
Abstract
Type 2 diabetes (T2D), cardiovascular disease (CVD) and chronic kidney disease (CKD), are recognized among the most disruptive public health issues of the current century. A large body of evidence from epidemiological and clinical research supports the existence of a strong interconnection between these conditions, such that the unifying term cardio-metabolic-renal (CMR) disease has been defined. This coexistence has remarkable epidemiological, pathophysiologic, and prognostic implications. The mechanisms of hyperglycemia-induced damage to the cardio-renal system are well validated, as are those that tie cardiac and renal disease together. Yet, it remains controversial how and to what extent CVD and CKD can promote metabolic dysregulation. The aim of this review is to recapitulate the epidemiology of the CMR connections; to discuss the well-established, as well as the putative and emerging mechanisms implicated in the interplay among these three entities; and to provide a pathophysiological background for an integrated therapeutic intervention aiming at interrupting this vicious crosstalks.
Similar content being viewed by others


Introduction
The International Diabetes Federation estimates that there are currently more than 500 million people living with diabetes worldwide, the vast majority of whom suffering from type 2 diabetes (T2D) [1]. Moreover, globally, there are about 64 million persons diagnosed with heart failure (HF) [2] and almost 700 million individuals affected by chronic kidney disease (CKD) [3], these three entities being the major pandemics of the twenty-first century. Taken individually, each of these three conditions are associated with relevant morbidity and mortality, but it is broadly recognized that they often coexist: patients with HF have a four-fold higher prevalence of T2D (20%) than patients without HF (4–6%) [4], and T2D is associated with a two-to four-fold higher risk of developing cardiovascular disease (CVD) [5]. Furthermore, recent statistics report a CKD prevalence close to 40% among individuals with T2D [6] and 50% among individuals with HF [7]. Conversely, CVD is diagnosed more frequently among patients with CKD than in the general population, being its prevalence inversely related with kidney function [8].
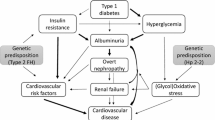
Since growing evidence supports the existence of a strong interplay among T2D, CVD and CKD, the unifying term cardio-metabolic-renal (CMR) disease has been introduced to describe the systemic interdependence of these conditions [9] (Fig. 1).

Cardio-metabolic-renal interconnections and therapeutic options. SNS, sympathetic nervous system; RAAS, renin–angiotensin–aldosterone system; NPs, natriuretic peptides; SGLT2i: sodium-glucose cotransporter 2 inhibitors; GLP1- RA: glucagon-like peptide 1 receptor agonists
While the mechanisms whereby T2D promotes the onset of CVD and CKD have been widely described in the literature, how to what extent HF and CKD promote the development of T2D or worsen T2D control is still much less appreciated. The aim of this review is to summarize the current knowledge of the multidirectional pathophysiological interactions that occur among these three entities.
Epidemiology of the cardio-renal-metabolic disease
Epidemiological studies have consistently demonstrated that cardiovascular, renal, and metabolic diseases often overlap and coexist in the same patients. According to a meta-analysis of 102 prospective studies, individuals with T2D have a two-fold higher risk for coronary heart disease, stroke and death attributed to other vascular causes [10]. In the study by Kodama et al., T2D was a significant risk factor for both new-onset (risk ratio [RR], 2.14 [95% CI 1.96–2.34]) and recurrent HF (RR, 1.39 [95% CI 1.33–1.45]), and the risk of HF associated with T2D was increased especially in young populations [11]. Conversely, T2D prevalence is greater in HF cohorts than in the general population, with reports of 24% among overall patients with HF and of 40% among those hospitalized with worsening HF [12]. Individuals with HF have an increased risk for subsequent onset of T2D after adjustment for multiple cardiovascular confounders [13], and an analysis from the Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity (CHARM) program reported an incidence of T2D of 28 per 1000 person-years among initially nondiabetic individuals with HF [14], which was substantially higher than that in the general population (7.1 per 1000 person-years) [15]. These associative findings are still insufficient to claim that HF plays a role in the development of T2D and further studies are needed to better dissect the HF-T2D bidirectional interplay. The coexistence of T2D and HF worsens the overall prognosis: diabetes is a predictor of poor clinical outcome, cardiovascular morbidity and mortality in HF cohorts [16, 17] and, reciprocally, incident HF is associated with a tenfold higher mortality risk in patients with T2D aged 65 years or older [18].
Furthermore, it is well validated that diabetes is the leading etiology of both CKD and end-stage kidney disease (ESKD) [19]. It is estimated that half of individuals diagnosed with type 2 diabetes and one-third of those with type 1 diabetes will develop CKD during their lifetime [19], and a meta-analysis including data from more than 5 million participants estimated the relative risk to develop CKD for females and males with T2D versus those without to be 3.34 (95% CI 2.27–4.93) and 2.84 (95% CI 1.73–4.68), respectively [20].
Unsurprisingly, not only diabetes is an established risk factor for CKD, but a high prevalence of diabetes has also been described among patients with CKD, ranging from 31 to 40% [21,22,23]. Although few studies have evaluated incident T2D in individuals with CKD, the Chronic Renal Insufficiency Cohort (CRIC) Study found an overall T2D incidence rate of 17.8 cases per 1000-persons years, markedly higher than that in the general population [24], supporting the bidirectionality of the interplay between these two conditions.
CKD is also considered a major risk factor for CVD, including HF [25], and the risk for cardiovascular events and death increases with declining estimated glomerular filtration rate (eGFR) [26]. An analysis from the Cardiovascular Health Study provides evidence that elevated serum creatinine is an independent predictor of CVD, HF, cardiovascular- and all-cause mortality [27]. Conversely, individuals with HF have more than two-fold higher risk of incident CKD and rapid eGFR decline [28].
The interconnections among these pathological entities assume even more relevance considering that T2D is per se associated with adverse cardiovascular outcomes, though the risk is further increased when renal dysfunction coexists, leading to a ninefold increase in relative cardiovascular mortality [29]. Altogether, these epidemiological data support the existence of a multidirectional link between T2D, HF and CKD, with each one independently increasing the incidence and worsening the prognosis of the others.
Pathophysiological mechanisms underlining the cardio-renal-metabolic connection
Mechanistic pathways linking T2D to cardio-renal damage
A detailed description of the molecular mechanisms through which diabetes affects the cardiovascular and renal systems is beyond the scope of this review and has been provided elsewhere [30, 31]. In brief, according to the “unifying hypothesis”, in hyperglycemic states, the excessive intracellular glucose flux leads to mitochondrial superoxide production and exacerbation of oxidative stress, postulated as the primary initiating event in diabetes-induced organ damage [32]. The increased reactive oxygen species (ROS) production causes tissue damage through several mechanisms: activation of the polyol and hexosamine pathways—which exacerbate oxidative stress in a vicious circle—activation of protein kinase C (PKC), formation of advanced glycation end-products (AGEs), resulting from non-enzymatic glycation of proteins, and upregulation of their cellular receptor RAGE [33]. In turn, AGEs can damage the heart, the vessels and the kidney both directly, causing cross-linking of matrix proteins and increasing tissue stiffness, and indirectly, via interaction with their receptor RAGE, activating signaling pathways that alter cellular function and promote oxidative stress, inflammation, and fibrosis [34]. Thus, AGEs are involved in the pathogenesis of diabetes-related organ damage, such as diabetic cardiomyopathy, diabetic kidney disease (DKD) and atherosclerosis [30, 34, 35]. AGEs and ROS are also closely associated with endothelial dysfunction, which is a major driver of diabetic microvascular and macrovascular complications [6].
