
Abstract
Background
Recombinant antibodies are highly successful in many different pathological conditions and currently enjoy overwhelming recognition of their potential. There are a wide variety of protein expression systems available, but almost all therapeutic antibodies are produced in mammalian cell lines, which mimic human glycosylation. The production of clinical-grade antibodies in mammalian cells is, however, extremely expensive. Compared to mammalian systems, protein production in yeast strains such as Pichia pastoris, is simpler, faster and usually results in higher yields.
Results
In this work, a trivalent single-chain fragment variable (scFv)-based N-terminal trimerbody, specific for the human carcinoembryonic antigen (CEA), was expressed in human embryonic kidney 293 cells and in Pichia pastoris. Mammalian- and yeast-produced anti-CEA trimerbody molecules display similar functional and structural properties, yet, the yield of trimerbody expressed in P. pastoris is about 20-fold higher than in human cells.
Conclusions
P. pastoris is an efficient expression system for multivalent trimerbody molecules, suitable for their commercial production.
Similar content being viewed by others


Background
Over the past decades, there has been growing interest in the use of recombinant antibodies in bioanalytical and medical applications [1]. In an attempt to improve the therapeutic efficacy of antibodies, new recombinant formats with modified properties have been generated [2]. Multivalent and multispecific antibodies capable of simultaneously blocking multiple growth and survival pathways have the potential to meet the current and future therapeutic challenges, and indeed many of them are advancing in clinical development [3]. The most common strategy to create multivalent IgG-like formats has been the fusion of antibody fragments with homodimerization sequences (e.g., ZIP miniantibody [4], minibody [5] or single-chain fragment variable (scFv)-Fc antibody [6]). A different strategy to multimerize antibody fragments is based on the modification of the interdomain linker length to generate bivalent, trivalent or tetravalent molecules [7,8]. Other protein-protein interactions have been also used to create multivalent non-IgG-like formats, such as the streptavidin-biotin system, the C-terminal multimerization domain of the tumor-suppressor protein p53 [9], and the ribonuclease barnase with its inhibitor, barstar [10], among others [2].
A variety of expression systems ranging from bacterial cells to mammalian cells have been used to express recombinant antibodies [11,12]. E. coli is the most commonly used host for the expression of antibody fragments, whereas mammalian cells are used for the expression of large, multidomain antibodies such as full-length monoclonal antibodies or complex recombinant antibody fragments [13]. In fact, almost all approved therapeutic antibodies for human use are produced in mammalian cell culture systems [14].
In previous studies, we reported the in vitro and in vivo characterization of a multivalent antibody generated by fusing a trimerization (TIE) domain to the C-terminus of a scFv antibody [15-17]. TIE domains are composed of the N-terminal trimerization region of collagen XVIII NC1 (TIEXVIII) or collagen XV NC1 (TIEXV) flanked by flexible linkers. The new antibody format, termed trimerbody, is trimeric in solution and exhibited excellent antigen binding capacity and multivalency [15-17]. Furthermore, by fusing scFv antibodies with the same or different specificity to both ends of a TIEXVIII domain, we have produced monospecific or bispecific hexavalent-binding molecules, expanding the scope of potential applications of trimerbody molecules [18].
To date, trivalent and hexavalent scFv-based trimerbodies have only been produced in mammalian cell cultures [15-18]. However, the generation of stable antibody-producing mammalian cell lines is an expensive and time-consuming procedure. Here, we evaluated the potential of the methylotrophic yeast P. pastoris [12,19,20] to produce with high yield a N-terminal trimerbody specific for the human carcinoembryonic antigen (CEA) [16]. The functional and biochemical properties of both mammalian- and yeast-derived trimerbodies were gauged demonstrating the functional equivalence of the two preparations. Our results demonstrate that P. pastoris is a viable alternative expression system for scFv-based N-terminal trimerbody molecules.
Results
Generation of anti-CEA scFv-based N-terminal trimerbody expression vectors
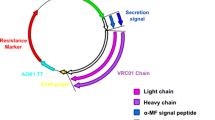
In this study we have generated a pPICZαA-based vector for the expression of the MFE-23 scFv-based N-terminal trimerbody (MFE-23N) in P. pastoris (Figure 1), and we demonstrated that MFE-23N molecules are efficiently secreted as soluble proteins by transformed P. pastoris cells. Western blot analysis shows that under reducing conditions, a single polypeptide chain with mass around 37 kDa was seen (Additional file 1: Figure S1B). As previously shown [16], the MFE-23N trimerbody is efficiently secreted as soluble functional protein by HEK-293 cells transfected (Additional file 1: Figure S1A) with the expression vector pCEP4-MFE-23-NC1ES- (Figure 1). Secreted MFE-23N trimerbodies from both sources are able to recognize immobilized human CEA with high affinity and specificity (Additional file 2: Figure S2).

