
Abstract
The epidemic of diabetes mellitus (DM) necessitates the development of novel therapeutic and preventative strategies to attenuate complications of this debilitating disease. Diabetic cardiomyopathy (DCM) is a frequent disorder affecting individuals diagnosed with DM characterized by left ventricular hypertrophy, diastolic and systolic dysfunction and myocardial fibrosis in the absence of other heart diseases. Progression of DCM is associated with impaired cardiac insulin metabolic signaling, increased oxidative stress, impaired mitochondrial and cardiomyocyte calcium metabolism, and inflammation. Various non-coding RNAs, such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), as well as their target genes are implicated in the complex pathophysiology of DCM. It has been demonstrated that miRNAs and lncRNAs play an important role in maintaining homeostasis through regulation of multiple genes, thus they attract substantial scientific interest as biomarkers for diagnosis, prognosis and as a potential therapeutic strategy in DM complications. This article will review the different miRNAs and lncRNA studied in the context of DM, including type 1 and type 2 diabetes and the contribution of pathophysiological mechanisms including inflammatory response, oxidative stress, apoptosis, hypertrophy and fibrosis to the development of DCM

.
Similar content being viewed by others


Background
Diabetes mellitus (DM) is a disease characterized by chronic hyperglycemia and impaired metabolism of carbohydrates, proteins, and lipids caused by impaired insulin secretion and action. Approximately 463 million people aged 20–79 years are currently living with DM. The number of affected people is estimated to reach 578 million by 2030 according to the International Diabetes Federation. Cardiovascular diseases (CVDs) are the most common cause of mortality of diabetic patients [1]. There are two different forms of diabetes. Type 1 diabetes mellitus (T1DM) is a chronic disease due to lack of insulin hormone production from pancreatic β-cells. On the other hand, type 2 diabetes (T2DM) is characterized by high blood glucose and ineffective insulin response [2, 3].
Abnormal cardiac structure and function in patients with DM with no other CVDs i.e. coronary artery disease (CAD), valvular heart disease, and hypertension, is known as diabetic cardiomyopathy (DCM) [4]. DCM is associated with myocardial fibrosis and cardiac remodeling leading to impaired diastolic and systolic function, and consequently to heart failure (HF). The Framingham Heart Study showed the prevalence of HF increase of 2.4-fold in men and fivefold in women with DM in comparison to controls [5]. The development and progression of DCM are closely related to abnormal cardiac insulin metabolic signaling, increased oxidative stress, impaired mitochondrial, cardiomyocyte calcium handling and inflammation. Typically DCM has a long subclinical period [6, 7].
Since clear diagnostic criteria are still lacking, the diagnosis of DCM remains a challenge [8, 9]. Currently, there is a lack of specific histological property, biomarker, or clinical manifestation for the definitive diagnosis of DCM [8, 10]. The earliest changes observed in patients relate to myocardial fibrosis and left ventricular hypertrophy with subsequent systolic dysfunction, which develop in the early stages of diabetes [11, 12]. Although there are dedicated guidelines for the management of DM and HF as isolated conditions, there is insufficient guidance on caring for patients with both DM and HF [9, 13]. Currently, the most widely used test for DCM diagnosis is echocardiography. This approach allows the simultaneous detection of structural and functional changes in the myocardium and the exclusion of other potential causes of the disorder [11, 12, 14, 15]. On the other hand, the routine use of echocardiography for screening purposes appears to be uneconomical. Therefore, the situation requires the development of new blood-based diagnostic tools that will allow identification of patients with increased risk of DCM.
The current strategy for treating DCM is adequate control of DM, improvement of cardiovascular risk factors, including treatment of obesity and hypertension, and standard treatment of HF when required. Unfortunately, although DM has been known as an independent predictor of HF for many years, there is currently no individualized therapy to prevent HF and reduce the economic costs associated with treating these patients [16].
Inflammation is a complex response of an organism to different stimuli, which has two phases—acute and chronic. In the acute phase granulocytes, cytokines and acute-phase proteins (APPs) play a role in removing the inflammatory stimuli. Continuous inflammatory stimulation or impaired reaction to self-molecules leads to a chronic phase, in which an immune response can cause tissue damage and fibrosis. Chronic inflammation is considered to contribute to numerous diseases such as cerebrovascular diseases, atherosclerosis, cardiomyopathy, and DM [17]. There is increasing evidence that chronic inflammation is one of the main factors implicated in DM pathophysiology [18, 19].
Oxidative stress is an imbalance in the generation and elimination of reactive oxygen species (ROS) [20, 21]. ROS are highly unstable free radicals, produced mostly within mitochondria and endoplasmic reticulum. At normal levels, ROS take part in biological processes like immune response or cellular components maturation. On the other hand, high levels of free radicals can cause cell damage [22]. Oxidative stress plays a crucial role in the pathophysiology of numerous diseases including DM, in which glucose overload in mitochondria causes excessive ROS generation and mitochondrial dysfunction [23].
There are many well-known markers of inflammatory processes such as cytokines or APPs—CRP, amyloid A (AA) and amyloid P (AP), complement compounds, ceruloplasmin, or α2M [17]. However, novel and more precise biomarkers that will allow the early diagnosis of DCM are needed [15].
Pathophysiological mechanisms leading to DCM may also include impairment in microRNA (miRNA, miR) and long non-coding RNA (lncRNA) regulatory networks [6, 7, 24]. MiRNAs are a class of small, endogenous, non-coding RNA (ncRNA) molecules. They play a role in numerous biological processes like cell proliferation, differentiation, apoptosis, and metabolism, through the suppression or activation of gene expression. MiRNAs are stable and abundant in different body fluids, which makes them useful as potential novel biomarkers for DM and its complications [25,26,27]. LncRNAs are cell and tissue-specific transcripts consisting of more than 200 nucleotides (nt) that are not translated into proteins. LncRNAs are predicted to have many functions including transcript regulation, nuclear domains organization, and regulation of proteins or RNA molecules like miRNA. LncRNA through base-pairing interactions influence miRNAs abundance and activity [7, 28].
Recent reports indicate participation of ncRNAs in the pathogenesis of oxidative stress and inflammation related to several human disorders including DM complications [29, 30]. Also, it was shown that ncRNA may act as a promising tool to facilitate the development of therapeutic strategies and clinical management of patients with cardiomyopathy [31]. Obesity often accompanies DM. Independently of other cardiometabolic risk factors, it dysregulates inflammation-related ncRNAs. It was demonstrated that therapeutic manipulations of inflammation-related ncRNAs expression can potentially treat obesity-induced vascular complication [32, 33].
This review aims to provide a comprehensive overview of the current knowledge of diagnostic, prognostic, and treatment value of miRNA and lncRNA in the pathophysiological processes of DCM.
The essential role of miRNAs in the regulation of DCM pathogenic processes
MiRNAs regulate protein synthesis, mostly through inhibition of mRNA. Synthesis of those molecules can be influenced by high glucose (HG) levels, thus miRNAs play a role in DM-related pathophysiological processes in the myocardium [34, 35]. Based on previous studies it was found that more than 300 different miRNAs are dysregulated in DCM. MiRNAs can modulate the oxidative stress response, influence the inflammatory processes and cardiomyocytes survival. Hence, they might be useful to treat and monitor DCM [36,37,38].
The synthesis of miRNAs can be altered on different levels in the diabetic model
The previous study used the insulin2 mutant (Ins2+/−) Akita, a genetic mice model of T1DM to investigate the role of Dicer, a crucial enzyme for miRNA maturation and several miRNAs in DCM. The study showed that Dicer, numerous miRNAs and inflammatory cytokines (TNFα and cardiac IL-10) are associated with diabetes-induced HF. Dicer is an RNase III endonuclease plays role on pre-miRNAs maturation. The study found the upregulation of mRNA and protein levels of Dicer in diabetic mouse hearts compared to wild type [39]. Besides, many miRNAs, such as Let-7a, miR-345-3p, miR-433 and miR-455 were downregulated, while the only upregulated miRNA was miR-295. It is important to note that the study used only microarray analysis with no qRT-PCR validation. MiR-295 is typical for the embryonic stage, thus its upregulation in diabetic myocardium may indicate adaptive mechanisms to pathological conditions. Let-7a was also found downregulated in diabetic nephropathy by targeting PI3K/Akt signaling, an important pathway in the pathogenesis of insulin resistance and DCM development [40]. Moreover, miR-345 family was identified as an important regulator in children with the recent onset of T1DM. Importantly, in silico pathway analysis based on inferred miRNA target genes showed that PI3K/Akt, MAPK, and Wnt signaling pathways are related to T1DM [41]. Moreover, miR-433 was identified as a novel regulator of doxorubicin-induced cardiac fibrosis both in an animal model and in cardiac tissue from patients with dilated cardiomyopathy. Therefore further analyzes are needed to clarify its importance in DCM [42].
Another study pointed out the importance of miR-373 related to the MAPK signaling pathway. MiR-373 was significantly downregulated in diabetic mice cardiac tissue. In vitro analysis in rat cardiomyocytes exposed to HG and transfected by miR-373 showed that overexpression of miR-373 decreased MEF2C and hypertrophy. MEF2C, a transcription factor commonly found in heart tissue, seems to be a target gene for miR-373. Moreover, gene ontology analysis revealed the MAPK signaling pathway is the one most associated with dysregulated miRNAs in the diabetic mouse heart. Further in vitro inhibition of p38 MAPK reduced miR-373 expression, which suggests that miR-373 transcription may be regulated by p38 MAPK. Thus, p38 MAPK/miR-373/MEF2C was suggested to be a regulatory pathway in glucose-dependent cardiomyocyte hypertrophy [43].
Upregulation of miR-19b, miR-27a, miR-34a, miR-125b, miR-146a, miR-155, miR-210, miR-221 and the downregulation of miR-1 in DCM by in vitro/in vivo analysis was demonstrated [44]. An interesting study performed by Costantino et al. [44] on a DM mouse myocardium, indicates the existence of hyperglycaemic memory, which means that even after normalization of glucose levels, the negative effects of hyperglycemia can persist. MiRNA profiling in heart samples revealed that despite intensive glycaemic control, miRNA signatures in diabetic myocardium were only partially reversible. Bioinformatic analysis showed that among dysregulated miRNAs, miR-221, miR-146a, miR-34a, miR-210, miR-19b, miR-125b, miR-27a, and miR-155 were associated with oxidative stress. Among them, miR-221 was upregulated in the diabetic myocardium suggesting a key role of miR-221 in the progression of diabetic myocardial damage after obtaining normoglycemia. The study results also suggest that miR-34a may be a mediator linking DM and cardiac aging [44]. Moreover, glycaemic control failed to restore the underexpressed antifibrotic miRNAs, including miR-1, which is known to play an important role in cardiac dysfunction under hyperglycemia [44, 45]. Additionally, it was previously shown that the expression of miR-210 is induced in diabetic ischaemic HF patients [46]. The existence of metabolic memory has been proposed previously, although its mechanisms were not clearly explained [47]. MiRNAs help to determine why the diabetic cardiovascular complications progression is not halted by the normalization of glucose levels. The results of the study done by Costantino et al. suggest oxidative stress-related miRNAs as potential novel therapeutic targets. Inhibition of those miRNAs (miR-221, miR-146a, miR-34a, miR-210, miR-19b, miR-125b, miR-27a, miR-155) may lead to the reduction of adverse effects of hyperglycaemic memory in the heart [44] (Table 1).
In conclusion, HG conditions can influence the synthesis of all miRNAs via the modulation of dicer. The function of this enzyme depends on the myocardial impairment stage. Additionally, changes in gene expression can be reversed only partially. These findings make miRNA-genes interactions even more complex and indicate the great importance of identifying novel therapeutic approaches that could potentially prevent the development of DCM.
MiRNAs can modulate macrophages function and phenotypes
Macrophages play an important role in the regulation of inflammatory processes. Classically, activated M1 cells can produce pro-inflammatory cytokines, while alternatively activated M2 cells are important for the resolution of inflammation [48]. In inflammatory disorders miR-155 is known to be upregulated and the administration of antagomiR-155 (miR-155 inhibitor) was observed to decrease the cardiac infiltration by inflammatory mediators, reduce myocardial damage, and improve cardiac function [49]. Estrogen deficiency increased inflammation in DCM mice due to the over-infiltration by M1 macrophages (pro-inflammatory type) [50]. However, the aggravation of DCM caused by estrogen deficiency was prevented by treatment with gold nanoparticle-conjugated antagomiR-155, which induced M2 macrophages infiltration and ameliorated the structure and function of the heart. It is suggested that miR-155 inhibition therapy could serve as a promising approach to improve cardiac function in DCM [50].