Additionally, hyperglycemia is associated with the activation of local renin–angiotensin–aldosterone system (RAAS) in the myocardium and in the kidney, promoting vasoconstriction, fibrosis, and exacerbation of organ dysfunction [36, 37].
Furthermore, diabetes is perceived as a state of “nutrient abundance”, characterized by an aberrant activation of nutrient-sensing pathways, such as AMPK, sirtuins and mTOR, downregulating cytoprotective responses and promoting organ impairment [38, 39]. It has been demonstrated that, in glomerular podocytes, the cells responsible for the integrity of the glomerular basement membrane (GBM) and the correct functioning of the glomerular capillary loop, mTOR activation recapitulates many characteristics of DKD, such as proteinuria and mesangial expansion [40].
In parallel to glucotoxicity, insulin resistance is associated with a cellular metabolic shift towards free fatty acid (FFA) oxidation, which is more oxygen-consuming than glucose oxidation. This leads to impaired metabolic flexibility and reduced energetic efficiency, which are findings of diabetes–associated organ alterations [4, 9]. The increased uptake of FFA, when excessive, leads to accumulation of intracellular triacylglycerols, promoting oxidative stress, lipotoxicity and apoptosis [12]. In the heart, epicardial adipose tissue (EAT), a visceral fat depot located between the myocardium and the epicardium endowed with paracrine properties of regulating the myocardium and coronary arteries [41], has been proposed to function as a buffer to provide energy for the myocardium while protecting it from FFA overload [42]. T2D has been associated with pathological changes in EAT volume [43], cytokine secretory profile [44], and FFA release [45], which are potential drivers of diabetes-associated cardiovascular dysfunctions, such as atherosclerosis, intramyocardial fatty infiltration, cardiac remodeling, and HF [46].
In addition to activating several pathways driving tissue damage, diabetes compromises tissue repair, at least in part by jeopardizing the contribution of bone marrow-derived hematopoietic stem/progenitor cells (HSPCs).
Solid evidence shows that T2D is associated with a reduction in the levels of circulating HSPCs [47], mainly driven by an impaired mobilization from the bone marrow (BM) [48]. The putative HSPC property of maintaining tissue homeostasis by contributing to vascular and tissue repair [49] can explain why their shortage promotes multi-organ damage [50]. Indeed, HSPC defect has been extensively linked to the development of microvascular and macrovascular complications in T2D population [51,52,53], and represents a risk factor for adverse cardiovascular outcomes and death [54]. The evidence that BM-derived cells contribute to renal parenchymal regeneration after damage [55] may point to a role of HSPC shortage in DKD. HSPCs might be particularly relevant for non-albuminuric DKD, given the association between HSPCs and several CVD risk factors involved in the development of this CKD phenotype [49, 56]. In this view, diabetes can be considered a disease of impaired damage control, with defects in the physiological processes of tissue repair [57].
Finally, it should be mentioned that several other mechanisms can impair cardio-renal function in T2D, including functional abnormalities driven by metabolic and hemodynamic impairment. Insulin resistance per se leads to peripheral microvascular dysfunction [58] and skeletal muscle dysfunction [59], both of which are linked to an increased risk of HF [60, 61]. In the kidney, hyperfiltration has long been considered the major early functional alteration paving the way to the subsequent development of later DKD stages [62], though its role has been recently challenged, especially in T2D [63].
Global impact of T2D on the cardiovascular system
Diabetes affects the cardiovascular system in different ways. It is strongly associated with the development of atherosclerosis, whose manifestations include coronary artery disease (CAD), peripheral artery disease (PAD) ad stroke. As aforementioned, hyperglycemia is closely linked with endothelial dysfunction, vascular abnormalities and inflammation, which are drivers for atherosclerotic plaque formation and progression [64]. Moreover, T2D coexists with well-known cardiovascular risk factors, including atherogenic dyslipidemia, characterized by high levels of small-dense LDL and low levels of HDL cholesterol [65]. LDL glycation occurring in hyperglycemic states increases their atherogenic potential, as glycated LDL are recognized by a scavenger receptor expressed on macrophages, resulting in a non-regulated intracellular cholesterol accumulation and enhanced plaque formation [66]. Diabetes is also associated with hypertension due to a predominance of endothelial vasoconstrictive over vasodilation signals in the diabetic milieu, a comorbidity that rises the risk of atherosclerosis and cardiovascular adverse outcomes [6]. Another frequent comorbidity which extensively contributes to the increased cardiovascular risk in T2D is obesity, which negatively impacts the cardiovascular system via different mechanisms, such as altered hemodynamic load, neurohormonal disturbances and low-grade systemic inflammation [67].
HF is a prominent diabetes-induced complication, and its onset can be promoted through different mechanisms. Atherosclerosis in the coronary arteries is strongly accelerated by diabetes, and plaque complications leading to myocardial ischemia can result in ischemic cardiomyopathy that eventually culminates in HF [68]. In parallel, metabolic alterations associated with diabetes can directly affect myocardial performance. Diabetic cardiomyopathy is defined as a diastolic or systolic dysfunction in the presence of a history of long-standing and/or poorly controlled diabetes with the exclusion of other causes of cardiomyopathy, such as coronary, congenital, valvular, and hypertensive heart disease [4, 68]. Diabetic cardiomyopathy can assume two different phenotypes: (i) restrictive, HFpEF-like phenotype, more commonly found in women with obesity and linked to coronary endothelial inflammation; (ii) dilated, HfrEF-phenotype, more closely associated with cardiomyocyte loss [69]. Although it is currently uncertain which factors are crucial for the development of one or the other phenotype, hyperglycemia, hyperinsulinemia, lipotoxicity and coronary endothelial dysfunction play a pivotal role in the development of diabetic cardiac abnormalities [4]. AGEs, maladaptive calcium homeostasis, and activation of the local RAAS, together with myocardial energetic inefficiency induced by insulin resistance, promote the development of impaired contraction, myocardial stiffness and fibrosis, which contribute to cardiac dysfunction in diabetes [35, 68, 70, 71].
Impact of T2D on kidney function
The kidney is a major target of microvascular diabetic damage and DKD is associated with adverse health outcomes and high mortality burden. The scientific community has moved from the concept of “diabetic nephropathy”—defined by a rise in urinary albumin excretion classically followed by a progressive decline in renal function and traditionally classified in five stages [72]—to the term “diabetic kidney disease”, including all possible renal abnormalities occurring in diabetes [6].