Schematic diagrams showing the genetic and domain structure of scFv-based N-terminal trimerbodies. (A) Diagrammatic representation of gene constructs. Both constructs bear the anti-CEA MFE-23 scFv gene (VH-linker-VL), a TIEXVIII domain and c-myc and His tags (hatched box), for subsequent purification and immunodetection. Signal peptides were from oncostatin M (OM) and α-factor for expression in HEK-293 cells (upper) and P. pastoris (lower) respectively. (B) Schematic representation of the domain structure of the scFv-based N-terminal trimerbody.
Purification and functional characterization of yeast- and mammalian- produced anti-CEA scFv-based N-terminal trimerbodies
For purification, the extracellular medium of P. pastoris cells after 72 hours of methanol induction, and the serum-free conditioned media from stably transfected HEK-293 cells were independently collected. Both MFE-23N trimerbodies were purified by immobilized metal affinity chromatography, which yielded >95% pure 37 kDa proteins as assessed by reducing SDS-PAGE (Figure 2A). Both systems produced soluble and functional MFE-23N molecules, but with significant differences in antibody yields from Pichia and HEK-293 cells, 6 and 0.35 mg/l, respectively. Importantly, the yeast-produced MFE-23N trimerbody was functional and recognized, as efficiently as the mammalian-produced MFE-23N trimerbody, human CEA either plastic immobilized (Figure 2B) or expressed on the tumor cell surface (Figure 2C).

Characterization of purified trimerbodies. (A) Reducing SDS-PAGE of anti-CEA scFv-based N-terminal trimerbody (MFE-23N) purified from HEK-293 cells or P. pastoris. The functionality of purified MFE-23N trimerbodies is demonstrated by ELISA against plastic immobilized CEA (B) and by FACS on CEA− and CEA+ tumor cells (C). The anti-CEA C6G9 mAb (IgG) was used as control.
Structural characterization of yeast and mammalian produced anti-CEA scFv-based N-terminal trimerbodies
Both mammalian- and yeast-produced trimerbodies elute from the analytical gel-filtration columns as major peaks at 13 ml with molar masses of 110 or 108 kDa, respectively. These masses are consistent with the calculated values for the trimeric molecules (110 and 113 kDa, respectively) (Figure 3A, B). A minor peak eluting at 11 ml is also seen in the chromatograms of both molecules, with molar mass of 214 and 210 kDa (yeast and mammalian cells, respectively) (Figure 3A, B). These minor peaks contain about 10% of the protein (relative to the major ones as estimated from absorbance at the corresponding maxima), and their masses are consistent with hexamers (possibly dimers of the corresponding trimmers). SDS-PAGE analysis of the two species separated in the gel filtration column showed a single band at the same position (Additional file 3: Figure S3), and at the expected position relative to the molecular weight markers (between the 45 kDa and 35 kDa markers, in agreement with the calculated values of 37 and 38 kDa for the yeast- and mammalian-produced MFE-23N, respectively). These results demonstrate that the purified MFE-23N trimerbodies behave predominantly as trimers with a small proportion of hexamers, independently of the producer organism.

Structural characterization of purified trimerbodies. Oligomeric analysis of MFE-23N purified from P. pastoris (A) or HEK-293 cells (B). Circular dichroism spectrum (C) and thermal denaturation (D) for MFE-23N molecules purified from P. pastoris (red line) and HEK-293 cells (black line).
The CD spectra of both trimerbodies were very similar, with minima at 217 nm and less negative minima at 228–230 nm (Figure 3C). This is consistent with the secondary structures of the scFv domain, mainly β-sheet and irregular loops, plus the contribution of the helical structures of the trimerization domains of collagen XVIII NC1 domain and the linker sequences (which probably are flexible random coils). The MFE-23N molecules produced in P. pastoris and in HEK-293 cells showed a major cooperative thermal transition, with essentially the same mid-point denaturation temperature of 48–49°C. At high temperatures another minor transition is observed, possibly due to aggregation phenomena of the denatured polypeptide chains. The same behavior was observed in experiments recorded at 210 nm with the trimeric molecules separated from hexameric ones by gel filtration (Figure 3D). These results show that the scFv-based N-terminal trimerbodies produced in P. pastoris and in HEK-293 cells have very similar structures and thermal stabilities.