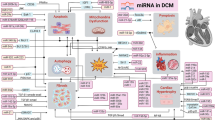
Efferocytosis, phagocytic clearance of the apoptotic cells, is impaired in macrophages of DCM patients. MiR-126 was found to modify macrophage-mediated phagocytosis of apoptotic myocytes [51]. Under hyperglycemic conditions, the expression of miR-126 in macrophages was downregulated, which was accompanied by increased expression of its target gene, ADAM9. ADAMs are membrane-anchored enzymes that are involved in a variety of biological processes including cytokine and growth factor shedding, cell migration, as well as inflammatory response [52]. Similarly, overexpression of miR-126 diminishes efferocytosis impairment. Thus, enhancing this pathway by pharmacological treatment that will induce the expression of miR-126, has a potential to improve cardiac muscle function after injury and under inflammatory conditions accompanying DM [51] (Fig. 1) (Table 1).

The possible therapeutic mechanism of miRNAs-contributed to the oxidative stress, inflammation and cardiomyocytes function process in diabetic cardiomyopathy. Bcl-2 B-cell lymphoma 2, CASP Caspase, Col4α1 collagen type IV alpha 1, CPDT 5,6 Dihydrocyclopenta-1,2-dithiole-3-thione, DCM diabetic cardiomyopathy, ELAVL1 ELAV like protein 1, FGF1 fibroblast growth factor 1, ERK1/2 extracellular signal-regulated kinases 1 and 2, FOXO3a Forkhead box O3, HOTAIR HOX transcript antisense intergenic RNA, IL interleukin, IRAK1 interleukin-1 receptor-associated kinase 1, LAZ3 lymphoma-associated zinc finger 3, LncRNA long non-coding RNA, MDA malondialdehyde, MiR MicroRNA, MMP-9 matrix metalloproteinase 9, NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells, Nrf2: nuclear factor erythroid 2-related factor 2, PI3K/Akt phosphoinositide 3-kinase and protein kinase B, PPARα peroxisome proliferator-activated receptor-alpha, ROS reactive oxygen species, SIRT-1 Sirtuin 1, SMAD7 SMAD family member 7, SOD superoxide dismutase, TGF-β1 transforming growth factor β1, TNFα tumor necrosis factor-alpha, TRAF6 TNF receptor associated factor 6
The importance of NF‐κB induced inflammation and TNFα mediated processes in the development of DCM
NF‐κB is a family of transcription factors that regulate multiple biological processes, including, inflammatory response, cell survival and cell cycle progression [53, 54]. PI3K/Akt pathway can activate NF‐κB induced inflammation [55]. PI3K/Akt signaling also represents a pivotal pathway in the pathogenesis of insulin resistance and DCM development. It regulates multiple biological processes, including apoptosis, cell growth, and proliferation of cardiomyocytes [56]. PI3KT/Akt activates platelets in response to multiple stimuli and subsequently may aggravate fibrosis of the heart muscle as activated platelets can have profibrotic action by releasing TGF-β1 and inducing platelet-fibroblast conjugation. TGF-β1 plays a key role in cardiac fibrosis and platelets can contain high concentrations of TGF-β1. Importantly, studies showed that platelet-derived TGF-β1 promoted ventricular fibrosis in a mouse model and atrial fibrosis in cell culture [57, 58].
Yang et al. [59] aimed to evaluate the role of miR-203 and PI3KT/Akt pathway in the progression of DCM in a rodent model. It was found that miR-203, which is downregulated in diabetic mice, directly targets PIK3CA and can downstream PI3KT/Akt pathway. In cardiomyocytes, miR-203-mediated inhibition of PI3K/Akt was found to be related to reduced cardiac hypertrophy, fibrosis, and myocardial apoptosis. Interestingly, the upregulation of miR-203 reduced the levels of oxidative stress biomarkers, such as MDA and ROS, in cardiomyocytes. The study suggests that the upregulation of miR-203 might be a promising treatment strategy for inhibiting the progression of DCM via PI3KT/Akt cascade [59].
In HG conditions cardiac fibroblasts exhibit markedly increased IL‐1β production and NF‐κB activity. This is accompanied by a significantly upregulated expression of miR-150-5p and downregulation of its target gene expression—Smad7 [60]. Smad7 is a shear-stress induced gene, the expression of which can be enhanced via NF‐κB signaling pathway. Smad7 suppresses TGF‐β1 signaling, which results in the suppression of its anti-inflammatory actions [61]. The inhibition of miR-150-5p attenuates cardiac muscle fibrosis and inflammation mediated by NF‐κB signaling TGF‐β1/Smad pathways. HG-treated cardiac fibroblasts manifest also significantly elevated fibrotic markers and extracellular matrix (ECM) proteins: CTGF, FN, α‐SMA, Col‐I, Col‐III [60]. Importantly, it was previously shown that miR‐150‐5p plays a crucial role in nonclassical monocyte generation, development of B and T lymphocytes, inflammatory cytokine production, and vascular remodeling and fibrosis [62,63,64,65]. In lymphocytes, a direct target of miR-150-5p—c-myb is responsible for the regulation of hematopoietic stem cells. It was also found that c-myb is involved in miR-150-mediated ROS-induced cardiomyocytes apoptosis and injury [65]. It was shown that cardiac remodeling may be reversed by miR-150-5p knockdown and aggravated by its overexpression, thus inhibition of miR-150-5p may become a promising target for DCM treatment [60].
Moreover, miR-142-3p was found to be a direct regulator of TGF-β1, a mediator essential for endothelial-to-mesenchymal transition (EMT) process, which plays an important role in myocardial fibrosis. The expression of miR-142-3p in human aortic endothelial cells (HAECs) exposed to HG levels was declining in a dose- and time-dependent manner. Further evaluation showed that miR-142-3p inhibits EMT induced by HG levels through blocking of TGF-β1/Smad pathway. MiR-142-3p/TGFβ1/Smad axis was suggested to become a possible target in DCM therapy [66]. Similarly, miR-700 was found to modulate cardiac fibrosis by interacting with TGF-β3 and Col1α1 in the heart [67]. Previously, Shen et al. demonstrated the upregulation of miR-700 in diabetes-induced cardiac hypertrophy by using microarray analysis in an STZ-induced T1DM animal model. Importantly, the study used a bioinformatic tool and found 28 putative target genes for miR-700. However, further analysis with qRT-PCR validations is needed to confirm the upregulation of this promising miRNA in DCM [43, 68].
MiR-146a has a regulatory relationship with components of the NF-κB signaling pathway, which acts as a key mediator in hyperglycemia and inflammation [69, 70]. Several studies have indicated the important role of miR-146a in the pathogenesis of myocardial injury and DCM [44, 71,72,73]. Importantly, miR-146a was suggested as a predictive biomarker of HF [73]. Myocardial injury increases the expression of several pro-inflammatory mediators (IL-6, MCP-1, TNFα), contributing to the progression of cardiac muscle remodeling, which leads to its irreversible dysfunction and in consequence to HF [74, 75]. Impaired miR-146a expression was found associated with subclinical inflammation and insulin resistance in T2DM patients [76]. Downregulated miR-146a levels were also found in the hearts of diabetic mice [71]. Interestingly, endothelial cells (ECs) were the main cell type that exhibited decreased miR-146a levels, while cardiomyocytes remained unaltered [71]. Along with diminished miR-146a expression, the level of cardiac functional abnormalities, including defective cardiac contractility as well as inflammatory markers and ECM proteins (IL-6, TNFα, IL-1β, MCP-1, NF-κB, Col1α1, Col4α1) were increased in the hearts of DM wild type mice. However, these changes were not observed in the diabetic transgenic mice with overexpression of miR-146a. In vitro studies with isolated human cardiac ECs revealed glucose-induced upregulation of IRAK1 and TRAF6, which are specific NF-κB regulators and targets of miR-146a [71]. An increased level of miR-146a transcripts is accompanied by a decreased level of c-Fos mRNA and diminished activity of AP-1, a c-Fos-containing transcription factor complex. Downregulation of the c-Fos/AP-1 signaling by miR-146a inhibits MMP-9 activity, which is involved in cardiac remodeling. Thus, the overexpression of miR-146a appears to play a protective role against cardiomyocytes injury and may be a novel therapeutic approach for the prevention of CVD, however further studies are needed [71, 72].
MiR-223, which is downregulated in DM, is associated with inflammatory response [39, 77]. MiR-223 targets Mef2c and, what follows, inhibits the proliferation of myeloid progenitor cells, suppresses granulocytes differentiation and activation. Thus, the miR-223 downregulation leads to an excessive inflammatory response [78]. The suppression of miR-223 could result from attenuated IL-10 transcription. Moreover, due to insufficient inhibitory function of IL-10, elevated levels of TNFα can be observed [39]. Upregulation of TNFα may lead to enhancing TNFα-induced tissue factor pro-coagulation activity in ECs [77]. Importantly, in left ventricular biopsies of patients with T2DM miR-223 was found to mediate cardiac function by regulating the Glut4 expression and cardiomyocyte glucose metabolism [79]. Moreover, miR-223 was found significantly downregulated in left ventricular cardiac biopsies in diabetic ischaemic HF patients [46] (Fig. 1) (Table 1).
MiRNAs can modulate cell response to oxidative stress acting through PPARα and Nrf2 transcription factors
There is a growing evidence that oxidative stress plays an important role in the progression of myocardial dysfunction in DM [34, 80]. Therefore, it is essential to search for mechanisms underpinning the association between oxidative stress and cardiac function. Nrf2 is an increasingly interesting transcriptional factor acting as a key regulator of oxidative stress genes. In DM models, the activation of Nrf2 is enhanced due to excessive production of oxidizing agents [81]. Fatty acids activated transcriptional factors such as PPARs can demonstrate anti-inflammatory activity and are confirmed to downregulate the expression of proinflammatory genes through transrepressive mechanisms [82]. Both Nrf2 and PPARs play a key role in establishing cellular antioxidative defense systems. Moreover, several studies strongly suggest the existence of reciprocal regulation of Nrf2 and PPARs signaling pathways, which mutually reinforces their expression [83, 84]. Activation of PPARα pathway may result in secondary changes in the oxidative stress state, which may lead to Nrf2 activation via PGC-1α [85].
Recent data indicate that miR-30c may be involved in transcriptional activity of PPARα [80]. Interestingly, in T1DM diabetic model, miR-30c levels were downregulated leading to the higher expression of its direct target—PGC-1β, which by acting on PPARα causes metabolic disturbances, lipotoxicity in the heart, and excessive ROS production [80]. Importantly, miR-30c expression was found reduced in cardiac tissue and plasma collected from DCM patients [86,87,88] Another study showed that forced overexpression of miR-30c in HG-induced cardiomyocytes was correlated with downregulation of prohypertrophic genes—Cdc42 and Pak1 and attenuation of cardiomyocyte hypertrophy, while anti-miR-30c treatment had the opposite effect [86]. Moreover, miR-30c may act synergistically with miR-181a in the modulation of the p53-p21 pathway important for cardiomyocyte apoptosis and hypertrophy in DCM. HG-induced cardiomyocytes transfection by miR-30c and miR-181a caused a decrease of p53 and p21 expression, ANP protein levels, and significantly attenuated hypertrophy. The effect was more potent when both miRNAs were overexpressed than for miR-30c or miR-181 alone [87]. Additionally, transfection of recombinant adeno-associated virus 9 (rAAV9)-mediated miR-30c in DCM mice resulted in cardiomyocyte miR-30c overexpression in DCM model resulted in the increased left ventricular ejection fraction, reduced left ventricle mass, and fractional shortening in comparison controls [88]. MiR-30c seems to be a multidirectional player in DCM. Therefore, the overexpression of miR-30c is hypothesized to attenuate cardiac dysfunction and appears to be a promising therapeutic target in DCM [80].
Another pathway regulating PPARα and Nrf2 activation is related to LAZ3, which is a protein-encoding gene that acts as a transcriptional repressor and regulates inflammation by interfering with NF-κB signaling [37, 89]. LAZ3 was found decreased in diabetic mouse hearts and cardiomyocytes of rats [37]. Upregulation of LAZ3 inhibits the miR-21 expression, which targets PPARα. Silencing of LAZ3 leads to the increased expression of miR-21 and subsequently to the decreased PPARα and Nrf2 activation, resulting in an impaired response to the oxidative stress. Thus, the downregulation of the PPARα-Nrf2 signaling pathway by the overexpression of miR-21 results in the impaired cardiomyocytes function. Therefore, treatment targeting miR-21 inhibition may be beneficial for DCM management [37]. In line with these observations, a cardiac release of miR-21 has been recently reported in an unselected cohort of patients with non-ischemic cardiomyopathy, including DCM [90].