DKD is the renal manifestation of the same hyperglycemia-induced damage pathways that target susceptible sites elsewhere in the body when exposed to the gluco- and lipotoxic diabetic milieu [38]. One of the earliest alterations in the diabetic kidney is glomerular hyperfiltration, whose pathogenesis has been linked to both altered tubule-glomerular feedback and glomerular hemodynamic abnormalities occurring in the diabetic milieu [73]. Persistent hyperfiltration results in progressive and irreversible damage to the nephron and eGFR decline, eventually terminating in ESKD. Renal tissue impairment is reflected by albuminuria and proteinuria, which are associated with functional and structural cellular alterations favored by dysregulated metabolic conditions [74]. In detail, tubular cells undergo maladaptive hypertrophy and hyperplasia in consequence of the increased glucose load delivered to the tubule and upregulate their sodium-glucose cotransporters to favor its reabsorption [75]. Consequently, the reduced amount of sodium delivered to the macula densa activates tubulo-glomerular feedback, which results in local activation of RAAS, hyperfiltration, and progressive damage to the glomeruli [31]. At later stages, tubular cells undergo atrophy, and their dysfunction leads to impaired protein reuptake and albuminuria [75], being tubulointerstitial fibrosis the final shared pathway for progressive renal impairment in DKD [38].
Podocytes undergo pathological changes as well, including de-differentiation, detachment, and foot-process effacement [38]. As podocytes control GBM matrix turnover, it is plausible that dysfunctional podocytes favor GBM thickening and alter its function, promoting glomerular damage and albuminuria [76]. Concurrently, the mechanical stress caused by accumulation of ECM proteins contributes to glomerular injury [77].
Mesangial cells constitute another target of diabetes-induced damage: they become hypertrophic, proliferate, and increase the synthesis of matrix proteins, leading to some of the typical structural features of diabetic glomerulopathy [78]. Thus, although hyperglycemia may be the primary initiating event in DKD, its pathogenesis is multifactorial, with diverse hemodynamic, mechanical, and structural processes contributing to the decline in kidney function [79]. Indeed, it should be mentioned that, in addition to hyperglycemia, several cardiovascular risk factors, including hypertension, obesity, hyperuricemia and inflammation, can promote renal injury in T2D [80]. Thus, these multifaceted mechanisms may sustain the development of the emerging “non-albuminuric DKD” phenotype, whose pathogenesis hypothetically relies on mechanisms operating at the macrovascular and tubule-interstital level [81], in contrast to the typical glomerular damage characterizing the classical albuminuric DKD [38].
The bidirectional cardio-renal interplay
There is broad evidence of a close interconnection between kidney and heart disease: the term cardiorenal syndrome (CRS) has been defined to underline bidirectionality of heart-kidney interplay, with acute or chronic dysfunction of one organ leading to acute or chronic dysfunction of the other [82], conferring relevant morbidity and mortality [83]. Since combined heart and kidney abnormalities can differ in their clinical presentation and time frame (acute vs chronic), a subdivision of CRS in five subtypes has been adopted [82]. A detailed discussion of pathophysiological mechanisms potentially responsible for CRS is beyond the scope of this article as such information can be found elsewhere [82,83,84]. It is worth mentioning that hemodynamic and neurohormonal abnormalities are putatively key players in the detrimental crosstalk between the failing heart and the failing kidney [83]. Briefly, HF-associated low cardiac output, effective hypovolemia, and excess of vasoconstrictive mediators lead to chronic renal hypoperfusion and decrease in eGFR, favoring CKD initiation and/or progression [82]. Conversely, sodium and water retention and chronic RAAS activation in CKD exacerbate hypertension and increase cardiac pre- and after-load. These hemodynamic abnormalities—together with CKD-associated uremic toxins retention and chronic inflammation—sustain pathological cardiac remodeling and the onset and worsening of cardiac dysfunction, completing a vicious cycle injurious to both organs [82]. The parallel pathways leading to cardiac and renal disease in the context of T2D are illustrated in Fig. 2.

Cardio-renal interplay in the context of T2D. The figure illustrates some of the mechanisms that sustain the bidirectional relationships between kidney disease and cardiac remodeling leading to heart failure in the context of type 2 diabetes. SNS, sympathetic nervous system; RAAS, renin-angiotensin-aldosterone system; eGFR: estimated glomerular filtration rate CKD, chronic kidney disease; ESKD: end-stage kidney disease
Mechanisms for the impact of cardiovascular dysfunction on T2D onset
Not only diabetes is traditionally considered a major cardiovascular risk factor, but, conversely, there is growing evidence that cardiovascular dysfunction can promote metabolic alterations and new-onset T2D. To explain this relationship, different mechanisms have been proposed, although not entirely elucidated. First, endothelial dysfunction has increasingly been recognized as a common soil between disorders of glucose and cardiovascular homeostasis [85]. This hypothesis is supported by a close bidirectional relationship between insulin resistance, a core metabolic abnormality in T2D, and endothelial dysfunction, which is one of the crucial initiating events in the pathogenesis of CVD [86]. Additionally, HF can per se be considered an “insulin resistant state” [87, 88]. One mechanism affecting insulin sensitivity in HF is neuro-hormonal activation [89, 90]: in particular, sympathetic nervous system (SNS) hyperactivation that occurs in HF impairs glucose homeostasis via stimulation of alpha-adrenergic receptors, resulting in skeletal muscle hypoperfusion and diminished tissue glucose uptake [91]. Insulin resistance is not the only mechanism through which catecholamine hypersecretion affects glucose homeostasis in HF: chronic SNS activation enhances lipolysis, and elevated FFA can deposit ectopically and have been shown to promote hepatic gluconeogenesis and impair insulin secretions from pancreatic β-cells [92].
The onset of T2D in individuals with HF may also be mediated by the overactivity of the RAAS: angiotensin II (AngII) induces skeletal muscle vasoconstriction and defective muscle glucose uptake, which are associated with diminished insulin sensitivity [93]. AngII has also been shown to directly interfere with insulin signaling pathway by inducing phosphorylation of insulin signal molecules, thereby inhibiting downstream signal transduction [94]. Additionally, AngII can per se induce β-cell dysfunction via an endoplasmic reticulum stress-mediated mechanism [95], impairing insulin secretion and favoring β-cell apoptosis [96].
Other neuro-hormonal factors involved are natriuretic peptides (NPs): they have recently emerged as metabolic hormones, improving insulin sensitivity, lipid oxidation and browning of the adipose tissue [97]. Additionally, there is evidence that NP exert a direct influence on β-cells, modulating their function and enhancing insulin secretion [98]. Metabolic dysregulated conditions, such as obesity and T2D, are associated with low NP levels, suggesting that NP defect may impair glucose homeostasis [99]. Although NP serum levels are increased in HF, their effectiveness is reduced [100]: this evidence suggests that HF is paradoxically similar to metabolic diseases characterized by NP deficiency, pointing to a role of NP in the development of HF-associated glucose abnormalities.
It is also broadly recognized that proinflammatory cytokines play a role in the development of insulin resistance by interfering with insulin signaling [101]. HF is a pro-inflammatory condition, and a significant association between inflammation-related biomarkers in HF and new-onset T2D has been described, suggesting that immuno-inflammatory mechanisms may be involved in the pathogenesis of HF-associated diabetes [102].
The influence of physical activity on T2D onset has been widely described across literature [103, 104], and exercise activity limitation experienced by patients with HF potentially contributes to the development of glucose abnormalities in this condition [89]. HF is associated with reduced skeletal muscle perfusion and loss of muscle mass, the main tissue where glucose is utilized, and both these processes lead to impaired peripheral glucose utilization and diminished sensitivity to insulin [90].