Serum stability study of yeast and mammalian produced N-terminal trimerbodies
Both MFE-23N trimerbodies were further analyzed to evaluate their long-term stability in serum, an important feature of recombinant antibodies for potential diagnostic or therapeutic applications. For this purpose, purified MFE-23 scFv N-terminal trimerbodies were incubated in human serum for 0 (control) to 4 days at 37°C (Figure 4). MFE-23N molecules purified from P. pastoris were more stable with 60% CEA binding activity after 4 days of incubation, whereas mammalian-produced MFE-23Nmolecules retained around 40% CEA binding activity. The stability was also analyzed by western blot (Figure 4B), and we found that after 4 days at 37°C 60% of the MFE-23N trimerbody produced in P. pastoris was structurally intact, while around 40% of the mammalian-produced MFE-23N trimerbody was functional at the end of the assay.

Serum stability of MFE-23 N purified from P. pastoris or HEK-293 cells. ELISA against plastic immobilized CEA (A) and western blot (B) were performed after incubation at 37°C for different time periods in human serum, as indicated in material and methods.
Discussion
In the present study we demonstrate that methylotrophic yeast P. pastoris secreted functional CEA-specific MFE-23 scFv-based N-terminal trimerbody at significant levels. Moreover, we demonstrate that both yeast- and mammalian-produced MFE-23N trimerbodies have similar functional and structural properties. Purified MFE-23N molecules were trimeric in solution, as unambiguously shown by the light scattering measurements. The anti-CEA scFv-based N-terminal trimerbodies produced in P. pastoris and in HEK-293 cells are highly efficient at recognizing antigen either immobilized in plastic, or associated to the cell surface. The dose-dependent binding curves of purified MFE-23N molecules to plastic immobilized human CEA were comparable. Furthermore, both scFv-based N-terminal trimerbodies specifically recognize CEA cancer cells. Additionally, we demonstrated that MFE-23N molecules produced in yeast are slightly more stable in human serum than MFE-23N molecules produced in mammalian cells.
P. pastoris is widely used for the secretion of properly folded proteins with high yields in a cost-efficient and rapid way [21]. It offers complex posttranslational modification pathways avoiding pyrogenic contamination. In this sense, P. pastoris holds a generally recognized as safe (GRAS) status [22]. The yield of anti-CEA scFv-based N-terminal trimerbody expressed in P. pastoris was of 6 mg of pure protein per liter of culture, which is about 20-fold higher than in mammalian cells. This is consistent with P. pastoris ability to reach very high cell densities, up to 100 OD600, allowing significantly increased amounts of secreted protein. P. pastoris has been widely used in the expression of recombinant antibodies, such as scFv [23,20], tandem scFvs, also known as (scFv)2 [24], diabodies [25], Fab fragments [26-28], tribodies [29], scFv-Fc [30], scFv-immunotoxins [31,32] and full-length IgG [33,34]. Moreover, favorable protein folding by P. pastoris seems to play a fundamental role in the stability and activity of a single domain antibody fragment against botulinum neurotoxin in comparison to the same produced in E. coli [35]. Another relevant issue is that P. pastoris displays both O- and N-linked glycosylation, but glycosylation patterns are different from those found in higher eukaryotes and may lead to reduction in activity and antigenic response. Moreover, P. pastoris is known to glycosylate proteins that are non-glycosylated in mammalian cells [14]. Therefore, although the prediction of potential glycosylation sites using the GlycoEP server [36] showed that the MFE-23N trimerbody does not contain putative N- and O-glycosylation sites, we can not rule out that yeast-produced trimerbodies may be “decorated” with some extra sugars, and this could be the explanation for the subtle difference in size observed between yeast- and mammalian-produced MFE-23N molecules. Importantly, we have demonstrated that even if trimerbody glycosylation takes place, it does not affect antigen binding. A current alternative is the use of the P. pastoris genetically engineered to produce humanized glycosylation patterns. In fact, anti-Her2 mAb produced in glyco-engineered P. pastoris exhibit features comparable to those of trastuzumab in preclinical assays [34]. New marketed therapeutic proteins produced in Pichia evidence the rise of P. pastoris as a producer organism. In 2009 the FDA approved ecallantide, a small recombinant protein acting as a potent, specific and reversible inhibitor of plasma kallikrein for the treatment of acute hereditary angioedema [37,38].