Downregulated miR-144 was found in diabetic cardiomyocytes in the regulation of oxidative stress. MiR-144 can directly target Nrf2 [34]. Interestingly, although miR-144 levels were reduced in T1DM diabetic mice model and in HG conditions in cultured cardiomyocytes, the administration of miR-144 mimic reduced the expression of Nrf2 proteins and augmented ROS formation, whereas miR-144 inhibitor enhanced Nrf2 expression and decreased ROS generation. This effect was not observed in normal glucose conditions. Thus, blocking miR-144 as a therapeutic intervention may decrease oxidative stress in HG condition [34]. On the other hand, Tao et al. [91] have found conflicting results. Plasma miR-144 decreased markedly in diabetic patients with cardiac dysfunction Although miR-144 was found also decreased in HG-induced cardiomyocytes and in an STZ-induced diabetic model, the overexpression of miR-144 was shown to protect the heart from the hyperglycemia-induced injury. To clarify the role of miR-144 in DCM, it may be necessary to generate mice with cardiomyocyte-specific miR-144 knockout or overexpression, at different stages of hyperglycemic cardiac injury [91].
The upregulation of miR-503 may be related to the increased diabetic cardiac dysfunction and Nrf2 activation can be enhanced through the phase II enzyme inducer –CPDT, an enzyme complex, which initiates the expression of antioxidative enzymes and plays a crucial role in the protection against oxidative stress [92, 93]. It has been hypothesized that CPDT may lead to a similar effect in diabetic heart dysfunction and that the downregulation of miR-503 can decrease the DCM development. For these purposes, Miao et al. [93] used CPDT as an intervention agent to investigate its correlation with miR-503 in DCM. CPTD treated diabetic rats presented with decreased expression of miR-503 and increased levels of its target—Nrf2 as well as other detoxification enzymes such as MDA and HO-1, when compared to a non-treated diabetic group. The data suggest that CPTD has a protective effect because of its ability to inhibit miR-503 expression and, as a result, to increase Nrf2 expression, which can result in the diminished myocardial cell apoptosis and reduced development of cardiomyopathy [93].
Cardioprotective role is postulated regarding downregulation of miR-22 which is also directly linked with oxidative stress [94]. Excessive oxidative stress leads to the production of ROS, such as superoxide or hydrogen peroxide, which can cause DNA mutation, consequently resulting in cell injuries. On the other hand, superoxide dismutase (SOD) is an enzyme and can be effective as a potent antioxidant [95, 96]. MiR-22, the levels of which are decreased in diabetic myocardium in T1DM STZ-induced diabetes model, was found to target SIRT1 leading to the upregulation of SIRT1 protein expression [94]. It was shown that the overexpression of miR-22 can decrease the levels of ROS, MDA, and increase SOD, indicating its cytoprotective properties. Administration of miR-22 had a positive effect on blood glucose levels, ejection fraction, left ventricular end-diastolic pressure, and cardiac tissue apoptosis in a diabetic animal model. Moreover, miR-22 was unable to inhibit the oxidative stress injury when SIRT1 was knocked down, suggesting its protective effect being mediated by SIRT1 expression. Overall, this study shows that miR-22 administration can decrease oxidative stress injury by upregulation of SIRT1, thus miR-22 can be a potential therapeutic target for diabetic patients with cardiac insufficiency [94].
Altogether, in DM the modulation of levels of different miRNAs in the myocardium can be observed. Downregulated expressions of miR-30c, miR-22 and miR-144 and upregulated expressions of miR-503 were observed in in vivo and in vitro models of DCM. Those miRNAs influence the antioxidative action of Nrf2 transcription factor and impair its ability to prevent the adverse effects of oxidative stress due to ROS, which are synthesized in excessive amounts in DM (Fig. 1) (Table 1).
MiRNAs can modulate the activation of cardiomyocyte pyroptosis and apoptosis via different pathways
Pyroptosis is an inflammatory form of programmed cell death triggered by CASP-1, which causes cardiac dysfunction due to decreased cell survival and increased pro-inflammatory cytokines, such as IL-1ß and IL-18 [97, 98]. Activation of these both interleukins is controlled by miR-30d, which is upregulated in HG conditions directly targeting FOXO3a [38]. MiR-30d mediates FOXO3a downregulation and results in decreased ARC and, what follows—increased CASP-1 activation and inflammatory cytokines (IL-1ß and IL-18) secretion. It subsequently leads to the formation of the inflammasome complex, which induces the cardiomyocyte pyroptosis. Conversely, the miR-30d knock-down by its antisense inhibitor may suppress the process. Therefore, it is suggested that treatment targeted at blocking miR-30d expression may prove to be advantageous in DCM management [38]. Moreover, ARC was reported to protect cardiomyocytes in oxidative stress through inhibition of CASP-2-mediated apoptosis [99]. ARC also seems to attenuate the ischemia/reperfusion (I/R) injury and drug-induced cardiotoxicity [100, 101]. Besides that, in a rodent model, miR-30d showed also an anti-autophagic effect by regulating the KLF9/VEGFA pathway. Its knockdown prevented the aggravation of cardiac dysfunction in diabetic rats. Furthermore, SGLT2 treatment was associated with decreased miR-30d expression and improved cardiac function in DCM rats [102].
Another protein that plays an important role in pyroptosis-induced inflammatory processes is ELAVL1, which stabilizes TNFα mRNA. An increased concentration of CASP-1 proinflammatory enzyme, IL-1β proinflammatory cytokine along with an overexpression of ELAVL1 was found in human diabetic cardiomyocytes [103]. It was shown that ELAVL1 deficiency counteracts TNFα induced canonical pyroptosis via NLRP3, IL-1β and CASP-1 suppression. Moreover, ELAVL1 is a direct target of miR-9, the expression of which is significantly downregulated under hyperglycaemic conditions in human diabetic hearts. Inhibition of miR-9 can lead to the upregulation of ELAVL1 and CASP-1. On the other hand, overexpression of miR-9 reduces ELAVL1, CASP-1 and IL-1β expression in human cardiomyocytes and prevents cardiomyocyte pyroptosis. Thus, miR-9 may act as a potential target to reduce the DCM progression [103].
Apoptosis is another crucial factor triggering HF. It is a programmed self-eliminating process of dead cells occurring also in injured cardiomyocytes under different conditions e.g. ischemia. In T1DM and T2DM models, the upregulation of miR-195 has been confirmed to impact signaling pathways involved in oxidative stress-induced apoptosis. Furthermore, miR-195 was found to target BCL2 and SIRT1, the expression of which was observed to be decreased in diabetic rat cardiomyocytes. Moreover, miR-195 levels are elevated in diabetic hearts in animal models, thus the inhibition of its targets and more intense apoptosis of cardiomyocytes can be observed. The knockout of miR-195 increased the levels of SIRT1 and BCL-2 and significantly improved cardiac function and coronary circulation but did not reduce myocardial fibrosis. Furthermore, inhibition of miR-195 reduced ROS production, protein oxidation and CASP-3 activity, indicating the role of miR-195 in oxidative stress-related cell injury. Moreover, the silencing of miR-195 inhibited TNFα mRNA and protein expression and improved insulin sensitivity [104]. Altogether, in HG conditions the downregulation of miR-9 and the upregulation of miR-30d and miR-195 can be observed. Modulations of their levels lead to enhanced pyroptosis and apoptosis in diabetic hearts and the progression of DCM (Fig. 1) (Table 1).
The role of miR-1, miR-133a and miR-21 in DCM
Over the past decade, miR-1 and miR-21 have been some of the most frequently studied miRNAs in the CVD area, especially in the CAD, acute coronary syndrome and HF field [105, 106]. Evaluations revealed that these molecules are confirmed to impact signaling pathways involved in atherosclerosis, hypertrophy, myocardial remodeling and fibrosis [107]. Additionally, both miR-1 and miR-21 are hypothesized to be crucial targets for a new CVD treatment approach [108].
Junctin is a component of the ryanodine receptor Ca2 + release channel complex. It has been proved that the overexpression of junctin induces cardiac hypertrophy and arrhythmia via adaptive changes in Ca2 + handling [109, 110]. MiR-1, which is significantly downregulated in diabetic cardiomyocytes in the T1DM model of DCM, directly targets junctin and suppresses its expression [45]. Decreased levels of miR-1 in HG-conditions result in increased expression of junctin. Thus, the study suggests that miR-1 plays an important role in cardiac dysfunction under hyperglycemia. Indeed, miR-1 has been shown to contribute not only to the regulation of DCM, but also it was found to play a role in arrhythmias, myocardial infarction, myocardial hypertrophy, cardiomyocyte differentiation, and cell reprogramming [111]. It may be hypothesized that myocardial-specific miRNAs significantly contribute to DM-induced cardiomyocytes injury, and the intervention with antioxidant treatment controls the level of miRNAs including miR-1 and its target protein junctin, which can have a cardioprotective effect against DM-induced injury [45].
Abundantly expressed in normal cardiac tissue miR-133a was significantly downregulated in DCM mice. An in vitro study showed that miR-133a inhibited SGK1 and IGF1R upregulation induced by HG levels leading to the depletion of ANP, BNP, and transcription factors MEF2A, and MEF2B as well as the attenuation of cardiomyocyte hypertrophy [112]. Furthermore, overexpressed miR-133a decreased mRNAs of fibrosis biomarkers such as fibronectin, FGF1, TGF-β, and COL4A1 and may prevent myocardial extracellular matrix (ECM) accumulation, presumably by preventing ERK1/2 and Smad2 phosphorylation. Thus, the lower expression of miR-133 associated with hyperglycemia may attenuate cardiac fibrosis [113]. In addition, miR-133a participates in cardiac contractility by targeting and downregulating tyrosine aminotransferase, an enzyme catabolizing tyrosine, a substrate for norepinephrine synthesis. In DCM rats, miR-133a deficiency was associated with decreased heart contractility. MiR-133a treatment resulted in increased expression of the β-adrenergic receptor, normalization of norepinephrine levels, and as a result, contractility improvement [114]. In transgenic diabetic mice with cardiac miR-133a overexpression (Akita/miR-133aTg) myocardial fibrosis, hypertrophy and impaired contractility were reduced when compared to standard Akita mice. Akita/miR-133aTg also had a normal lipid accumulation in heart tissue, whereas standard Akita exhibited lipid deposits [115]. Thus, miR-133a may prevent metabolic heart remodeling related to DM-induced lipotoxicity. In the plasma of DM patients and a mimic model of insulin resistance, increased miR-133a levels were associated with higher myocardial steatosis. It was shown that the lipid-loaded cardiomyocytes release exosomes rich in miR-133a. Therefore, miR-133a was proposed as a diagnostic biomarker of subclinical DCM [116].
MiR-21 is recognized as one of the most studied miRNAs that control myocardial remodeling [117]. However, it should be noted that the role of miR-21 in cardiac disease appears controversial. It was shown that miR-21 is upregulated in failing myocardium in humans and in an animal model, while interfering miR-21 expression with inhibitors prevents cardiac fibrosis in a mouse model of pressure overload [117, 118]. Moreover, upon pressure overload, cardiac dysfunction was only prevented in mice with miR-21 deficiency in nonmyocyte cardiac cells, but not in mice with global or myocyte targeted deletion of miR-21 [117]. Additionally, as mentioned above in neonatal rat cardiomyocytes incubated in HG conditions, it was shown that LAZ3 protects against cardiac remodeling by decreasing miR-21 [37]. Additionally, another in vitro study showed that HG conditions promoted the proliferation and collagen synthesis of rat cardiac fibroblasts, which was mediated by increased expression of miR-21 [119],
It appears that the effect of miR-21 differs depending on cell type and disease condition. In cardiac myocytes the overexpression of miR-21 protects against ROS-induced damage via its target gene PDCD4, whereas miR-21 deficiency in fibroblasts protects against cardiac dysfunction and myocardial fibrosis [117, 120]. Also, the effect of miR-21 on ROS production and its role on cardiac myocytes in DCM associated diastolic dysfunction was studied [121]. Increased levels of cardiac oxidative stress biomarkers, which were found in diabetic mice model cardiomyocytes, were reversed by miR-21 treatment and overexpression of phospho-Akt and phospho-eNOS. Therefore, miR-21 inhibits cardiac hypertrophy via decreasing ROS levels and increasing bioavailable nitric oxide via gelsolin and thus may have potential therapeutic role [121]. Study conducted by Dai et al. suggested that the overexpression of miR-21 may be a promising therapeutic target for treatment of DCM [121]. On the other hand, due to the conflicted results, it can be concluded that miR-21 may have a different role depending on the tissue or cell type and disease condition, thus further human studies are needed to clear out this discrepancy (Table 1).