Mechanisms for the impact of kidney dysfunction on T2D onset
While diabetes is a prominent risk factor for kidney disease, it becomes to be appreciated that kidney disease may, in turn, promote metabolic dysregulation and new-onset or worsening T2D. Although some studies suggest that the incident rate of T2D in patients with CKD is significantly higher than that in the general population [24, 105], the pathophysiology underlying glucose abnormalities in CKD remains largely unclear.
There is evidence that insulin resistance is an early finding in people diagnosed with CKD [106], and kidney dysfunction seems to be associated per se with defective insulin-signaling pathway, since insulin resistance is a frequent abnormality in CKD regardless of its etiology [107]. The skeletal muscle is recognized as the primary site of CKD-associated insulin resistance, which is caused by “post-receptor defects” involving signal transduction proteins [107]. Several mechanisms are potentially involved in the genesis of CKD-associated glucose abnormalities. CKD is a condition characterized by chronic inflammation and enhanced oxidative stress [108], and it is well validated that proinflammatory cytokines promote insulin resistance through post-translational modifications of signal-transduction proteins [107, 109].
Furthermore, metabolic acidosis—a common CKD complication—has been associated with decreased sensitivity to insulin in both healthy and CKD individuals [110, 111]. Although only few studies have been conducted, there is evidence that acidosis correction by administration of sodium bicarbonate ameliorates insulin resistance, supporting the causal involvement of metabolic acidosis in suboptimal biological response to insulin [112].
Another mechanism potentially affecting glucose homeostasis in CKD is vitamin D deficiency, a very frequent finding in individuals with impaired kidney function [113]. Vitamin D status may directly influence glucose metabolism: vitamin D regulates insulin release through the modulation of intracellular calcium in β-cells [114], increases the expression of the insulin receptor [115], and its deficiency is associated with secondary hyperparathyroidism, which can diminish insulin secretion [116]. A body of evidence supports the existence of an association between vitamin D deficiency and glucose metabolism abnormalities in populations with CKD [117, 118]. A randomized controlled-study including non-diabetic individuals with CKD found that insulin resistance incidence was significantly higher in the vitamin D-deficient than in the vitamin D-normal group; in the same study, the supplementation with activated vitamin D analogs significantly ameliorated insulin sensitivity and β-cell function, supporting the hypothesis of a direct role of vitamin D in regulating metabolic homeostasis in CKD [119].
Other factors favoring abnormalities in glucose metabolism are toxins accumulating as the renal function critically diminishes, such as blood urea, p-cresyl sulfate, and asymmetric dimethylarginine [120,121,122]. Although these compounds can alter glucose homeostasis via inflammation-mediated insulin resistance [107], urea can directly induce β-cell dysfunction: elevated levels of urea increase islet protein O-GlcNAcylation and impair glycolysis, resulting in insulin secretory defects [123].
Understanding the pathogenesis of CKD-associated glucose abnormalities becomes even more crucial considering that CKD is a condition associated with high cardiovascular risk [8] and insulin resistance is a predictor of CVD and cardiovascular mortality in several CKD cohorts [124, 125].
Integrated management of CMR disease
Since T2D, HF and CKD share a common pathophysiologic background and often coexist, adopting a holistic therapeutic strategy targeting CMR comorbidities would have a synergistic effect on patient health, resulting in significant outcome improvements. Data from large-scale clinical trials have consistently shown that the beneficial effects of novel glucose-lowering drugs, such as sodium-glucose cotransporter 2 inhibitors (SGLT2i) and glucagon-like peptide 1 receptor agonists (GLP-1 RA), extend far beyond glycemic control, reducing important cardiovascular and renal endpoints in populations with T2D [126,127,128]. Similarly, the selective, non-steroidal mineralocorticoid receptor antagonist Finerenone has exhibited cardiorenal protective effects in individuals with T2D and CKD [129, 130]. The CMR abnormalities on which these classes of drugs have been shown to have favorable impact are summarized in Table 1. The putative mechanisms responsible for CMR benefits of these medications have been deeply revised elsewhere [131,132,133,134,135]. Briefly, while SGLT2i are believed to exert cardiorenal protection prominently via a hemodynamic action sustained by their natriuretic and osmotic effects [136], anti-atherogenic and immune-modulating mechanisms may be responsible for GLP-1 RA-mediated protective effects [133, 137]. The novel bireceptor agonist Tirzepatide simultaneously activates two incretin-dependent pathways [138], and this duality acts synergistically on glycemic and weight control, significantly improving metabolic outcomes when compared to selective GLP-1 RA [139]. On the other hand, Finerenone exerts its cardiorenal protection at a different level, targeting MR overactivation, a major pro-inflammatory and pro-fibrotic driver of cardiorenal complications in T2D [135].
While SGLT2i are now popular drugs for the management of T2D, they appear to exert their beneficial effects against HF and ESKD largely independently from glycemic control. Indeed, protection from hospitalization for HF, as well as from the progression of CKD have been demonstrated even in people without diabetes. In addition, the glucose lowering capacity of SGLT2i reduces together with the decline in renal function, though the protection against ESKD is preserved until the later CKD stages [140]. On the other side, the cardiovascular or renal benefits of GLP-1RA in non-diabetic individuals still needs to be demonstrated and mediation analyses found the change in HbA1c to be a predictor of the end-organ protection [141].
The use of single glucose-lowering agents with manifold protective effects on CMR system, as well as concomitant treatment with multiple drugs having complementary mechanisms of action are promising for the management of CMR disease spectrum. Thus, there is potential to use multifactorial intervention to fully take advantages of complementary pharmacological effects and simultaneously target comorbid conditions.
Conclusions
A large amount of epidemiological data from observational and clinical trials supports the existence of a substantial overlap between metabolic, cardiovascular, and renal diseases, with the onset of one increasing the risk and worsening the outcome of the others. These three entities share common pathophysiological mechanisms, whose activation results in a vicious cycle of perpetuation of diseases processes, increasing morbidity and mortality. The identification of the pathophysiological interconnections among these comorbidities is key to unravel common therapeutic approaches. A better understanding of the shared core mechanisms underlining CMR disease can provide targets for pharmacological intervention aiming at interrupting the detrimental crosstalk, thereby ameliorating clinical outcomes.
Adopting a tailored therapeutic approach addressing overall patient’s comorbid conditions becomes even more essential considering the availability of novel glucose-lowering drugs with proven renal and cardiovascular protection. Further research is needed to gain deeper insight into CMR pathophysiology, elucidate the benefits from an integrated management of comorbid T2D, CVD and CKD and guide individualized treatment choices in clinical practice (Table 2).
Availability of data and materials
All the data used to write this manuscript are presented in the text, tables or references.
Change history
06 October 2023
The error in the funding note has been corrected
References
Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183:109119.
Groenewegen A, Rutten FH, Mosterd A, Hoes AW. Epidemiology of heart failure. Eur J Heart Fail. 2020;22:1342–56.
Bikbov B, Purcell CA, Levey AS, Smith M, Abdoli A, Abebe M, et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2020;395:709–33.