Conclusions
Wave demonstrated that scFv-based N-terminal trimerbodies can be efficiently produced in P. pastoris in a trimeric fully functional active form. These results illustrate the potential of Pichia pastoris for the secretion of multivalent antibodies.
Methods
Reagents and antibodies
The mAb used include: C6G9 (Sigma-Aldrich, St. Louis, MO, USA) anti-human CEA (CD66e) and Tetra-His (Qiagen, GmbH, Hilden, Germany). The polyclonal antibodies included: phycoerytrin (PE)-conjugated goat F(ab’)2 fragment anti-mouse IgG (Fc fragment specific, Jackson Immuno Research, Newmarket, UK), horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Fc specific) (Sigma-Aldrich), and IRDye800-conjugated donkey anti-mouse IgG (H&L) (Rockland Immunochemicals, Gilbertsville, PA, USA). Human CEA was obtained from Calbiochem (Merck, Darmstadt, Germany) and bovine serum albumin (BSA) was from Sigma-Aldrich.
Cells and culture conditions
HEK-293 (CRL-1573) and HeLa (CCL-2) cells were obtained from the American Type Culture Collection (Rockville, MD, USA). They were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Lonza, Walkersville, MD, USA) supplemented with 10% (vol/vol) heat inactivated fetal calf serum (FCS) (Thermo Fisher, MA, USA). The HeLaCEA cell line [39] was cultured in medium containing 750 μg/ml G418 (Promega, Madison, WI; USA). The methylotrophic yeast P. pastoris strain KM71 was obtained from Invitrogen (Life Technologies, Carlsbad, CA, USA). Cells were grown on yeast extract peptone dextrose (YPD) plates or YPD medium at 30°C. When harbouring an expression vector the cells were grown on YPD plates with zeocin.
Construction of expression vectors
The mammalian expression vector pCEP4-MFE-23-NC1ES-encoding the CEA-specific MFE-23 scFv-based N-terminal trimerbody, containing a murine TIEXVIII domain, has been previously reported [16]. To generate the P. pastoris expression vector the DNA fragment encoding the MFE-23 scFv was PCR amplified from pCEP4-MFE-23-NC1ES- with primers EcoRI FW and NotI RV (Table 1). The EcoRI /NotI-digested PCR fragment was ligated into the EcoRI/NotI-digested backbone of plasmid pPICZαA (Life Technologies) to generate the intermediate plasmid pPICZαA-MFE-23. The DNA encoding human TIEXVIII was PCR amplified from plasmid pCR3.1-L36-hNC1 [18] with primers NotI FW and SalI RV (Table 1). The NotI/SalI-digested PCR fragment was ligated into the NotI/SalI-digested backbone of plasmid pPICZαA-MFE-23 to obtain pPICZαA-MFE-23-TIE. The sequence was verified using primers 5’ AOX1 and 3’AOX1 (Table 1).
Stable expression in mammalian cells
HEK-293 cells were transfected with pCEP4-MFE-23-NC1ES- vector using calcium phosphate [40], and selected in DMEM with 150 μg/ml hygromycin B (Life Technologies) to generate stable cell lines. Supernatants from stably transfected cell populations were analyzed for protein expression by ELISA, SDS-PAGE and western blotting using Tetra-His mAb.
Stable expression in yeast cells
Electrocompetent P. pastoris KM71 cells were electroporated with linearized pPICZαA-MFE-23-TIE plasmid, as previously described [31,32], using a Bio-Rad Gene pulser apparatus (Bio-Rad, Hercules, CA, USA). Cells harboring the desired construct were selected after plating the transformation mixture on YPDS (Yeast Peptone Dextrose Sorbitol) media containing different amounts (100 to 750 μg/ml) of zeocin (Life Technologies), and three independent clones were tested by small-scale production. The colony that showed better results was selected for larger scale production, which was performed by inoculating 2 l baffled flasks containing 250 ml of buffered methanol-complex (BMMY) medium [1% yeast extract, 2% peptone, 100 m K3PO4 (pH 6.0), 1.34% yeast nitrogen base (NYD), 4.5x10−5% biotin, 0.5% methanol] for induction at 25°C and 250 rpm shaking for 72 h. Every 24 h, methanol was added to the medium, to afford a final methanol concentration of 0.5% (v/v).