LncRNAs
LncRNAs are non-protein-coding RNAs, that are at least 200 nt in length [7]. LncRNAs participate in many biological processes, such as modulation of various pathways linked with the oxidative stress and inflammatory processes, which influence DCM development, severity of myocardial I/R injury, cardiac hypertrophy, HF or diabetic vascular complications [7, 122]. Moreover, lncRNAs can act as miRNA sponges, meaning that they prevent the regulatory functions of miRNAs by binding to them and hindering interactions with their target [122].
LncRNAs mediate cardiomyocytes apoptosis induced by high glucose
Cardiomyocyte apoptosis occurs in response to different pathological stimuli in DCM. As a consequence, it leads to the remodeling and fibrosis of the heart muscle, thus impairing its contractile function [123]. Several studies indicated the role of lncRNAs in pathomechanisms associated with deteriorating heart function in diabetic models [124, 125].
LncRNA H19, which plays a role in maintaining cardiac function and development of DCM, was known previously for its involvement in carcinogenesis [126]. It was shown that the expression of H19 was markedly downregulated in the myocardium of diabetic rats [127]. Moreover, transfection with H19 siRNA decreases the expression of miR-675. LncRNA H19 and H19-derived miR-675 downregulates its target gene VDAC1, which influences cardiomyocyte apoptosis and plays a crucial role in the progression of cardiac muscle dysfunction. Additionally, LV systolic and diastolic functions were found to be impaired in diabetic models along with exacerbated inflammation and oxidative stress. However, the overexpression of H19 reverses this effect [127].
In another study [128], authors sequenced 400 lncRNAs associated with ROS generation, which may potentially lead to the deterioration of cardiac function and apoptosis. In HG-treated primary rat cardiomyocytes ROS production was upregulated along with an increased level of apoptotic cardiomyocytes. However, among sequenced lncRNA, in further functional studies it was only the silencing of lncRNA NON-RATT007560.2 that decreased the ROS formation and apoptosis [128]. Importantly, it was found that NON-RATT007560.2 may have binding sites for miR-208a [128] which is known for its association with maladaptive cardiac remodelling in diabetic myocardium of T2DM patients [129].
Those studies suggest that the modulation of lncRNA expression may ameliorate cardiac dysfunction and apoptosis-related progression of cardiomyopathy, and may be a promising therapeutic strategy for the treatment of DCM.
LncRNAs regulate cardiac remodeling via TGF-β-mediated NLRP3 pathway
Kcnq1ot1, which is a lncRNA, has been linked with pathophysiological mechanisms of multiple disorders associated with cardiac dysfunction [130, 131]. The Kcnq1ot1 expression was found to be elevated in the blood serum of diabetic patients, cardiac tissue of a T1DM STZ-induced diabetes mice model, and HG-induced cardiomyocytes along with activation of pyroptosis and fibrosis in DCM models [132, 133]. The increased expression of Kcnq1ot1 is followed by collagen deposition and induced TGF-β1, p-Smad2 and p-Smad3 expression was found. As a consequence, the activation of fibrotic formation and cardiac remodeling lead to the deterioration of LV function. Moreover, immunohistochemical analysis of mouse cardiac tissue showed that high Kcnq1ot1 levels contribute to NLRP3 inflammasome activation as well as significantly elevated expression of proinflammatory mediator-IL-1β and pro-apoptotic mediators-CASP-1 and GSDMD-N, thereby demonstrating the role of Kcnq1ot1 in mediating pyroptosis under HG conditions. On the other hand, the silencing of Kcnq1ot1 expression significantly improved cardiac function, reduced remodeling and pyroptosis via miR-214-3p and CASP-1 axis, and TGF-β1/Smads pathway [132].
In another study authors obtained replicated results [133]. In line with the above-mentioned findings, Kcnq1ot1 was enhanced in AC16 human myocardial cells in HG conditions, and followed by an increase of NLRP3, CASP-1, IL-1β, and IL-18 expression. It was shown that Kcnq1ot1 impacted post-acute myocardial infarction (AMI) I/R myocardial injury by regulating Adipor1. Additionally, Kcnq1ot1 knockdown promoted QT interval prolongation via Kcnq1 gene inhibition. Transfection with small interfering RNA (siRNA) downregulated the level of those pyroptosis markers, reversed DNA fracture, ameliorated vimentin expression, and reduced Ca2+ overload. Silencing Kcnq1ot1 promoted the decrease of CASP-1 mRNA and protein expression, as well as the increase of miR-214-3p level. What is more, the inhibition of CASP-1 in primary cardiomyocytes resulted in reduced Ca2+ overload and increased miR-214-3p expression. In the animal model silencing Kcnq1ot1 and CASP-1 produced results consistent with in vitro outcome followed by improved cardiac function as ameliorated ejection fraction and fractional shortening [133].
HOTAIR is one of the first identified lncRNAs and it has a crucial role in the pathophysiology of CVDs [134]. Expression of HOTAIR was found significantly downregulated in myocardial tissues and serum of patients with DCM in comparison to patients with DM and healthy controls [135]. In the diabetic model of T1DM, the expression of HOTAIR was also found decreased, whereas its target miR-34a was found to be overexpressed [125]. On the other hand, the overexpression of HOTAIR protected against diabetes‐induced cardiac hypertrophy and dysfunction. Furthermore, diminished levels of fibrotic markers, such as TGF-β, col-I, col-III showed that HOTAIR treatment decreased cardiac fibrosis. Importantly, the inhibition of HOTAIR enhanced HG‐induced inflammation, oxidative stress, and apoptosis, whereas enhancing HOTAIR expression negated inflammation of STZ-induced DCM [125]. HOTAIR was found to function as a molecular sponge of miR‐34a which directly targeted SIRT1, a promising potential target for the treatment of CVD, and in particular DCM [125, 136].
The above-mentioned studies indicate that the influence of LncRNAs on CASP-1, NLRP3, SIRT1 and TGF-β pathways presents as a new significant pathophysiology mechanism of cardiac remodeling in DCM and stands as a possible future therapeutic target (Fig. 2) (Table 2).

The possible therapeutic mechanism of lncRNAs-contributed to the oxidative stress, inflammation and cardiac function process in diabetic cardiomyopathy. CASP-1 Caspase 1, CK-MB creatine kinase myocardial band, GSDMD-N N-terminal of gasdermin D, HMGB1 high mobility group box 1, HOTAIR HOX transcript antisense intergenic RNA, IL interleukin, Kcnq1ot1 KCNQ1 overlapping transcript 1, LDH lactate dehydrogenase, LncRNA long non-coding RNA, MALAT1 metastasis-associated lung adenocarcinoma 1, miR microRNA, mRNA messenger RNA, NLRP3 nod-like receptor protein 3, NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells, ROS reactive oxygen species, SAA3 serum amyloid antigen 3, SIRT1 Sirtuin 1, TLR4 toll-like receptor 4, TNFα transforming growth factor β, VDAC1 voltage-dependent anion channel 1
NF-κB and TNF signaling pathways are involved in cardiomyocyte injury mediated by LncRNAs
Myocardial damage resulting from an imbalance in the ROS generation and inflammation might be a consequence of DM associated hyperglycemia or hyperlipidemia [137]. For example, the excess saturated fatty acids, especially palmitic acid (PA), found in patients with DM may deposit in cardiomyocytes and cause lipotoxic injury [138]. Cardiomyocytes treated with PA were found to produce significantly upregulated inflammatory factors TNFα and IL-1β, along with MALAT1, which belongs to the lncRNA family. Recently MALAT1 has been reported to play a crucial role in cardiomyocytes ischemia reperfusion damage [139]. On the other hand, the silencing of MALAT1 expression decreased the range of myocardial injury by reducing biomarkers of myocardial damage, lactate dehydrogenase (LDH) and CK-MB, as well as by increasing sponge-miR-26 expression [124]. What is important, miR-26 inhibits TLR4/NF-κB inflammatory signaling pathway by binding to its target gene, HMGB1. Furthermore, manipulation of MALAT1/miR-26 expression revealed the potential role of these molecules in ameliorating PA-induced cell death via TNFα apoptosis pathway. MALAT1 inhibition results in the alleviation of SFA-induced myocardial inflammatory injury via the miR-26a/HMGB1/TLR4/NF-κB axis [124].
In HG conditions ECs exhibit markedly upregulated both MALAT1 expression and SAA3, inflammatory ligand, and target of MALAT1 [140]. Those changes are followed by the increased expression of inflammatory markers, i.e. IL-6 and TNFα. The silencing of MALAT1 was found to result in the reduction of IL-6 and TNFα expression, even in the presence of SAA3, but cytokine levels were reversed only partially. It indicates that there are other pathways than MALAT1-SAA3 that play a role in HG stress regulation [140].
ANRIL is located at locus with the strongest genetic susceptibility for CVD in the chromosome 9p21 region and was shown to control the expression of CDH5 and HBEGF gene involved in vascular permeability, leukocyte transmigration and inflammation [141]. ANRIL was found to be upregulated along with elevated levels of TNFα in the myocardium of diabetic rats, which suggests its association with the development of DCM [142]. ANRIL silencing leads to the decreased levels of CK-MB and LDH, which indicates that interference of ANRIL expression can improve cardiac function index. Moreover, plasma levels of TNFα, IL-6 and IL-1β levels were found to be significantly elevated in the diabetic model, though ANRIL expression interference diminished the level of these cytokines. Novel ANRIL-based therapeutic strategies can offer a promising approach to inhibit cardiomyocytes fibrosis, apoptosis and ROS generation in DCM treatment [142].
By and large, the downregulation of MALAT-1 decreases the range of myocardial lipotoxic injury and reduces inflammation under HG conditions. Interfering ANRIL expression with siRNA alleviates myocardial remodeling in diabetic rats, and decreases the level of inflammatory cytokines including TNFα. Subsequent studies should answer whether MALAT1 and ANRIL can serve as a drug target in chronic diabetic complications (Fig. 2) (Table 2).
LncRNAs targeting HMGA1
High-mobility group AT-hook 1 (HMGA1) is a key partaker in cardiovascular complications of diabetes, both at the vascular and the cardiac level [143, 144]. HMGA1 expression is modulated by miRNAs known to be involved in cardiovascular disease, such as Let-7 and miR-26a [145]. More recently, a number of lncRNAs are emerging as regulators of HMGA1. SNHG1, the expression of which is induced by oxygen deprivation, mediates cardiomyocyte hypertrophy via targeting miR-15a/HMGA1 [146,147,148]. HOTTIP has been shown to regulate the miR-218/HMGA1 axis [149, 150]. This is particularly interesting, as both HMGA1 and miR-218 are involved in the development of cardiovascular complications in diabetes [24, 151]. LncRNA GACAT3 acts as a competing endogenous RNA of HMGA1 [152]. The increased understanding of the modulation of HMGA1 expression might pave the way to both diagnostic and therapeutic applications (Fig. 2) (Table 2).
Current perspectives and limitations
Numerous studies have shown that miRNAs and lncRNAs could act as potential biomarkers as well as novel treatments in DCM. In Fig. 3 miRNAs/lncRNAs as potential therapeutics was presented.