Maack C, Lehrke M, Backs J, Heinzel FR, Hulot JS, Marx N, et al. Heart failure and diabetes: metabolic alterations and therapeutic interventions: a state-of-the-art review from the translational research committee of the heart failure association-European society of cardiology. Eur Heart J. 2018;39:4243–54.
Seferović PM, Petrie MC, Filippatos GS, Anker SD, Rosano G, Bauersachs J, et al. Type 2 diabetes mellitus and heart failure: a position statement from the heart failure association of the European society of cardiology. Eur J Heart Fail. 2018;20:853–72.
Usman MS, Khan MS, Butler J. The interplay between diabetes, cardiovascular disease, and kidney disease. ADA Clin Compend. 2021;2021:13–8.
Damman K, Valente MAE, Voors AA, O’Connor CM, van Veldhuisen DJ, Hillege HL. Renal impairment, worsening renal function, and outcome in patients with heart failure: an updated meta-analysis. Eur Heart J. 2014;35:455–69.
Jankowski J, Floege J, Fliser D, Böhm M, Marx N. Cardiovascular disease in chronic kidney disease. Circulation. 2021;143:1157–72.
Kadowaki T, Maegawa H, Watada H, Yabe D, Node K, Murohara T, et al. Interconnection between cardiovascular, renal and metabolic disorders: a narrative review with a focus on Japan. Diabetes Obes Metab. 2022;24:2283–96.
The Emerging Risk Factors Collaboration. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215–22.
Kodama S, Fujihara K, Horikawa C, Sato T, Iwanaga M, Yamada T, et al. Diabetes mellitus and risk of new-onset and recurrent heart failure: a systematic review and meta-analysis. ESC Heart Fail. 2020;7:2146–74.
Dei Cas A, Khan SS, Butler J, Mentz RJ, Bonow RO, Avogaro A, et al. Impact of diabetes on epidemiology, treatment, and outcomes of patients with heart failure. JACC Heart Fail. 2015;3:136–45.
Amato L, Paolisso G, Cacciatore F, Ferrara N, Ferrara P, Canonico S, et al. Congestive heart failure predicts the development of non-insulin-dependent diabetes mellitus in the elderly. The Osservatorio Geriatrico Regione Campania Group. Diabetes Metab. 1997;23:213–8.
Preiss D, Zetterstrand S, McMurray JJV, Östergren J, Michelson EL, Granger CB, et al. Predictors of development of diabetes in patients with chronic heart failure in the candesartan in heart failure assessment of reduction in mortality and morbidity (CHARM) program. Diabetes Care. 2009;32:915–20.
Geiss LS, Wang J, Cheng YJ, Thompson TJ, Barker L, Li Y, et al. Prevalence and incidence trends for diagnosed diabetes among adults aged 20 to 79 years, United States, 1980–2012. JAMA. 2014;312:1218–26.
MacDonald MR, Petrie MC, Varyani F, Ostergren J, Michelson EL, Young JB, et al. Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: an analysis of the candesartan in heart failure: assessment of reduction in mortality and morbidity (CHARM) programme. Eur Heart J. 2008;29:1377–85.
Shindler DM, Kostis JB, Yusuf S, Quinones MA, Pitt B, Stewart D, et al. Diabetes mellitus, a predictor of morbidity and mortality in the studies of left ventricular dysfunction (SOLVD) trials and registry. Am J Cardiol. 1996;77:1017–20.
Bertoni AG, Hundley WG, Massing MW, Bonds DE, Burke GL, Goff DC. Heart failure prevalence, incidence, and mortality in the elderly with diabetes. Diabetes Care. 2004;27:699–703.
Koye DN, Magliano DJ, Nelson RG, Pavkov ME. The global epidemiology of diabetes and kidney disease. Adv Chronic Kidney Dis. 2018;25:121–32.
Shen Y, Cai R, Sun J, Dong X, Huang R, Tian S, et al. Diabetes mellitus as a risk factor for incident chronic kidney disease and end-stage renal disease in women compared with men: a systematic review and meta-analysis. Endocrine. 2017;55:66–76.
Kuznik A, Mardekian J, Tarasenko L. Evaluation of cardiovascular disease burden and therapeutic goal attainment in US adults with chronic kidney disease: an analysis of national health and nutritional examination survey data, 2001–2010. BMC Nephrol. 2013;14:132.
Titze S, Schmid M, Kottgen A, Busch M, Floege J, Wanner C, et al. Disease burden and risk profile in referred patients with moderate chronic kidney disease: composition of the German Chronic Kidney Disease (GCKD) cohort. Nephrol Dial Transplant. 2015;30:441–51.
Nitta K, Iimuro S, Imai E, Matsuo S, Makino H, Akizawa T, et al. Risk factors for increased left ventricular hypertrophy in patients with chronic kidney disease: findings from the CKD-JAC study. Clin Exp Nephrol. 2019;23:85–98.
Jepson C, Hsu JY, Fischer MJ, Kusek JW, Lash JP, Ricardo AC, et al. Incident type 2 diabetes among individuals with CKD: findings from the chronic renal insufficiency cohort (CRIC) study. Am J Kidney Dis. 2019;73:72–81.
Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL, et al. Kidney disease as a risk factor for development of cardiovascular disease. Hypertension. 2003;42:1050–65.
Go AS, Chertow GM, Fan D, McCulloch CE, Hsu C-Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. New England J Med. 2004;351:1296–305.
Fried LF, Shlipak MG, Crump C, Kronmal RA, Bleyer AJ, Gottdiener JS, et al. Renal insufficiency as a predictor of cardiovascular outcomes and mortality in elderly individuals. J Am Coll Cardiol. 2003;41:1364–72.
George LK, Koshy SKG, Molnar MZ, Thomas F, Lu JL, Kalantar-Zadeh K, et al. Heart failure increases the risk of adverse renal outcomes in patients with normal kidney function. Circ Heart Fail. 2017. https://doi.org/10.1161/CIRCHEARTFAILURE.116.003825.
Opie LH, Parving HH. Diabetic nephropathy. Circulation. 2002;106:643–5.
Li Y, Liu Y, Liu S, Gao M, Wang W, Chen K, et al. Diabetic vascular diseases: molecular mechanisms and therapeutic strategies. Signal Transduct Target Ther. 2023;8:152.
Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137–88.
Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20.
Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–70.
Singh VP, Bali A, Singh N, Jaggi AS. Advanced glycation end products and diabetic complications. Korean J Physiol Pharmacol. 2014;18:1–14.
Lombardi C, Spigoni V, Gorga E, Dei CA. Novel insight into the dangerous connection between diabetes and heart failure. Herz. 2016;41:201–7.
Lim HS, MacFadyen RJ, Lip GYH. Diabetes mellitus, the renin-angiotensin-aldosterone system, and the heart. Arch Intern Med. 2004;164:1737–48.
Giacchetti G, Sechi LA, Rilli S, Carey RM. The renin-angiotensin-aldosterone system, glucose metabolism and diabetes. Trends Endocrinol Metab. 2005;16:120–6.
Thomas MC, Brownlee M, Susztak K, Sharma K, Jandeleit-Dahm KAM, Zoungas S, et al. Diabetic kidney disease. Nat Rev Dis Primers. 2015;1:15018.