Purification
Harvested serum-free conditioned mammalian medium was centrifuged, 0.22 μm filtered (Nalgene, Neerijse, Belgium), concentrated (10x) with a 10.000 MWCO Vivaflow 50 filter (Vivascience GmbH, Hannover, Germany), dialyzed against PBS (pH7.4) and loaded onto a HisTrap HP 1 ml column using and ÄKTA Prime plus system (GE Healthcare, Uppsala, Sweden). The purified trimerbody was dialyzed against PBS, analyzed by SDS-PAGE under reducing conditions, and stored at −80°C. Harvested yeast medium was dialyzed against 50 mM Na3PO4 buffer, containing 100 mM NaCl (pH 8.0), 0.22 μm filtered and loaded onto a HisTrap HP 1 ml column using and ÄKTA Prime plus system. The purified trimerbody, was dialyzed against Na3PO4 buffer, analyzed by SDS-PAGE under reducing conditions, and stored at −80°C. For lyophilization, samples were dialyzed with 50 mM (NH4)HCO3 (pH 8.0), and lyophilized protein was stored at −20°C.
Western blotting
Samples were separated under reducing conditions on 12% Tris-glycine gels and transferred to nitrocellulose membranes (Life Technologies) and reacted with Tetra-His mAb, followed by incubation with an IRDye800-conjugated donkey anti-mouse IgG. Visualization and quantitative analysis of protein bands were carried out with the Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA).
ELISA
The ability of scFv-based N-terminal trimerbodies to bind human CEA was studied by ELISA as previously described [16]. Briefly, Maxisorp plates (Nunc A/S, Roskilde, Denmark) were coated with CEA (0.25 μg/well) and after washing and blocking with 5% BSA in PBS, 100 μl with indicated amount of purified protein or supernatant were added for 1 hour at room temperature. After three washes, 100 μl of Tetra-His mAb (10 μg/ml) were added for 1 hour at room temperature. After three washes, 100 μl of HRP-conjugated goat anti-mouse IgG were added for 1 hour at room temperature, after which the plate was washed and developed. Antigen titration was performed with serial dilutions of the purified trimerbodies.
Flow cytometry
The ability of purified antibodies to bind to cell surface CEA was studied by FACS as described previously [16]. Briefly, cells were incubated with anti-CEA mAb (10 μg/ml) or purified trimerbodies (10 μg/ml) and Tetra-His mAb for 30 min. After washing, the cells were treated with appropriate dilutions of PE-conjugated goat F(ab’)2 anti-mouse IgG. All samples were analyzed with a Beckman-Coulter FC-500 Analyzer (Beckman-Coulter, Brea, CA, USA).
Size exclusion chromatography-multi-angle laser light scattering (SEC-MALLS)
Static light scattering experiments were performed at room temperature using a Superdex 200 10/300 GL column (GE HealthCare) connected to a DAWN-HELEOS light scattering detector and an Optilab rEX differential refractive index detector (Wyatt Technology, Santa Barbara, CA, USA). The column was equilibrated with running buffer (PBS pH 7.0 + 0.03% NaN3, 0.1 μm filtered) and the SEC-MALLS system was calibrated with a sample of BSA at 1 g/l in the same buffer. Samples of 100 μl of the MFE-23N molecules at 0.55 g/l were injected into the column at a flow rate of 0.5 mL/min. Data acquisition and analysis employed ASTRA software (WyattTechnology). Based on numerous measurements on BSA samples at 1 g/l under the same or similar conditions we estimate that the experimental error in molar mass is around 5%.
Circular dichroism and thermal denaturation studies
Circular dichroism (CD) measurements were performed with a Jasco J-810 spectropolarimeter equipped with Peltier thermal control unit (Jasco, MD, USA). The spectra were recorded at 25°C on protein samples at 0.05 g/l in PBS using a 0.2 cm path length stoppered quartz cuvette a response of 8 s and a band width of 2 nm. The spectra were baseline corrected by subtraction of the buffer spectrum recorded in same cuvette under identical conditions. The thermal denaturations were recorded on the same samples increasing the temperature from 10 to 95°C at a rate of 1°C/min and measuring the ellipticity at 210 nm every 1°C with a 32 second response and 4 nm bandwidth. For the graphical representation of the melting curves of both samples the ellipticity values were normalized from 0 (at 10°C) to 1 (at 95°C). The CD data were processed with the program Origin (OriginLab, MA, USA). We estimate that the uncertainty in the molar ellipticity is about 5% and the uncertainty in the mid-point denaturation temperature is 0.5°C.