MiRNAs and lncRNAs as therapeutics in diabetes-induced cardiomyopathy. ↑ indicates the mimic-use as treatment, ↓ indicates inhibitor-use as treatment against diabetic cardiomyopathy. ANRIL antisense non-coding RNA in the INK4 locus, H19 imprinted maternally expressed transcript, HOTAIR HOX transcript antisense intergenic RNA, IL interleukin, Kcnq1ot1 KCNQ1 overlapping transcript 1, lncRNA long non-coding RNA, MALAT1 metastasis-associated lung adenocarcinoma 1, miRNA microRNA, miR; mRNA messenger RNA, TGFβ transforming growth factor β, TNFα tumor necrosis factor-alpha
To summarize and present the published data of miRNAs and lncRNAs involved in pathophysiology of DCM, we have generated Fig. 4 regarding ncRNAs and their target genes, affecting biological processes. Literature data (Tables 1 and 2) was transformed into a tabular network file. Cardiac fibrosis, hypertrophy, oxidative stress, inflammation, apoptosis, autophagy, and pyroptosis were presented. The miRNA, lncRNA and their targets are shown as nodes and connections between them as edges. Visualization of the network was performed using Cytoscape v3.7.2. Additional information regarding model organisms and ncRNAs expression changes shown in the studies was used for visual mapping of the nodes. According to this network (Fig. 4), we can conclude that miR-146a and miR-195 appear to be the most promising miRNAs as regulators in DCM, since those miRNAs placed in the center of the network, can target at least six different genes and can regulate different biological processes involved in DCM, such as inflammation, hypertrophy, apoptosis, or oxidative stress (confirmed in the literature by experimental analysis). Furthermore, to date most of the studies have aimed to analyze the impact of lncRNAs MALAT1 and Kcnq1ot1 in DCM. MALAT1 was found to be associated with inflammation and apoptosis processes, whereas Kcnq1ot1 association was identified with pyroptosis, cardiac fibrosis, and inflammation. Importantly, as it is presented in Fig. 4 those lncRNAs can regulate at least 6 different genes and can sponge miRNAs. Important to note that miR-146a and those lncRNAs (MALAT1 and Kcnq1ot1) were studied not only in the in vitro and in vivo analysis, but were also demonstrated in human studies [71, 124, 132, 133].

Summarizing graph showing information from the studies evaluating miRNAs and lncRNAs as potential biomarkers in diabetic cardiomyopathy. The figure was generated using data from the published literature regarding ncRNAs, their targets and affected biological processes. Literature data (Figs. 1, 2, Tables 1 and 2) were transformed into a tabular network file. The miRNA, lncRNA and their targets are shown as nodes and connections between them as edges. Visualization of the network was performed using Cytoscape v3.7.2. and the Perfuse force layout algorithm. Additional information regarding affected biological processes retrieved from publications, model organisms and ncRNAs expression changes shown in the studies was used for visual mapping for the nodes
On the other hand, as it is summarized in Fig. 4, several studies have shown that the upregulation of ANRIL and downregulation of HOTAIR may have consequences in the form of cardiac fibrosis, oxidative stress, apoptosis and inflammation processes in DCM. Therefore, future studies should focus more on these lncRNAs (HOTAIR and ANRIL) as their therapeutic potential may be higher than that demonstrated in the current literature.
Measuring their expression in blood components and heart tissue may improve the diagnosis and prediction of adverse cardiac outcomes. However, the use of lncRNAs and miRNAs as biomarkers in clinical practice still faces many limitations: (i) a small number of human studies describing the role of ncRNAs in the processes of oxidative stress and inflammation in DCM; (ii) many of the studies described in this review require further validation and assessment of their results reproducibility; (iii) a number of studies that analyzed the importance of ncRNAs in DCM used STZ-induced diabetes, which resembles T1DM more closely than T2DM. Due to the different pathophysiology of T1 and T2 diabetes, further research is needed to clarify the differences in expression pattern of individual miRNAs/lncRNAs related to processes underlying DCM in T1DM and T2DM. (iv) individual molecules examined in diabetic patients such as miR-223 and miR-126 are not specific to this disease only; (v) although similar expression of single lncRNAs and miRNAs in DCM has been confirmed independently by various authors, describing and validation of specific biomarker signature patterns in DCM remain challenging.
Compared to other types of drugs, ncRNA therapies excel in several aspects. Due to the development of bioinformatics tools including in silico prediction analysis and the simple structure of ncRNAs, the mechanism of action of these molecules can be predictable. The use of miRNAs in therapy allows for the simultaneous targeting of several protein-coding genes. Moreover, upregulation or downregulation of miRNAs expression to their physiological concentrations allows the restoration of homeostasis [153]. Importantly, ncRNAs are able to target genes that are still “undruggable” and unlike regular medications used today, they have not been found to be affected by drug resistance [154].
The use of ncRNA as a therapeutic agent is a promising approach with the possibility of treating a wide range of human diseases at the molecular level [155]. It is worth mentioning that the delivery systems that are successful in the in vivo studies differ structurally and chemically. In each particular case, special forms of delivery of ncRNAs may be necessary to achieve the best efficacy without causing harmful side effects. Lipid nanoparticles (LNP) seem to be a promising and effective way for ncRNA therapy [153]. For example, inclisiran in LNP formulation as a small interfering RNAs (siRNA) against PCSK9 is used for a gene therapy of primary hyperlipidemias [156, 157]. Alternatively, neutral or synthetic polymers may be applied for ncRNA therapy. It was shown on an animal model that in post-infarcted heart intracoronary injection of an antagomiR-92 encapsulated in poly(lactic-co-glycolic acid) stimulated angiogenesis, improved myocardial function and prevented against adverse infarct remodeling [158]. Moreover, the use of exosomes, which are extracellular vesicles released by cells, appears to be promising as well. It has been shown that due to their favorable pharmacokinetic properties, exosomes can serve as an attractive carrier of nucleic acids capable of penetrating physiological barriers inaccessible to other drug carriers [159, 160]. However, much work is still needed in this field. Nevertheless, the potential clinical impact is undoubtedly worth the investment.
Conclusion
Several studies highlighted the promising role of these molecules as potential therapeutic targets in miRNA- and lncRNA-based novel treatments. As described above, this therapeutic approach may consist in the inhibition by means of antagonists or restoration of loss of function molecules with mimics that are similar to endogenous molecules. Yet, the detailed mechanism of action of the described miRNAs and lncRNAs on cardiomyocytes oxidative stress and inflammation has not been fully explained and more studies need to be conducted. Importantly, a single miRNA or lncRNA may target multiple genes, thus understanding the miRNA–lncRNA interaction network and functions, as well as creating an effective and inexpensive way of delivering therapeutics are prerequisites to apply therapy in the future. A subsequent comprehensive in silico analysis may provide novel information of signaling pathways involved in pathological changes in DCM and identify miRNAs and lncRNAs that could potentially serve as therapeutic targets [25, 27, 161].
Availability of data and materials
Not applicable.
Abbreviations
- α2M:
-
Alpha-2-macroglobulin
- α‐SMA:
-
Alpha-smooth muscle actin
- AA:
-
Amyloid A
- ADAM9:
-
ADAM metallopeptidase domain 9
- ADAMs:
-
Human A disintegrin and metalloproteases
- Adipor1:
-
Adiponectin receptor 1
- AMI:
-
Acute myocardial infarction
- ANRIL:
-
Antisense non-coding RNA in the INK4 locus
- AP:
-
Amyloid P
- AP-1:
-
Activator protein 1
- APPs:
-
Acute-phase proteins
- ARC:
-
Activity regulated cytoskeleton associated protein
- Bcl-2:
-
B-cell lymphoma 2
- CAD:
-
Coronary artery disease
- CASP:
-
Caspase
- CDH5:
-
Cadherin 5
- CK-MB:
-
Creatine kinase myocardial band
- Col1α1:
-
Collagen type I alpha 1
- Col4α1:
-
Collagen type IV alpha 1
- Col‐I:
-
Collagen type-I
- Col‐III:
-
Collagen type-III
- CPDT:
-
5,6 Dihydrocyclopenta-1,2-dithiole-3-thione
- CRP:
-
C-reactive protein
- CTGF:
-
Connective tissue growth factor
- CVDs:
-
Cardiovascular diseases
- DCM:
-
Diabetic cardiomyopathy
- DM:
-
Diabetes mellitus
- DNA:
-
Deoxyribonucleic acid
- ECM:
-
Extracellular matrix
- ECs:
-
Endothelial cells
- ELAVL1:
-
ELAV like protein 1
- EMT:
-
Endothelial-to-mesenchymal transition
- ERK1/2:
-
Extracellular signal-regulated kinases 1 and 2
- FGF1:
-
Fibroblast growth factor 1
- FN:
-
Fibronectin
- FOXO3a:
-
Forkhead box O3
- GACAT3:
-
Gastric cancer-associated transcript 3
- GSDMD-N:
-
N-terminal of gasdermin D
- H19:
-
Imprinted maternally expressed transcript
- HAECs:
-
Human aortic endothelial cells
- HBEGF:
-
Heparin binding EGF like growth factor
- HF:
-
Heart failure
- HG:
-
High glucose
- HMGA1:
-
High-mobility group AT-hook 1
- HMGB1:
-
High mobility group box 1
- HO-1:
-
Heme oxygenase 1
- HOTAIR:
-
HOX transcript antisense intergenic RNA
- HOTTIP:
-
HOXA transcript at the distal tip
- I/R:
-
Ischemia/reperfusion
- IL:
-
Interleukin
- IRAK1:
-
Interleukin-1 receptor-associated kinase 1
- Kcnq1:
-
Potassium voltage-gated channel subfamily Q member 1
- Kcnq1ot1:
-
Potassium voltage-gated channel subfamily Q member 1 overlapping transcript 1
- LAZ3:
-
Lymphoma-associated zinc finger 3
- LDH:
-
Lactate dehydrogenase,
- lncRNAs:
-
Long non-coding RNAs
- LNP:
-
Lipid nanoparticle
- MALAT1:
-
Metastasis-associated lung adenocarcinoma 1
- MAPK:
-
Mitogen-activated protein kinase
- MCP-1:
-
Monocyte chemoattractant protein-1
- MDA:
-
Malondialdehyde
- Mef2c:
-
Myocyte enhancer factor 2C
- MMP-9:
-
Matrix metalloproteinase 9
- mRNA:
-
Messenger RNA
- miRs:
-
MicroRNAs, miRNAs
- ncRNA:
-
Non-coding RNA
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NLRP3:
-
Nod-like receptor protein 3
- NLRP3:
-
NLR family pyrin domain containing 3
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- Nt:
-
Nucleotides
- PA:
-
Palmitic acid
- PCSK9:
-
Proprotein convertase subtilisin/kexin type 9
- PGC-1α:
-
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PGC-1β:
-
Peroxisome proliferator-activated receptor gamma coactivator 1-bet
- phospho-Akt:
-
Phosphorylated protein kinase B
- phospho-eNOS:
-
Phosphorylated endothelial nitric oxide synthase
- PI3K/Akt:
-
Phosphoinositide 3-kinase and protein kinase B
- PIK3CA:
-
Phosphoinositide 3-kinase catalytic subunit alpha
- PPARα:
-
Peroxisome proliferator-activated receptor-alpha
- p-Smad2:
-
Phosphorylated SMAD family member 2
- p-Smad3:
-
Phosphorylated SMAD family member 3
- PDCD4:
-
Programmed cell death 4
- RNA:
-
Ribonucleic acid
- ROS:
-
Reactive oxygen species
- SAA3:
-
Serum amyloid antigen 3
- siRNA:
-
Small interfering RNA
- Sirt1:
-
Sirtuin 1
- Smad7:
-
SMAD family member 7
- SNHG1:
-
Small nucleolar RNA host gene 1
- SOD:
-
Superoxide dismutase
- STZ:
-
Streptozocin
- T1DM:
-
Type 1 diabetes mellitus
- T2DM:
-
Type 2 diabetes mellitus
- TGF-β1:
-
Transforming growth factor β1
- TGF-β3:
-
Transforming growth factor β3
- TLR4:
-
Toll-like receptor 4
- TNFα:
-
Tumor necrosis factor-alpha
- TRAF6:
-
TNF Receptor associated factor 6
- VDAC1:
-
Voltage-dependent anion channel 1
References
Website. International Diabetes Federation. IDF Diabetes Atlas, 9th edn. Brussels, Belgium: 2019. https://www.diabetesatlas.org
Jarosz-Popek J, Wolska M, Gasecka A, Czajka P, Jakubik D, Sharif L, et al. The importance of non-coding RNAs in neurodegenerative processes of diabetes-related molecular pathways. J Clin Med Res. 2020. https://doi.org/10.3390/jcm10010009.
American Diabetes Association. (2) Classification and diagnosis of diabetes. Diabetes Care. 2015;38(Suppl):S8-16. https://doi.org/10.2337/dc15-S005.
Borghetti G, von Lewinski D, Eaton DM, Sourij H, Houser SR, Wallner M. Diabetic cardiomyopathy: current and future therapies. Beyond glycemic control. Front Physiol. 2018. https://doi.org/10.3389/fphys.2018.01514.
Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA. 1979;241:2035–8. https://doi.org/10.1001/jama.241.19.2035.
Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy. Circ Res. 2018;122:624–38. https://doi.org/10.1161/circresaha.117.311586.
Pant T, Dhanasekaran A, Fang J, Bai X, Bosnjak ZJ, Liang M, et al. Current status and strategies of long noncoding RNA research for diabetic cardiomyopathy. BMC Cardiovasc Disord. 2018. https://doi.org/10.1186/s12872-018-0939-5.
Paolillo S, Marsico F, Prastaro M, Renga F, Esposito L, De Martino F, et al. Diabetic cardiomyopathy. Heart Fail Clin. 2019. https://doi.org/10.1016/j.hfc.2019.02.003.
Lee MMY, McMurray JJV, Lorenzo-Almorós A, Kristensen SL, Sattar N, Jhund PS, et al. Diabetic cardiomyopathy. Heart. 2019;105:337–45. https://doi.org/10.1136/heartjnl-2016-310342.
Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. 2018;122:624–38. https://doi.org/10.1161/CIRCRESAHA.117.311586.
Seferović PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J. 2015;36(1718–27):1727a–1727c. https://doi.org/10.1093/eurheartj/ehv134.
Murtaza G, Virk HUH, Khalid M, Lavie CJ, Ventura H, Mukherjee D, et al. Diabetic cardiomyopathy—a comprehensive updated review. Prog Cardiovasc Dis. 2019;62:315–26. https://doi.org/10.1016/j.pcad.2019.03.003.
Dunlay SM, Givertz MM, Aguilar D, Allen LA, Chan M, Desai AS, et al. Type 2 Diabetes Mellitus and Heart Failure: A Scientific Statement From the American Heart Association and the Heart Failure Society of America: This statement does not represent an update of the 2017 ACC/AHA/HFSA heart failure guideline update. Circulation. 2019;140:e294-324. https://doi.org/10.1161/CIR.0000000000000691.
Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. 2020;17:585–607. https://doi.org/10.1038/s41569-020-0339-2.
Lorenzo-Almorós A, Tuñón J, Orejas M, Cortés M, Egido J, Lorenzo Ó. Diagnostic approaches for diabetic cardiomyopathy. Cardiovasc Diabetol. 2017. https://doi.org/10.1186/s12933-017-0506-x.
Tate M, Grieve DJ, Ritchie RH. Are targeted therapies for diabetic cardiomyopathy on the horizon? Clin Sci. 2017. https://doi.org/10.1042/cs20160491.
Germolec DR, Shipkowski KA, Frawley RP, Evans E. Markers of Inflammation. Methods Mol Biol. 2018. https://doi.org/10.1007/978-1-4939-8549-4_5.
Tsalamandris S, Antonopoulos AS, Oikonomou E, Papamikroulis GA, Vogiatzi G, Papaioannou S, et al. The role of inflammation in diabetes: current concepts and future perspectives. Eur Cardiol. 2019;14:50–9. https://doi.org/10.15420/ecr.2018.33.1
Biesinger BS, Gasecka A, Perkmann T, Wojta J, Lesiak M, Grygier M, et al. Inflammatory state does not affect the antiplatelet efficacy of potent P2Y12 inhibitors in ACS. Platelets. 2020. https://doi.org/10.1080/09537104.2020.1766670.
Komosa A, Rzymski P, Perek B, Ropacka-Lesiak M, Lesiak M, Siller-Matula JM, et al. Platelets redox balance assessment: current evidence and methodological considerations. Vasc Pharmacol. 2017;93–95:6–13. https://doi.org/10.1016/j.vph.2017.06.002.
Komosa A, Perek B, Rzymski P, Lesiak M, Siller-Matula JM, Grygier M, et al. Transcatheter aortic valve replacement is associated with less oxidative stress and faster recovery of antioxidant capacity than surgical aortic valve replacement. J Clin Med Res. 2019. https://doi.org/10.3390/jcm8091364.
Rosa S, Cirillo P, Paglia A, Sasso L, Palma V, Chiariello M. Reactive oxygen species and antioxidants in the pathophysiology of cardiovascular disease: does the actual knowledge justify a clinical approach? Curr Vasc Pharmacol. 2010. https://doi.org/10.2174/157016110790887009.
Burgos-Morón, Burgos-Morón, Abad-Jiménez, Marañón, Iannantuoni, Escribano-López, et al. Relationship between oxidative stress, ER stress, and inflammation in type 2 diabetes: the battle continues. J Clin Med. 2019. https://doi.org/10.3390/jcm8091385
De Rosa S, Arcidiacono B, Chiefari E, Brunetti A, Indolfi C, Foti DP. Type 2 diabetes mellitus and cardiovascular disease: genetic and epigenetic links. Front Endocrinol. 2018;9:2. https://doi.org/10.3389/fendo.2018.00002.
Pordzik J, Jakubik D, Jarosz-Popek J, Wicik Z, Eyileten C, De Rosa S, et al. Significance of circulating microRNAs in diabetes mellitus type 2 and platelet reactivity: bioinformatic analysis and review. Cardiovasc Diabetol. 2019. https://doi.org/10.1186/s12933-019-0918-x.
Eyileten C, Sharif L, Wicik Z, Jakubik D, Jarosz-Popek J, Soplinska A, et al. The relation of the brain-derived neurotrophic factor with MicroRNAs in neurodegenerative diseases and ischemic stroke. Mol Neurobiol. 2020. https://doi.org/10.1007/s12035-020-02101-2.
Sabatino J, Wicik Z, De Rosa S, Eyileten C, Jakubik D, Spaccarotella C, et al. MicroRNAs fingerprint of bicuspid aortic valve. J Mol Cell Cardiol. 2019;134:98–106. https://doi.org/10.1016/j.yjmcc.2019.07.001.
Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018. https://doi.org/10.1016/j.cell.2018.01.011.
Ghafouri-Fard S, Shoorei H, Taheri M. Non-coding RNAs are involved in the response to oxidative stress. Biomed Pharmacother. 2020. https://doi.org/10.1016/j.biopha.2020.110228.
Marques-Rocha JL, Samblas M, Milagro FI, Bressan J, Martínez JA, Marti A. Noncoding RNAs, cytokines, and inflammation-related diseases. FASEB J. 2015;29:3595–611. https://doi.org/10.1096/fj.14-260323.
Calderon-Dominguez M, Belmonte T, Quezada-Feijoo M, Ramos-Sánchez M, Fernández-Armenta J, Pérez-Navarro A, et al. Emerging role of microRNAs in dilated cardiomyopathy: evidence regarding etiology. Transl Res. 2020;215:86–101. https://doi.org/10.1016/j.trsl.2019.08.007.
Hijmans JG, Diehl KJ, Bammert TD, Kavlich PJ, Lincenberg GM, Greiner JJ, et al. Influence of overweight and obesity on circulating inflammation-related microRNA. Microrna. 2018;7:148–54. https://doi.org/10.2174/2211536607666180402120806.
Ait-Aissa K, Nguyen QM, Gabani M, Kassan A, Kumar S, Choi S-K, et al. MicroRNAs and obesity-induced endothelial dysfunction: key paradigms in molecular therapy. Cardiovasc Diabetol. 2020. https://doi.org/10.1186/s12933-020-01107-3.
Yu M, Liu Y, Zhang B, Shi Y, Cui L, Zhao X. Inhibiting microRNA-144 abates oxidative stress and reduces apoptosis in hearts of streptozotocin-induced diabetic mice. Cardiovasc Pathol. 2015. https://doi.org/10.1016/j.carpath.2015.06.003.
Hu X, Bai T, Xu Z, Liu Q, Zheng Y, Cai L. Pathophysiological fundamentals of diabetic cardiomyopathy. Comprehensive Physiol. 2017. https://doi.org/10.1002/cphy.c160021.
Evangelista I, Nuti R, Picchioni T, Dotta F, Palazzuoli A. Molecular dysfunction and phenotypic derangement in diabetic cardiomyopathy. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20133264.
Gao L, Liu Y, Guo S, Xiao L, Wu L, Wang Z, et al. LAZ3 protects cardiac remodeling in diabetic cardiomyopathy via regulating miR-21/PPARa signaling. Biochimica et Biophysica Acta (BBA) Mol Basis Dis. 2018. https://doi.org/10.1016/j.bbadis.2018.07.019.
Li X, Du N, Zhang Q, Li J, Chen X, Liu X, et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. 2014. https://doi.org/10.1038/cddis.2014.430.
Chavali V, Tyagi SC, Mishra PK. Differential expression of dicer, miRNAs, and inflammatory markers in diabetic Ins2 /− Akita hearts. Cell Biochem Biophys. 2014. https://doi.org/10.1007/s12013-013-9679-4.
Wang T, Zhu H, Yang S, Fei X. Let-7a-5p may participate in the pathogenesis of diabetic nephropathy through targeting HMGA2. Mol Med Rep. 2019;19:4229–37. https://doi.org/10.3892/mmr.2019.10057.
Erener S, Marwaha A, Tan R, Panagiotopoulos C, Kieffer TJ. Profiling of circulating microRNAs in children with recent onset of type 1 diabetes. JCI Insight. 2017;2:e89656. https://doi.org/10.1172/jci.insight.89656.
Tao L, Bei Y, Chen P, Lei Z, Fu S, Zhang H, et al. Crucial role of miR-433 in regulating cardiac fibrosis. Theranostics. 2016;6:2068–83. https://doi.org/10.7150/thno.15007.
Shen E, Diao X, Wang X, Chen R, Hu B. MicroRNAs involved in the mitogen-activated protein kinase cascades pathway during glucose-induced cardiomyocyte hypertrophy. Am J Pathol. 2011;179:639–50. https://doi.org/10.1016/j.ajpath.2011.04.034.
Costantino S, Paneni F, Lüscher TF, Cosentino F. MicroRNA profiling unveils hyperglycaemic memory in the diabetic heart. Eur Heart J. 2016;37:572–6. https://doi.org/10.1093/eurheartj/ehv599.
Yildirim SS, Akman D, Catalucci D, Turan B. Relationship between downregulation of miRNAs and increase of oxidative stress in the development of diabetic cardiac dysfunction: Junctin as a target protein of miR-1. Cell Biochem Biophys. 2013. https://doi.org/10.1007/s12013-013-9672-y.
Greco S, Fasanaro P, Castelvecchio S, D’Alessandra Y, Arcelli D, Di Donato M, et al. MicroRNA dysregulation in diabetic ischemic heart failure patients. Diabetes. 2012;61:1633–41. https://doi.org/10.2337/db11-0952.
Ceriello A. Hypothesis: the “metabolic memory”, the new challenge of diabetes. Diabetes Res Clin Pract. 2009;86(Suppl 1):S2-6. https://doi.org/10.1016/S0168-8227(09)70002-6.
Martinez FO. Macrophage activation and polarization. Front Biosci. 2008. https://doi.org/10.2741/2692.
Corsten MF, Papageorgiou A, Verhesen W, Carai P, Lindow M, Obad S, et al. MicroRNA profiling identifies MicroRNA-155 as an adverse mediator of cardiac injury and dysfunction during acute viral myocarditis. Circ Res. 2012. https://doi.org/10.1161/circresaha.112.267443.
Jia C, Chen H, Wei M, Chen X, Zhang Y, Cao L, et al. Gold nanoparticle-based miR155 antagonist macrophage delivery restores the cardiac function in ovariectomized diabetic mouse model. Int J Nanomed. 2017. https://doi.org/10.2147/ijn.s138400.
Babu SS, Thandavarayan RA, Joladarashi D, Jeyabal P, Krishnamurthy S, Bhimaraj A, et al. MicroRNA-126 overexpression rescues diabetes-induced impairment in efferocytosis of apoptotic cardiomyocytes. Sci Rep. 2016. https://doi.org/10.1038/srep36207.
Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 2003;17:7–30. https://doi.org/10.1101/gad.1039703.
Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transducti Target Therapy. 2017. https://doi.org/10.1038/sigtrans.2017.23.
Palomer X, Álvarez-Guardia D, Davidson MM, Chan TO, Feldman AM, Vázquez-Carrera M. The interplay between NF-kappaB and E2F1 coordinately regulates inflammation and metabolism in human cardiac cells. PLoS ONE. 2011;6:e19724. https://doi.org/10.1371/journal.pone.0019724.