Suhara T, Baba Y, Shimada BK, Higa JK, Matsui T. The mTOR signaling pathway in myocardial dysfunction in type 2 diabetes mellitus. Curr Diab Rep. 2017;17:38.
Gödel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Investig. 2011;121:2197–209.
Li Y, Liu B, Li Y, Jing X, Deng S, Yan Y, et al. Epicardial fat tissue in patients with diabetes mellitus: a systematic review and meta-analysis. Cardiovasc Diabetol. 2019;18:3.
Iacobellis G. Epicardial adipose tissue in endocrine and metabolic diseases. Endocrine. 2014;46:8–15.
Iacobellis G, Bianco AC. Epicardial adipose tissue: emerging physiological, pathophysiological and clinical features. Trends Endocrinol Metab. 2011;22:450–7.
Sacks HS, Fain JN, Cheema P, Bahouth SW, Garrett E, Wolf RY, et al. Inflammatory genes in Epicardial fat contiguous with coronary atherosclerosis in the metabolic syndrome and type 2 diabetes. Diabetes Care. 2011;34:730–3.
Iacobellis G, Barbaro G. Epicardial adipose tissue feeding and overfeeding the heart. Nutrition. 2019;59:1–6.
Christensen RH, von Scholten BJ, Lehrskov LL, Rossing P, Jørgensen PG. Epicardial adipose tissue: an emerging biomarker of cardiovascular complications in type 2 diabetes? Ther Adv Endocrinol Metab. 2020;11:1–16.
Fadini GP, Boscaro E, de Kreutzenberg S, Agostini C, Seeger F, Dimmeler S, et al. Time course and mechanisms of circulating progenitor cell reduction in the natural history of type 2 diabetes. Diabetes Care. 2010;33:1097–102.
Fadini GP, Albiero M, de Vigili KS, Boscaro E, Cappellari R, Marescotti M, et al. Diabetes impairs stem cell and proangiogenic cell mobilization in humans. Diabetes Care. 2013;36:943–9.
Fadini GP, Mehta A, Dhindsa DS, Bonora BM, Sreejit G, Nagareddy P, et al. Circulating stem cells and cardiovascular outcomes: from basic science to the clinic. Eur Heart J. 2020;41:4271–82.
Fadini GP, Albiero M. Impaired hematopoietic stem/progenitor cell traffic and multi-organ damage in diabetes. Stem Cells. 2022;40:716–23.
Rigato M, Bittante C, Albiero M, Avogaro A, Fadini GP. Circulating progenitor cell count predicts microvascular outcomes in type 2 diabetic patients. J Clin Endocrinol Metab. 2015;100:2666–72.
Fadini GP, Rigato M, Cappellari R, Bonora BM, Avogaro A. Long-term prediction of cardiovascular outcomes by circulating CD34+ and CD34+CD133+ stem cells in patients with type 2 diabetes. Diabetes Care. 2017;40:125–31.
Fadini GP, Sartore S, Albiero M, Baesso I, Murphy E, Menegolo M, et al. Number and function of endothelial progenitor cells as a marker of severity for diabetic Vasculopathy. Arterioscler Thromb Vasc Biol. 2006;26:2140–6.
Rigato M, Avogaro A, Fadini GP. Levels of circulating progenitor cells, cardiovascular outcomes and death. Circ Res. 2016;118:1930–9.
Poulsom R, Forbes SJ, Hodivala-Dilke K, Ryan E, Wyles S, Navaratnarasah S, et al. Bone marrow contributes to renal parenchymal turnover and regeneration. J Pathol. 2001;195:229–35.
Berezin AE, Kremzer AA, Samura TA, Berezina TA, Martovitskaya YV. Serum uric acid predicts declining of circulating proangiogenic mononuclear progenitor cells in chronic heart failure patients. J Cardiovasc Thorac Res. 2014;6:153–62.
Fadini GP. A reappraisal of the role of circulating (progenitor) cells in the pathobiology of diabetic complications. Diabetologia. 2014;57:4–15.
Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction Implications for the syndrome of insulin resistance. J Clin Investig. 1996;97:2601–10.
Yokota T, Kinugawa S, Yamato M, Hirabayashi K, Suga T, Takada S, et al. Systemic oxidative stress is associated with lower aerobic capacity and impaired skeletal muscle energy metabolism in patients with metabolic syndrome. Diabetes Care. 2013;36:1341–6.
Bilak JM, Alam U, Miller CA, McCann GP, Arnold JR, Kanagala P. Microvascular dysfunction in heart failure with preserved ejection fraction: pathophysiology assessment, prevalence and prognosis. Card Fail Rev. 2022. https://doi.org/10.15420/cfr.2022.12.
Kinugawa S, Takada S, Matsushima S, Okita K, Tsutsui H. Skeletal muscle abnormalities in heart failure. Int Heart J. 2015;56:475–84.
Moriconi D, Sacchetta L, Chiriacò M, Nesti L, Forotti G, Natali A, et al. Glomerular hyperfiltration predicts kidney function decline and mortality in type 1 and type 2 diabetes: a 21-year longitudinal study. Diabetes Care. 2023;46:845–53.
Gérard AO, Laurain A, Favre G, Drici MD, Esnault VLM. Activation of the tubulo-glomerular feedback by SGLT2 inhibitors in patients with type 2 diabetes and advanced chronic kidney disease: toward the end of a myth? Diabetes Care. 2022;45:148–9.
Sena CM, Pereira AM, Seiça R. Endothelial dysfunction—a major mediator of diabetic vascular disease. Biochimica et Biophysica Acta BBA Molr Basis of Dis. 2013;1832:2216–31.
Musunuru K. Atherogenic dyslipidemia: cardiovascular risk and dietary intervention. Lipids. 2010;45:907–14.
Aronson D, Rayfield EJ. How hyperglycemia promotes atherosclerosis: molecular mechanisms. Cardiovasc Diabetol. 2002. https://doi.org/10.1186/1475-2840-1-1.
Piché ME, Tchernof A, Després JP. Obesity phenotypes, diabetes, and cardiovascular diseases. Circ Res. 2020;126:1477–500.
Dunlay SM, Givertz MM, Aguilar D, Allen LA, Chan M, Desai AS, et al. Type 2 diabetes mellitus and heart failure: a scientific statement from the American heart association and the heart failure society of America: this statement does not represent an update of the 2017 ACC/AHA/HFSA heart failure guideline update. Circulation. 2019;140:e294-324.
Seferović PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J. 2015;36:1718–27.
Lebeche D, Davidoff AJ, Hajjar RJ. Interplay between impaired calcium regulation and insulin signaling abnormalities in diabetic cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2008;5:715–24.
Basta G, Schmidt A, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res. 2004;63:582–92.
Fineberg D, Jandeleit-Dahm KAM, Cooper ME. Diabetic nephropathy: diagnosis and treatment. Nat Rev Endocrinol. 2013;9:713–23.
Premaratne E, Verma S, Ekinci EI, Theverkalam G, Jerums G, MacIsaac RJ. The impact of hyperfiltration on the diabetic kidney. Diabetes Metab. 2015;41:5–17.
Jefferson JA, Shankland SJ, Pichler RH. Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney Int. 2008;74:22–36.
Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1009–22.
