isolated Pseudomonas aeruginosa ZJB1125. J Biotechnol. 2013;167:271–8.
Dewanti AR, Xu Y, Mitra B. Role of glycine 81 in (S)-mandelate dehydrogenase from Pseudomonas putida in substrate specificity and oxidase activity. Biochemistry. 2004;43:10692–700.
Dewanti AR, Mitra B. A transient intermediate in the reaction catalyzed by (S)-mandelate dehydrogenase from Pseudomonas putida. Biochemistry. 2003;42:12893–901.
Rydzanicz R, Zhao XS, Johnson PE. Assembly PCR oligo maker: a tool for designing oligodeoxynucleotides for constructing long DNA molecules for RNA production. Nucleic Acids Res. 2005;33:W521–5.
Streitenberger SA, Lopez-Mas JA, Sanchez-Ferrer A, Garcia-Carmona F. A study on the kinetic mechanism of apoenzyme reconstitution from Aerococcus viridans lactate oxidase. J Enzym Inhib Med Chem. 2003;18:285–8.
Chen CS, Fujimoto Y, Girdaukas G, Sih CJ. Quantitative analyses of biochemical kinetic resolutions of enantiomers. J Am Chem Soc. 1982;104:7294–9.
Authors’ contributions
YPX, HZ and YGZ designed the study. HZ carried out the bulk of the experiments. YPX, HZ, XLJ, ZQL and YGZ analyzed and interpreted the data. HZ and YPX wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Sequences data used in this study have been deposited in the GenBank with accession number KU612124 for the gene of (S)-2-HADH from P. aeruginosa CCTCC M 2011394 and KU612125 for the gene of (R)-2-HADH from L. mesenteroides CCTCC M 2016063.
Funding
This work was funded by the National Natural Science Foundation of China (No. 21676254).
Author information
Authors and Affiliations
Corresponding author
Additional file
12934_2016_560_MOESM1_ESM.docx
Additional file 1: Figure S1. Amino acid sequences multiple alignment of 2-HADH from P. aeruginosa CCTCC M 2011394, B. xenovorans LB400, P. putida ATCC 12633, P. aeruginosa NUST, P. fluorescens strain EBC191. Figure S2. Optimization of biooxidation reaction conditions by resting cells of recombinant E. coli BL21(DE3)/pET28b-HADH. (A) Effect of temperature on the biooxidation. The optimum temperature was determined over the range from 25 and 65 °C. The recombinant E. coli BL21(DE3)/pET28b-HADH showed high activity at 35–55 °C, and at higher temperatures the activity began to decrease significantly. (B) Effect of pH on the biooxidation; The optimum pH on the oxidation reaction was determined over the range from 6.0 and 9.0. When the pH was below 7.5 or over 8.5, the enzyme activity decreased dramatically. Figure S3. SDS-PAGE analysis of the expressed (R)-2-KAR in recombinant E. coli BL21(DE3)/pET28b-KAR. 1. Markers; 2. Cell-free extract of recombinant E. coli BL21(DE3)/pET28b-KAR; 3. Purified (R)-2-KAR from E. coli BL21(DE3)/pET28b-KAR (~32 kDa). Figure S4. SDS-PAGE analysis of the expressed GDH in recombinant E. coli BL21(DE3)/pET28b-GDH and coexpressed (R)-2-KAR and GDH in recombinant E. coli BL21(DE3)/pCDFDuet-KAR-GDH. 1. E. coli BL21(DE3)/pCDFDuet-KAR-GDH with 0.1 mM IPTG. The upper arrow indicated (R)-2-KAR (~32 kDa) and the lower arrow represented GDH (~28 kDa).2. E. coli BL21(DE3)/pCDFDuet-KAR-GDH with 0 mM IPTG; 3. E. coli BL21(DE3)/pET28b-GDH with 0.1 mM IPTG; 4. E. coli BL21(DE3)/pET28b-GDH with 0 mM IPTG; 5.Empty plasmid of pET28b with 0 mM IPTG. 6. Markers. Figure S5. SDS-PAGE analysis of the coexpressed (S)-2-HADH, (R)-2-KAR and GDH in recombinant E. coli BL21(DE3)/pET28b-HADH/pCDFDuet-KAR-GDH. 1. Markers; 2. Cell-free extract of recombinant E. coli BL21(DE3)/pET28b-HADH/pCDFDuet-KAR-GDH. The upper arrow indicated (S)-2-HADH (~42 kDa), the middle arrow indicated (R)-2-KAR (~32 kDa) and the lower arrow represented GDH (~28 kDa).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xue, YP., Zeng, H., Jin, XL. et al. Enantioselective cascade biocatalysis for deracemization of 2-hydroxy acids using a three-enzyme system. Microb Cell Fact 15, 162 (2016). https://doi.org/10.1186/s12934-016-0560-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-016-0560-1