Lin K, Baritaki S, Militello L, Malaponte G, Bevelacqua Y, Bonavida B. The role of B-RAF mutations in melanoma and the induction of EMT via dysregulation of the NF-κB/Snail/RKIP/PTEN circuit. Genes Cancer. 2010;1:409–20. https://doi.org/10.1177/1947601910373795.
Huang X, Liu G, Guo J, Su Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci. 2018;14:1483–96. https://doi.org/10.7150/ijbs.27173.
Liu Y, Lv H, Tan R, An X, Niu X-H, Liu Y-J, et al. Platelets promote Ang II (Angiotensin II)-induced atrial fibrillation by releasing TGF-β1 (transforming growth factor-β1) and interacting with fibroblasts. Hypertension. 2020;76:1856–67. https://doi.org/10.1161/HYPERTENSIONAHA.120.15016.
Meyer A, Wang W, Qu J, Croft L, Degen JL, Coller BS, et al. Platelet TGF-β1 contributions to plasma TGF-β1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood. 2012;119:1064–74. https://doi.org/10.1182/blood-2011-09-377648.
Yang X, Li X, Lin Q, Xu Q. Up-regulation of microRNA-203 inhibits myocardial fibrosis and oxidative stress in mice with diabetic cardiomyopathy through the inhibition of PI3K/Akt signaling pathway via PIK3CA. Gene. 2019. https://doi.org/10.1016/j.gene.2019.143995.
Che H, Wang Y, Li Y, Lv J, Li H, Liu Y, et al. Inhibition of microRNA-150–5p alleviates cardiac inflammation and fibrosis via targeting Smad7 in high glucose-treated cardiac fibroblasts. J Cell Physiol. 2019. https://doi.org/10.1002/jcp.29386.
Freudlsperger C, Bian Y, Contag Wise S, Burnett J, Coupar J, Yang X, et al. TGF-β and NF-κB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene. 2013. https://doi.org/10.1038/onc.2012.171.
Chen Z, Stelekati E, Kurachi M, Yu S, Cai Z, Manne S, et al. miR-150 regulates memory CD8 T cell differentiation via c-Myb. Cell Rep. 2017;20:2584–97. https://doi.org/10.1016/j.celrep.2017.08.060.
Li Y, Ren W, Wang X, Yu X, Cui L, Li X, et al. MicroRNA-150 relieves vascular remodeling and fibrosis in hypoxia-induced pulmonary hypertension. Biomed Pharmacother. 2019;109:1740–9. https://doi.org/10.1016/j.biopha.2018.11.058.
Sang W, Wang Y, Zhang C, Zhang D, Sun C, Niu M, et al. MiR-150 impairs inflammatory cytokine production by targeting ARRB-2 after blocking CD28/B7 costimulatory pathway. Immunol Lett. 2016;172:1–10. https://doi.org/10.1016/j.imlet.2015.11.001.
Li X, Kong M, Jiang D, Qian J, Duan Q, Dong A. MicroRNA-150 aggravates H2O2-induced cardiac myocyte injury by down-regulating c-myb gene. Acta Biochim Biophys Sin. 2013;45:734–41. https://doi.org/10.1093/abbs/gmt067.
Zhu G-H, Li R, Zeng Y, Zhou T, Xiong F, Zhu M. MicroRNA-142-3p inhibits high-glucose-induced endothelial-to-mesenchymal transition through targeting TGF-β1/Smad pathway in primary human aortic endothelial cells. Int J Clin Exp Pathol. 2018;11:1208–17.
Ruiz MA, Chakrabarti S. MicroRNAs: the underlying mediators of pathogenetic processes in vascular complications of diabetes. Can J Diabetes. 2013;37:339–44. https://doi.org/10.1016/j.jcjd.2013.07.003.
Diao X, Shen E, Wang X, Hu B. Differentially expressed microRNAs and their target genes in the hearts of streptozotocin-induced diabetic mice. Mol Med Rep. 2011;4:633–40. https://doi.org/10.3892/mmr.2011.489.
Emadi SS, Soufi FG, Khamaneh AM, Alipour MR. MicroRNA-146a expression and its intervention in NF-кB signaling pathway in diabetic rat aorta. Endoc Regul. 2014. https://doi.org/10.4149/endo_2014_02_103.
Wang Q, Bozack SN, Yan Y, Boulton ME, Grant MB, Busik JV. Regulation of retinal inflammation by rhythmic expression of MiR-146a in diabetic retina. Investig Opthalmol Vis Sci. 2014. https://doi.org/10.1167/iovs.13-13076.
Feng B, Chen S, Gordon AD, Chakrabarti S. miR-146a mediates inflammatory changes and fibrosis in the heart in diabetes. J Mol Cell Cardiol. 2017. https://doi.org/10.1016/j.yjmcc.2017.03.002.
Palomer X, Capdevila-Busquets E, Botteri G, Davidson MM, Rodríguez C, Martínez-González J, et al. miR-146a targets Fos expression in human cardiac cells. Dis Model Mech. 2015;8:1081–91. https://doi.org/10.1242/dmm.020768.
Watson CJ, Gupta SK, O’Connell E, Thum S, Glezeva N, Fendrich J, et al. MicroRNA signatures differentiate preserved from reduced ejection fraction heart failure. Eur J Heart Fail. 2015;17:405–15. https://doi.org/10.1002/ejhf.244.
Palomer X, Capdevila-Busquets E, Alvarez-Guardia D, Barroso E, Pallàs M, Camins A, et al. Resveratrol induces nuclear factor-κB activity in human cardiac cells. Int J Cardiol. 2013;167:2507–16. https://doi.org/10.1016/j.ijcard.2012.06.006.
Palomer X, Salvadó L, Barroso E, Vázquez-Carrera M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int J Cardiol. 2013;168:3160–72. https://doi.org/10.1016/j.ijcard.2013.07.150.
Balasubramanyam M, Aravind S, Gokulakrishnan K, Prabu P, Sathishkumar C, Ranjani H, et al. Impaired miR-146a expression links subclinical inflammation and insulin resistance in Type 2 diabetes. Mol Cell Biochem. 2011;351:197–205. https://doi.org/10.1007/s11010-011-0727-3.
Li S, Chen H, Ren J, Geng Q, Song J, Lee C, et al. MicroRNA-223 inhibits tissue factor expression in vascular endothelial cells. Atherosclerosis. 2014;237:514–20. https://doi.org/10.1016/j.atherosclerosis.2014.09.033.
Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451:1125–9. https://doi.org/10.1038/nature06607.
Lu H, Buchan RJ, Cook SA. MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc Res. 2010;86:410–20. https://doi.org/10.1093/cvr/cvq010.
Yin Z, Zhao Y, He M, Li H, Fan J, Nie X, et al. MiR-30c/PGC-1β protects against diabetic cardiomyopathy via PPARα. Cardiovasc Diabetol. 2019. https://doi.org/10.1186/s12933-019-0811-7.
da Costa RM, da Costa RM, Rodrigues D, Pereira CA, Silva JF, Alves JV, et al. Nrf2 as a potential mediator of cardiovascular risk in metabolic diseases. Front Pharmacol. 2019. https://doi.org/10.3389/fphar.2019.00382.
Dubois V, Eeckhoute J, Lefebvre P, Staels B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J Clin Investig. 2017. https://doi.org/10.1172/jci88894.
Reddy RC, Standiford TJ. Nrf2 and PPAR{gamma}: PPARtnering against oxidant-induced lung injury. Am J Respir Crit Care Med. 2010. https://doi.org/10.1164/rccm.201004-0457ED.
Polvani S, Tarocchi M, Galli A. PPAR and oxidative stress: Con() catenating NRF2 and FOXO. PPAR Res. 2012. https://doi.org/10.1155/2012/641087.
Li D, Du Y, Yuan X, Han X, Dong Z, Chen X, et al. Hepatic hypoxia-inducible factors inhibit PPARα expression to exacerbate acetaminophen induced oxidative stress and hepatotoxicity. Free Radic Biol Med. 2017;110:102–16. https://doi.org/10.1016/j.freeradbiomed.2017.06.002.
Raut SK, Kumar A, Singh GB, Nahar U, Sharma V, Mittal A, et al. miR-30c mediates upregulation of Cdc42 and Pak1 in diabetic cardiomyopathy. Cardiovasc Ther. 2015;33:89–97. https://doi.org/10.1111/1755-5922.12113.
Raut SK, Singh GB, Rastogi B, Saikia UN, Mittal A, Dogra N, et al. miR-30c and miR-181a synergistically modulate p53–p21 pathway in diabetes induced cardiac hypertrophy. Mol Cell Biochem. 2016;417:191–203. https://doi.org/10.1007/s11010-016-2729-7.
Chen C, Yang S, Li H, Yin Z, Fan J, Zhao Y, et al. Mir30c is involved in diabetic cardiomyopathy through regulation of cardiac autophagy via BECN1. Mol Ther Nucleic Acids. 2017;7:127–39. https://doi.org/10.1016/j.omtn.2017.03.005.
Barish GD, Yu RT, Karunasiri M, Ocampo CB, Dixon J, Benner C, et al. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 2010;24:2760–5. https://doi.org/10.1101/gad.1998010.
De Rosa S, Eposito F, Carella C, Strangio A, Ammirati G, Sabatino J, et al. Transcoronary concentration gradients of circulating microRNAs in heart failure. Eur J Heart Fail. 2018;20:1000–10. https://doi.org/10.1002/ejhf.1119.
Tao L, Huang X, Xu M, Yang L, Hua F. MiR-144 protects the heart from hyperglycemia-induced injury by regulating mitochondrial biogenesis and cardiomyocyte apoptosis. FASEB J. 2020;34:2173–97. https://doi.org/10.1096/fj.201901838R.
Wang L, Wang M, Hu J, Shen W, Hu J, Yao Y, et al. Protective effect of 3H–1, 2-dithiole-3-thione on cellular model of Alzheimer’s disease involves Nrf2/ARE signaling pathway. Eur J Pharmacol. 2017. https://doi.org/10.1016/j.ejphar.2016.12.013.
Miao Y, Wan Q, Liu X, Wang Y, Luo Y, Liu D, et al. miR-503 is involved in the protective effect of phase II enzyme inducer (CPDT) in diabetic cardiomyopathy via Nrf2/ARE signaling pathway. Biomed Res Int. 2017. https://doi.org/10.1155/2017/9167450.
Tang Q, Len Q, Liu Z, Wang W. Overexpression of miR-22 attenuates oxidative stress injury in diabetic cardiomyopathy via Sirt 1. Cardiovasc Ther. 2018. https://doi.org/10.1111/1755-5922.12318.
Mao C, Yuan J-Q, Lv Y-B, Gao X, Yin Z-X, Kraus VB, et al. Associations between superoxide dismutase, malondialdehyde and all-cause mortality in older adults: a community-based cohort study. BMC Geriatr. 2019;19:104. https://doi.org/10.1186/s12877-019-1109-z.
Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–28. https://doi.org/10.1152/ajplung.2000.279.6.L1005.
Byrne BG, Dubuisson J-F, Joshi AD, Persson JJ, Swanson MS. Inflammasome components coordinate autophagy and pyroptosis as macrophage responses to infection. MBio. 2013;4:e00620-e712. https://doi.org/10.1128/mBio.00620-12.
Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907–16. https://doi.org/10.1128/IAI.73.4.1907-1916.2005.
Zhang Y-Q, Herman B. ARC protects rat cardiomyocytes against oxidative stress through inhibition of caspase-2 mediated mitochondrial pathway. J Cell Biochem. 2006;99:575–88. https://doi.org/10.1002/jcb.20946.
An J, Li P, Li J, Dietz R, Donath S. ARC is a critical cardiomyocyte survival switch in doxorubicin cardiotoxicity. J Mol Med. 2009;87:401–10. https://doi.org/10.1007/s00109-008-0434-z.
Donath S, Li P, Willenbockel C, Al-Saadi N, Gross V, Willnow T, et al. Apoptosis repressor with caspase recruitment domain is required for cardioprotection in response to biomechanical and ischemic stress. Circulation. 2006;113:1203–12. https://doi.org/10.1161/CIRCULATIONAHA.105.576785.
Zhang WY, Wang J, Li AZ. A study of the effects of SGLT-2 inhibitors on diabetic cardiomyopathy through miR-30d/KLF9/VEGFA pathway. Eur Rev Med Pharmacol Sci. 2020;24:6346–59. https://doi.org/10.26355/eurrev_202006_21533.