Marshall CB. Rethinking glomerular basement membrane thickening in diabetic nephropathy: adaptive or pathogenic? Am J Physiol Renal Physiol. 2016;311:F831–43.
Lewko B, Stepinski J. Hyperglycemia and mechanical stress: targeting the renal podocyte. J Cell Physiol. 2009;221:288–95.
Qian Y, Feldman E, Pennathur S, Kretzler M, Brosius FC. From fibrosis to sclerosis. Diabetes. 2008;57:1439–45.
DeFronzo RA, Reeves WB, Awad AS. Pathophysiology of diabetic kidney disease: impact of SGLT2 inhibitors. Nat Rev Nephrol. 2021;17:319–34.
Porrini E, Ruggenenti P, Mogensen CE, Barlovic DP, Praga M, Cruzado JM, et al. Non-proteinuric pathways in loss of renal function in patients with type 2 diabetes. Lancet Diabetes Endocrinol. 2015;3:382–91.
Pugliese G, Penno G, Natali A, Barutta F, Di Paolo S, Reboldi G, et al. Diabetic kidney disease: new clinical and therapeutic issues. Joint position statement of the Italian diabetes society and the Italian society of nephrology on “The natural history of diabetic kidney disease and treatment of hyperglycemia in patients with type 2 diabetes and impaired renal function. J Nephrol. 2020;33:9–35.
Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52:1527–39.
Braam B, Joles JA, Danishwar AH, Gaillard CA. Cardiorenal syndrome—current understanding and future perspectives. Nat Rev Nephrol. 2014;10:48–55.
Raina R, Nair N, Chakraborty R, Nemer L, Dasgupta R, Varian K. An update on the pathophysiology and treatment of cardiorenal syndrome. Cardiol Res. 2020;11:76–88.
Kim J-A, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction. Circulation. 2006;113:1888–904.
Cersosimo E, DeFronzo RA. Insulin resistance and endothelial dysfunction: the road map to cardiovascular diseases. Diabetes Metab Res Rev. 2006;22:423–36.
Parsonage W, Hetmanski D, Cowley A. Differentiation of the metabolic and vascular effects of insulin in insulin resistance in patients with chronic heart failure. Am J Cardiol. 2002;89:696–703.
Swan J, Walton C, Godsland I, Clark A, Coats A, Oliver M. Insulin resistance in chronic heart failure. Eur Heart J. 1994;15:1528–32.
Palazzuoli A, Iacoviello M. Diabetes leading to heart failure and heart failure leading to diabetes: epidemiological and clinical evidence. Heart Fail Rev. 2022;28:585–96.
Tenenbaum A, Fisman EZ. Impaired glucose metabolism in patients with heart failure. Am J Cardiovasc Drugs. 2004;4:269–80.
Hayden MR, Tyagi SC. Myocardial redox stress and remodeling in metabolic syndrome, type 2 diabetes mellitus, and congestive heart failure. Med Sci Monit. 2003;9:SR35-52.
Kostis J, Sanders M. The association of heart failure with insulin resistance and the development of type 2 diabetes. Am J Hypertens. 2005;18:731–7.
Jandeleit-Dahm KA, Tikellis C, Reid CM, Johnston CI, Cooper ME. Why blockade of the renin–angiotensin system reduces the incidence of new-onset diabetes. J Hypertens. 2005;23:463–73.
Andreozzi F, Laratta E, Sciacqua A, Perticone F, Sesti G. Angiotensin II impairs the insulin signaling pathway promoting production of nitric oxide by inducing phosphorylation of insulin receptor substrate-1 on Ser 312 and Ser 616 in human umbilical vein endothelial cells. Circ Res. 2004;94:1211–8.
Chan SMH, Lau YS, Miller AA, Ku JM, Potocnik S, Ye JM, et al. Angiotensin II causes β-cell dysfunction through an ER stress-induced proinflammatory response. Endocrinology. 2017;158:3162–73.
Hayden MR, Sowers JR. Isletopathy in type 2 diabetes mellitus: implications of islet RAS, islet fibrosis, islet amyloid, remodeling, and oxidative stress. Antioxid Redox Signal. 2007;9:891–910.
Coué M, Moro C. Natriuretic peptide control of energy balance and glucose homeostasis. Biochimie. 2016;124:84–91.
Undank S, Kaiser J, Sikimic J, Düfer M, Krippeit-Drews P, Drews G. Atrial natriuretic peptide affects stimulus-secretion coupling of pancreatic β-cells. Diabetes. 2017;66:2840–8.
Schlueter N, de Sterke A, Willmes DM, Spranger J, Jordan J, Birkenfeld AL. Metabolic actions of natriuretic peptides and therapeutic potential in the metabolic syndrome. Pharmacol Ther. 2014;144:12–27.
Díez J. Chronic heart failure as a state of reduced effectiveness of the natriuretic peptide system: implications for therapy. Eur J Heart Fail. 2017;19:167–76.
de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. 2008;582:97–105.
Suthahar N, Meijers WC, Brouwers FP, Heerspink HJL, Gansevoort RT, van der Harst P, et al. Heart failure and inflammation-related biomarkers as predictors of new-onset diabetes in the general population. Int J Cardiol. 2018;250:188–94.
Perry IJ, Wannamethee SG, Walker MK, Thomson AG, Whincup PH, Shaper AG. Prospective study of risk factors for development of non-insulin dependent diabetes in middle aged British men. BMJ. 1995;310:560–4.
Helmrich SP, Ragland DR, Leung RW, Paffenbarger RS. Physical activity and reduced occurrence of non-insulin-dependent diabetes mellitus. N Engl J Med. 1991;325:147–52.
Thornley-Brown D. Differing effects of antihypertensive drugs on the incidence of diabetes mellitus among patients with hypertensive kidney disease. Arch Intern Med. 2006;166:797.
Fliser D, Pacini G, Engelleiter R, Kautzky-Willer A, Prager R, Franek E, et al. Insulin resistance and hyperinsulinemia are already present in patients with incipient renal disease. Kidney Int. 1998;53:1343–7.
Thomas SS, Zhang L, Mitch WE. Molecular mechanisms of insulin resistance in chronic kidney disease. Kidney Int. 2015;88:1233–9.
Spoto B, Leonardis D, Parlongo RM, Pizzini P, Pisano A, Cutrupi S, et al. Plasma cytokines, glomerular filtration rate and adipose tissue cytokines gene expression in chronic kidney disease (CKD) patients. Nutr Metab Cardiovasc Dis. 2012;22:981–8.
Spoto B, Pisano A, Zoccali C. Insulin resistance in chronic kidney disease: a systematic review. Am J Physiol Renal Physiol. 2016;311:F1087–108.
Kopple JD, Kalantar-Zadeh K, Mehrotra R. Risks of chronic metabolic acidosis in patients with chronic kidney disease. Kidney Int. 2005;67:S21–7.
DeFronzo RA, Beckles AD. Glucose intolerance following chronic metabolic acidosis in man. Am J Physiol Endocrinol Metabolism. 1979;236:E328–34.
Bellasi A, Di Micco L, Santoro D, Marzocco S, De Simone E, Cozzolino M, et al. Correction of metabolic acidosis improves insulin resistance in chronic kidney disease. BMC Nephrol. 2016. https://doi.org/10.1186/s12882-016-0372-x.