Serum stability
One microgram of each purified scFv-based N-terminal trimerbody was incubated in 60% human serum at 37°C for up to 96 h. Samples were removed for analysis at 3, 24, 48 and 96 hours and frozen at −80°C until the entire study was completed. As a control, a second set of serum-exposed samples was frozen immediately to represent a zero time point. Aliquots were then subjected to western blot, using Tetra-His mAb, and tested for their capability to bind human CEA by ELISA.
Abbreviations
- CEA:
-
Carcinoembryonic antigen
- mAb:
-
Monoclonal antibody
- scFv:
-
Single-chain variable fragment
- SEC-MALLS:
-
Size exclusion chromatography-multi-angle laser light scattering
- TIE:
-
Trimerization domain
- OM:
-
Oncostatin M
References
Li J, Zhu Z: Research and development of next generation of antibody-based therapeutics. Acta Pharmacol Sin 2010, 31:1198–1207.
Cuesta AM, Sainz-Pastor N, Bonet J, Oliva B, Alvarez-Vallina L: Multivalent antibodies: when design surpasses evolution. Trends Biotechnol 2010, 28:355–362.
Fitzgerald J, Lugovskoy AA: Rational engineering of antibody therapeutics targeting multiple oncogene pathways. MAbs 2011, 3:299–309.
Pack P, Plückthun A: Miniantibodies: use of amphipathic helices to produce functional, flexibly linked dimeric FV fragments with high avidity in Escherichia coli. Biochemistry (Mosc) 1992, 31:1579–1584.
Hu S, Shively L, Raubitschek A, Sherman M, Williams LE, Wong JYC, Shively JE, Wu AM: Minibody: a novel engineered anti-carcinoembryonic antigen antibody fragment (Single-Chain Fv-CH3) which exhibits rapid, high-level targeting of xenografts. Cancer Res 1996, 56:3055–3061.
Li S-L, Liang S-J, Guo N, Wu AM, Fujita-Yamaguchi Y: Single-chain antibodies against human insulin-like growth factor I receptor: expression, purification, and effect on tumor growth. Cancer Immunol Immunother 2000, 49:243–252.
Asano R, Hagiwara Y, Koyama N, Masakari Y, Orimo R, Arai K, Ogata H, Furumoto S, Umetsu M, Kumagai I: Multimerization of anti-(epidermal growth factor receptor) IgG fragments induces an antitumor effect: the case for humanized 528 scFv multimers. FEBS J 2013, 280:4816–4826.
Holliger P, Prospero T, Winter G: “Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci U S A 1993, 90:6444–6448.
Deyev SM, Lebedenko EN: Multivalency: the hallmark of antibodies used for optimization of tumor targeting by design. BioEssays News Rev Mol Cell Dev Biol 2008, 30:904–918.
Deyev SM, Waibel R, Lebedenko EN, Schubiger AP, Plückthun A: Design of multivalent complexes using the barnase · barstar module. Nat Biotech 2003, 21:1486–1492.
Gopal GJ, Kumar A: Strategies for the production of recombinant protein in Escherichia coli. Protein J 2013, 32:419–425.
Frenzel A, Kügler J, Wilke S, Schirrmann T, Hust M: Construction of human antibody gene libraries and selection of antibodies by phage display. Methods Mol Biol 2014, 1060:215–243.
Butler M, Meneses-Acosta A: Recent advances in technology supporting biopharmaceutical production from mammalian cells. Appl Microbiol Biotechnol 2012, 96:885–894.
Frenzel A, Hust M, Schirrmann T: Expression of recombinant antibodies. Front Immunol 2013, 4:217.
Sánchez-Arevalo Lobo VJ, Cuesta AM, Sanz L, Compte M, García P, Prieto J, Blanco FJ, Alvarez-Vallina L: Enhanced antiangiogenic therapy with antibody-collagen XVIII NC1 domain fusion proteins engineered to exploit matrix remodeling events. Int J Cancer 2006, 119:455–462.
Cuesta ÁM, Sánchez-Martín D, Sanz L, Bonet J, Compte M, Kremer L, Blanco FJ, Oliva B, Álvarez-Vallina L: In vivo tumor targeting and imaging with engineered trivalent antibody fragments containing collagen-derived sequences. PLoS ONE 2009, 4:e5381.