Levin A, Bakris GL, Molitch M, Smulders M, Tian J, Williams LA, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: Results of the study to evaluate early kidney disease. Kidney Int. 2007;71:31–8.
Sergeev IN, Rhoten WB. 1,25-dihydroxyvitamin D3 evokes oscillations of intracellular calcium in a pancreatic beta-cell line. Endocrinology. 1995;136:2852–61.
Maestro B, Campiòn J, Dàvlia N, Calle C. Stimulation by 1,25-dihydroxyvitamin D3 of insulin receptor expression and insulin responsiveness for glucose transport in U-937 human promonocytic cells. Endocr J. 2000;47:383–91.
Fadda GZ, Akmal M, Premdas FH, Lipson LG, Massry SG. Insulin release from pancreatic islets: effects of CRF and excess PTH. Kidney Int. 1988;33:1066–72.
Petchey WG, Hickman IJ, Duncan E, Prins JB, Hawley CM, Johnson DW, et al. The role of 25-hydroxyvitamin D deficiency in promoting insulin resistance and inflammation in patients with chronic kidney disease: a randomised controlled trial. BMC Nephrol. 2009;10:41.
Stefíková K, Spustová V, Krivošíková Z, Okša A, Gazdíková K, Fedelešová V, et al. Insulin resistance and vitamin D deficiency in patients with chronic kidney disease stage 2–3. Physiol Res. 2011;60:149–55.
Lu Y, Wang Y, Sun Y, Li Y, Wang J, Zhao Y, et al. Effects of active vitamin D on insulin resistance and islet β-cell function in non-diabetic chronic kidney disease patients: a randomized controlled study. Int Urol Nephrol. 2022;54:1725–32.
Lee W, Lee HJ, Jang HB, Kim HJ, Ban HJ, Kim KY, et al. Asymmetric dimethylarginine (ADMA) is identified as a potential biomarker of insulin resistance in skeletal muscle. Sci Rep. 2018;8:2133.
Koppe L, Pillon NJ, Vella RE, Croze ML, Pelletier CC, Chambert S, et al. p-cresyl sulfate promotes insulin resistance associated with CKD. J Am Soc Nephrol. 2013;24:88–99.
D’Apolito M, Du X, Zong H, Catucci A, Maiuri L, Trivisano T, et al. Urea-induced ROS generation causes insulin resistance in mice with chronic renal failure. J Clin Investig. 2010;120:203–13.
Koppe L, Nyam E, Vivot K, Fox JEM, Dai XQ, Nguyen BN, et al. Urea impairs β cell glycolysis and insulin secretion in chronic kidney disease. J Clin Investig. 2016;126:3598–612.
Shinohara K, Shoji T, Emoto M, Tahara H, Koyama H, Ishimura E, et al. Insulin resistance as an independent predictor of cardiovascular mortality in patients with end-stage renal disease. J Am Soc Nephrol. 2002;13:1894–900.
Li Y, Zhang L, Gu Y, Hao C, Zhu T. Insulin resistance as a predictor of cardiovascular disease in patients on peritoneal dialysis. Perit Dial Int. 2013;33:411–8.
Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834–44.
Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380:347–57.
Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117–28.
Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P, et al. Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes. N Engl J Med. 2020;383:2219–29.
Pitt B, Filippatos G, Agarwal R, Anker SD, Bakris GL, Rossing P, et al. Cardiovascular events with Finerenone in kidney disease and type 2 diabetes. N Engl J Med. 2021;385:2252–63.
Nagahisa T, Saisho Y. Cardiorenal protection: potential of SGLT2 inhibitors and GLP-1 receptor agonists in the treatment of type 2 diabetes. Diabetes Ther. 2019;10:1733–52.
DeFronzo RA, Norton L, Abdul-Ghani M. Renal, metabolic and cardiovascular considerations of SGLT2 inhibition. Nat Rev Nephrol. 2017;13:11–26.
Alicic RZ, Cox EJ, Neumiller JJ, Tuttle KR. Incretin drugs in diabetic kidney disease: biological mechanisms and clinical evidence. Nat Rev Nephrol. 2021;17:227–44.
Cherney DZI, Udell JA, Drucker DJ. Cardiorenal mechanisms of action of glucagon-like-peptide-1 receptor agonists and sodium-glucose cotransporter 2 inhibitors. Med. 2021;2:1203–30.
González-Juanatey JR, Górriz JL, Ortiz A, Valle A, Soler MJ, Facila L. Cardiorenal benefits of finerenone: protecting kidney and heart. Ann Med. 2023;55:502–13.
Zelniker TA, Braunwald E. Mechanisms of cardiorenal effects of sodium-glucose cotransporter 2 inhibitors. J Am Coll Cardiol. 2020;75:422–34.
Ussher JR, Drucker DJ. Glucagon-like peptide 1 receptor agonists: cardiovascular benefits and mechanisms of action. Nat Rev Cardiol. 2023. https://doi.org/10.1038/s41569-023-00849-3.
Fisman EZ, Tenenbaum A. The dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist tirzepatide: a novel cardiometabolic therapeutic prospect. Cardiovasc Diabetol. 2021;20:225.
Frías JP, Davies MJ, Rosenstock J, Pérez Manghi FC, Fernández Landó L, Bergman BK, et al. Tirzepatide versus Semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021;385:503–15.
Panico C, Bonora B, Camera A, Chilelli NC, Da PG, Favacchio G, et al. Pathophysiological basis of the cardiological benefits of SGLT-2 inhibitors: a narrative review. Cardiovasc Diabetol. 2023;22:164.
Mann JFE, Buse JB, Idorn T, Leiter LA, Pratley RE, Rasmussen S, et al. Potential kidney protection with liraglutide and semaglutide: exploratory mediation analysis. Diabetes Obes Metab. 2021;23:2058–66.
Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med. 2016;375:323–34.
Wilson JM, Lin Y, Luo MJ, Considine G, Cox AL, Bowsman LM, et al. The dual glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 receptor agonist tirzepatide improves cardiovascular risk biomarkers in patients with type 2 diabetes: a post hoc analysis. Diabetes Obes Metab. 2022;24:148–53.
Heerspink HJL, Sattar N, Pavo I, Haupt A, Duffin KL, Yang Z, et al. Effects of tirzepatide versus insulin glargine on kidney outcomes in type 2 diabetes in the SURPASS-4 trial: post-hoc analysis of an open-label, randomised, phase 3 trial. Lancet Diabetes Endocrinol. 2022;10:774–85.
Acknowledgements
None.
Funding
Open access funding provided by Università degli Studi di Padova. Supported by the University of Padova and the Italian Diabetes Society.
Author information
Authors and Affiliations
Contributions
MM and GPF conceived the article content, collected and reviewed data and wrote the manuscript. Both authors approved the final version to be submitted.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
GPF received funding, honoraria or lecture fees from Abbott, AstraZeneca, Boehringer, Lilly, MSD, Mundipharma, Novo Nordisk, Sanofi, Servier. MM has nothing to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Marassi, M., Fadini, G.P. The cardio-renal-metabolic connection: a review of the evidence. Cardiovasc Diabetol 22, 195 (2023). https://doi.org/10.1186/s12933-023-01937-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-023-01937-x