Cuesta AM, Sánchez-Martín D, Blanco-Toribio A, Villate M, Enciso-Álvarez K, Alvarez-Cienfuegos A, Sainz-Pastor N, Sanz L, Blanco FJ, Alvarez-Vallina L: Improved stability of multivalent antibodies containing the human collagen XV trimerization domain. MAbs 2012, 4:226–232.
Blanco-Toribio A, Sainz-Pastor N, Álvarez-Cienfuegos A, Merino N, Cuesta ÁM, Sánchez-Martín D, Bonet J, Santos-Valle P, Sanz L, Oliva B, Blanco FJ, Álvarez-Vallina L: Generation and characterization of monospecific and bispecific hexavalent trimerbodies. MAbs 2013, 5:70–79.
Cereghino JL, Cregg JM: Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev 2000, 24:45–66.
Damasceno LM, Huang C-J, Batt CA: Protein secretion in Pichia pastoris and advances in protein production. Appl Microbiol Biotechnol 2012, 93:31–39.
Gonçalves AM, Pedro AQ, Maia C, Sousa F, Queiroz JA, Passarinha LA: Pichia pastoris: a recombinant microfactory for antibodies and human membrane proteins. J Microbiol Biotechnol 2013, 23:587–601.
Mattia A, Merker R: Regulation of probiotic substances as ingredients in foods: premarket approval or “generally recognized as safe” notification. Clin Infect Dis Off Publ Infect Dis Soc Am 2008, 46(Suppl 2):S115–S118. discussion S144–151.
Ridder R, Schmitz R, Legay F, Gram H: Generation of rabbit monoclonal antibody fragments from a combinatorial phage display library and their production in the yeast Pichia pastoris. Biotechnol Nat Publ Co 1995, 13:255–260.
Wang Z, Duran-Struuck R, Crepeau R, Matar A, Hanekamp I, Srinivasan S, Neville DM, Sachs DH, Huang CA: Development of a diphtheria toxin based anti-porcine CD3 recombinant immunotoxin. Bioconjug Chem 2011, 22:2014–2020.
FitzGerald K, Holliger P, Winter G: Improved tumour targeting by disulphide stabilized diabodies expressed in Pichia pastoris. Protein Eng 1997, 10:1221–1225.
Lange S, Schmitt J, Schmid RD: High-yield expression of the recombinant, atrazine-specific Fab fragment K411B by the methylotrophic yeast Pichia pastoris. J Immunol Methods 2001, 255:103–114.
Lin S, Houston-Cummings NR, Prinz B, Moore R, Bobrowicz B, Davidson RC, Wildt S, Stadheim TA, Zha D: A novel fragment of antigen binding (Fab) surface display platform using glycoengineered Pichia pastoris. J Immunol Methods 2012, 375:159–165.
Takahashi K, Yuuki T, Takai T, Ra C, Okumura K, Yokota T, Okumura Y: Production of humanized fab fragment against human high affinity IgE receptor in Pichia pastoris . Biosci Biotechnol Biochem 2000, 64:2138–2144.
Schoonooghe S: Engineering and expression of bibody and tribody constructs in mammalian cells and in the yeast Pichia pastoris. Methods Mol Biol Clifton NJ 2012, 899:157–175.
Liu J, Wei D, Qian F, Zhou Y, Wang J, Ma Y, Han Z: pPIC9-Fc: a vector system for the production of single-chain Fv-Fc fusions in Pichia pastoris as detection reagents in vitro. J Biochem (Tokyo) 2003, 134:911–917.
Carreras-Sangrà N, Tomé-Amat J, García-Ortega L, Batt CA, Oñaderra M, Martínez-del-Pozo A, Gavilanes JG, Lacadena J: Production and characterization of a colon cancer-specific immunotoxin based on the fungal ribotoxin α-sarcin. Protein Eng Des Sel PEDS 2012, 25:425–435.
Tomé-Amat J, Menéndez-Méndez A, García-Ortega L, Batt CA, Oñaderra M, Martínez-del-Pozo A, Gavilanes JG, Lacadena J: Production and characterization of scFvA33T1, an immunoRNase targeting colon cancer cells. FEBS J 2012, 279:3022–3032.
Potgieter TI, Cukan M, Drummond JE, Houston-Cummings NR, Jiang Y, Li F, Lynaugh H, Mallem M, McKelvey TW, Mitchell T, Nylen A, Rittenhour A, Stadheim TA, Zha D, d’ Anjou M: Production of monoclonal antibodies by glycoengineered Pichia pastoris. J Biotechnol 2009, 139:318–325.
Zhang N, Liu L, Dumitru CD, Cummings NRH, Cukan M, Jiang Y, Li Y, Li F, Mitchell T, Mallem MR, Ou Y, Patel RN, Vo K, Wang H, Burnina I, Choi B-K, Huber H, Stadheim TA, Zha D: Glycoengineered Pichia produced anti-HER2 is comparable to trastuzumab in preclinical study. MAbs 2011, 3:289–298.
Baghban R, Gargari SLM, Rajabibazl M, Nazarian S, Bakherad H: Camelid-derived heavy chain nanobody against Clostridium botulinum neurotoxin E in Pichia pastoris. Biotechnol Appl Biochem 2014. doi:10.1002/bab.1226.
Chauhan JS, Rao A, Raghava GPS: In silico platform for prediction of N-, O- and C-glycosites in eukaryotic protein sequences. PLoS ONE 2013, 8:e67008.
Bos IGA, de Bruin EC, Karuntu YA, Modderman PW, Eldering E, Hack CE: Recombinant human C1-inhibitor produced in Pichia pastoris has the same inhibitory capacity as plasma C1-inhibitor. Biochim Biophys Acta 2003, 1648:75–83.
Lehmann A: Ecallantide (DX-88), a plasma kallikrein inhibitor for the treatment of hereditary angioedema and the prevention of blood loss in on-pump cardiothoracic surgery. Expert Opin Biol Ther 2008, 8:1187–1199.
Hefta LJF, Chen F-S, Ronk M, Sauter SL, Sarin V, Oikawa S, Nakazato H, Hefta S, Shively JE: Expression of carcinoembryonic antigen and its predicted immunoglobulin-like domains in HeLa cells for epitope analysis. Cancer Res 1992, 52:5647–5655.
Compte M, Blanco B, Serrano F, Cuesta AM, Sanz L, Bernad A, Holliger P, Alvarez-Vallina L: Inhibition of tumor growth in vivo by in situ secretion of bispecific anti-CEA [times] anti-CD3 diabodies from lentivirally transduced human lymphocytes. Cancer Gene Ther 2007, 14:380–388.
Acknowledgements
This study was supported by grants from Ministerio de Economía y Competitividad (BIO2011–22738), and Comunidad de Madrid (S-BIO-0236–2006 and S2010/BMD-2312) to L.A-V.; from Fondo de Investigación Sanitaria/Instituto de Salud Carlos III (PI13/00090) to L.S.; and from Ministerio de Economía y Competitividad (BFU2012-32404) to J.L. A.B-T. was supported by Programa Torres Quevedo from Ministerio de Economía y Competitividad, cofounded by the European Social Fund (PTQ11–04604).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AB-T, JL, FJB and LA-V designed research; AB-T, JL, NN-P, AA-C, MV and MC performed research; AB-T, JL, LS, FJB and LA-V analyzed data; AB-T, JL, LS, FJB and LA-V wrote the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1: Figure S1.
MFE-23N secretion in HEK-293 cells and P. pastoris. Western blot analysis of conditioned media from transfected HEK-293 cells (A), and of culture supernatants after the induction of three different clones (1–3) in P. pastoris (B).
Additional file 2: Figure S2.
ELISA against plastic immobilized CEA with conditioned media (MFE-23N SN) from transfected HEK-293 cells (A), and culture supernatants after the induction of three different clones in P. pastoris (B). In both cases MFE-23N purified (10 μg/ml) from HEK-293 cells was used as a control.
Additional file 3: Figure S3.
Reducing SDS-PAGE analysis of the samples used and separated in the SEC-MALLS experiments. Input corresponds to the sample injected in the column and the peaks correspond to fractions of the two peaks separated in the column. Peak1 and peak2 correspond to the peaks eluting at approximately volumes of 11 and 13 ml, respectively, as shown in Figures 3A and 3B. The total amount of protein loaded in each lane is different because for some of the samples eluted from the column a large volume was collected and the total protein was precipitated before loading in the gel.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Blanco-Toribio, A., Lacadena, J., Nuñez-Prado, N. et al. Efficient production of single-chain fragment variable-based N-terminal trimerbodies in Pichia pastoris . Microb Cell Fact 13, 116 (2014). https://doi.org/10.1186/s12934-014-0116-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-014-0116-1