Jeyabal P, Thandavarayan RA, Joladarashi D, Babu SS, Krishnamurthy S, Bhimaraj A, et al. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem Biophys Res Commun. 2016. https://doi.org/10.1016/j.bbrc.2016.02.065.
Zheng D, Ma J, Yu Y, Li M, Ni R, Wang G, et al. Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia. 2015. https://doi.org/10.1007/s00125-015-3622-8.
Xie L, Du R, Niu J, Liu H. Role of miRNAs in coronary artery disease. Curr Signal Transduct Ther. 2015. https://doi.org/10.2174/1574362410666141224210818.
Romaine SPR, Tomaszewski M, Condorelli G, Samani NJ. MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart. 2015. https://doi.org/10.1136/heartjnl-2013-305402.
Zhou S-S, Jin J-P, Wang J-Q, Zhang Z-G, Freedman JH, Zheng Y, et al. miRNAS in cardiovascular diseases: potential biomarkers, therapeutic targets and challenges. Acta Pharmacol Sin. 2018. https://doi.org/10.1038/aps.2018.30.
Kura B, Kalocayova B, Devaux Y, Bartekova M. Potential clinical implications of miR-1 and miR-21 in Heart disease and cardioprotection. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21030700.
Hong C-S, Kwon S-J, Cho M-C, Kwak Y-G, Ha K-C, Hong B, et al. Overexpression of junctate induces cardiac hypertrophy and arrhythmia via altered calcium handling. J Mol Cell Cardiol. 2008;44:672–82. https://doi.org/10.1016/j.yjmcc.2008.01.012.
Kirchhefer U, Hanske G, Jones LR, Justus I, Kaestner L, Lipp P, et al. Overexpression of junctin causes adaptive changes in cardiac myocyte Ca(2+) signaling. Cell Calcium. 2006;39:131–42. https://doi.org/10.1016/j.ceca.2005.10.004.
Song Z, Gao R, Yan B. Potential roles of microRNA-1 and microRNA-133 in cardiovascular disease. Rev Cardiovasc Med. 2020;21:57–64. https://doi.org/10.31083/j.rcm.2020.01.577.
Feng B, Chen S, George B, Feng Q, Chakrabarti S. miR133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes/Metab Res Rev. 2010. https://doi.org/10.1002/dmrr.1054.
Chen S, Puthanveetil P, Feng B, Matkovich SJ, Dorn GW, Chakrabarti S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J Cell Mol Med. 2014. https://doi.org/10.1111/jcmm.12218.
Nandi SS, Zheng H, Sharma NM, Shahshahan HR, Patel KP, Mishra PK. Lack of miR-133a decreases contractility of diabetic hearts: a role for novel cross talk between tyrosine aminotransferase and tyrosine hydroxylase. Diabetes. 2016;65:3075–90. https://doi.org/10.2337/db16-0023.
Kambis TN, Shahshahan HR, Kar S, Yadav SK, Mishra PK. Transgenic expression of miR-133a in the diabetic akita heart prevents cardiac remodeling and cardiomyopathy. Front Cardiovasc Med. 2019. https://doi.org/10.3389/fcvm.2019.00045.
de Gonzalo-Calvo D, van der Meer RW, Rijzewijk LJ, Smit JWA, Revuelta-Lopez E, Nasarre L, et al. Serum microRNA-1 and microRNA-133a levels reflect myocardial steatosis in uncomplicated type 2 diabetes. Sci Rep. 2017;7:47. https://doi.org/10.1038/s41598-017-00070-6.
Ramanujam D, Sassi Y, Laggerbauer B, Engelhardt S. Viral vector-based targeting of miR-21 in cardiac nonmyocyte cells reduces pathologic remodeling of the heart. Mol Ther. 2016;24:1939–48. https://doi.org/10.1038/mt.2016.166.
Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4. https://doi.org/10.1038/nature07511.
Liu S, Li W, Xu M, Huang H, Wang J, Chen X. Micro-RNA 21Targets dual specific phosphatase 8 to promote collagen synthesis in high glucose-treated primary cardiac fibroblasts. Can J Cardiol. 2014;30:1689–99. https://doi.org/10.1016/j.cjca.2014.07.747.
Cheng Y, Liu X, Zhang S, Lin Y, Yang J, Zhang C. MicroRNA-21 protects against the H(2)O(2)-induced injury on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol. 2009;47:5–14. https://doi.org/10.1016/j.yjmcc.2009.01.008.
Dai B, Li H, Fan J, Zhao Y, Yin Z, Nie X, et al. MiR-21 protected against diabetic cardiomyopathy induced diastolic dysfunction by targeting gelsolin. Cardiovasc Diabetol. 2018. https://doi.org/10.1186/s12933-018-0767-z.
López-Urrutia E, Bustamante Montes LP, Ladrón de Guevara Cervantes D, Pérez-Plasencia C, Campos-Parra AD. Crosstalk between long non-coding RNAs, Micro-RNAs and mRNAs: deciphering molecular mechanisms of master regulators in cancer. Front Oncol. 2019;9:669. https://doi.org/10.3389/fonc.2019.00669.
Zhang X, Pan L, Yang K, Fu Y, Liu Y, Chi J, et al. H3 relaxin protects against myocardial injury in experimental diabetic cardiomyopathy by inhibiting myocardial apoptosis, fibrosis and inflammation. Cell Physiol Biochem. 2017;43:1311–24. https://doi.org/10.1159/000481843.
Jia P, Wu N, Jia D, Sun Y. Downregulation of MALAT1 alleviates saturated fatty acid-induced myocardial inflammatory injury via the miR-26a/HMGB1/TLR4/NF-κB axis. Diabetes Metab Syndr Obes. 2019;12:655–65. https://doi.org/10.2147/DMSO.S203151.
Gao L, Wang X, Guo S, Xiao L, Liang C, Wang Z, et al. LncRNA HOTAIR functions as a competing endogenous RNA to upregulate SIRT1 by sponging miR-34a in diabetic cardiomyopathy. J Cell Physiol. 2019. https://doi.org/10.1002/jcp.27296.
Zhu M, Chen Q, Liu X, Sun Q, Zhao X, Deng R, et al. lncRNA H19/miR-675 axis represses prostate cancer metastasis by targeting TGFBI. FEBS J. 2014. https://doi.org/10.1111/febs.12902.
Li X, Wang H, Yao B, Xu W, Chen J, Zhou X. lncRNA H19/miR-675 axis regulates cardiomyocyte apoptosis by targeting VDAC1 in diabetic cardiomyopathy. Sci Rep. 2016. https://doi.org/10.1038/srep36340.
Yu M, Shan X, Liu Y, Zhu J, Cao Q, Yang F, et al. RNA-Seq analysis and functional characterization revealed lncRNA NONRATT007560.2 regulated cardiomyocytes oxidative stress and apoptosis induced by high glucose. J Cell Biochem. 2019;120:18278–87. https://doi.org/10.1002/jcb.29134.
Rawal S, Nagesh PT, Coffey S, Van Hout I, Galvin IF, Bunton RW, et al. Early dysregulation of cardiac-specific microRNA-208a is linked to maladaptive cardiac remodelling in diabetic myocardium. Cardiovasc Diabetol. 2019;18:13. https://doi.org/10.1186/s12933-019-0814-4.
Li X, Dai Y, Yan S, Shi Y, Han B, Li J, et al. Down-regulation of lncRNA KCNQ1OT1 protects against myocardial ischemia/reperfusion injury following acute myocardial infarction. Biochem Biophys Res Commun. 2017;491:1026–33. https://doi.org/10.1016/j.bbrc.2017.08.005.
Coto E, Calvo D, Reguero JR, Morís C, Rubín JM, Díaz-Corte C, et al. Differential methylation of lncRNA KCNQ1OT1 promoter polymorphism was associated with symptomatic cardiac long QT. Epigenomics. 2017;9:1049–57. https://doi.org/10.2217/epi-2017-0024.
Yang F, Qin Y, Lv J, Wang Y, Che H, Chen X, et al. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis. 2018;9:1000. https://doi.org/10.1038/s41419-018-1029-4.
Yang F, Qin Y, Wang Y, Li A, Lv J, Sun X, et al. LncRNA KCNQ1OT1 mediates pyroptosis in diabetic cardiomyopathy. Cell Physiol Biochem. 2018. https://doi.org/10.1159/000494576.
Gao L, Liu Y, Guo S, Yao R, Wu L, Xiao L, et al. Circulating long noncoding RNA HOTAIR is an essential mediator of acute myocardial infarction. Cell Physiol Biochem. 2017;44:1497–508. https://doi.org/10.1159/000485588.
Qi K, Zhong J. LncRNA HOTAIR improves diabetic cardiomyopathy by increasing viability of cardiomyocytes through activation of the PI3K/Akt pathway. Exp Ther Med. 2018;16:4817–23. https://doi.org/10.3892/etm.2018.6755.
Yang Y, Cheng H-W, Qiu Y, Dupee D, Noonan M, Lin Y-D, et al. MicroRNA-34a plays a key role in cardiac repair and regeneration following myocardial infarction. Circ Res. 2015;117:450–9. https://doi.org/10.1161/CIRCRESAHA.117.305962.
Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. 2010;11:31–9. https://doi.org/10.1007/s11154-010-9131-7.
Zlobine I, Gopal K, Ussher JR. Lipotoxicity in obesity and diabetes-related cardiac dysfunction. Biochim Biophys Acta. 2016;1861:1555–68. https://doi.org/10.1016/j.bbalip.2016.02.011.
Zhao Z-H, Hao W, Meng Q-T, Du X-B, Lei S-Q, Xia Z-Y. Long non-coding RNA MALAT1 functions as a mediator in cardioprotective effects of fentanyl in myocardial ischemia-reperfusion injury. Cell Biol Int. 2017;41:62–70. https://doi.org/10.1002/cbin.10701.
Puthanveetil P, Chen S, Feng B, Gautam A, Chakrabarti S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J Cell Mol Med. 2015. https://doi.org/10.1111/jcmm.12576.
Congrains A, Kamide K, Katsuya T, Yasuda O, Oguro R, Yamamoto K, et al. CVD-associated non-coding RNA, ANRIL, modulates expression of atherogenic pathways in VSMC. Biochem Biophys Res Commun. 2012. https://doi.org/10.1016/j.bbrc.2012.02.050.
Dai W, Lee D. Interfering with long chain noncoding RNA ANRIL expression reduces heart failure in rats with diabetes by inhibiting myocardial oxidative stress. J Cell Biochem. 2019. https://doi.org/10.1002/jcb.29162.
Rosa SD, De Rosa S, Chiefari E, Salerno N, Ventura V, D’Ascoli GL, et al. HMGA1 is a novel candidate gene for myocardial infarction susceptibility. Int J Cardiol. 2017. https://doi.org/10.1016/j.ijcard.2016.11.088.
Wu Q-Q, Liu C, Cai Z, Xie Q, Hu T, Duan M, et al. High-mobility group AT-hook 1 promotes cardiac dysfunction in diabetic cardiomyopathy via autophagy inhibition. Cell Death Dis. 2020;11:160. https://doi.org/10.1038/s41419-020-2316-4.
De Martino M, Esposito F, Pellecchia S, Cortez Cardoso Penha R, Botti G, Fusco A, et al. HMGA1-regulating microRNAs Let-7a and miR-26a are downregulated in human seminomas. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21083014.
Liu L, Shi Y, Shi J, Wang H, Sheng Y, Jiang Q, et al. The long non-coding RNA SNHG1 promotes glioma progression by competitively binding to miR-194 to regulate PHLDA1 expression. Cell Death Dis. 2019. https://doi.org/10.1038/s41419-019-1698-7.
Zhang L, Luo X, Chen F, Yuan W, Xiao X, Zhang X, et al. LncRNA SNHG1 regulates cerebrovascular pathologies as a competing endogenous RNA through HIF-1α/VEGF signaling in ischemic stroke. J Cell Biochem. 2018;119:5460–72. https://doi.org/10.1002/jcb.26705.
Yan S, Li H, Shu Q, Wu W, Luo X, Lu L. LncRNA SNHG1 exerts a protective role in cardiomyocytes hypertrophy via targeting miR-15a-5p/HMGA1 axis. Cell Biol Int. 2020. https://doi.org/10.1002/cbin.11298.
Wang J, Lv B, Su Y, Wang X, Bu J, Yao L. Exosome-mediated transfer of lncRNA HOTTIP promotes cisplatin resistance in gastric cancer cells by regulating HMGA1/miR-218 Axis. OncoTargets Ther. 2019;12:11325–38. https://doi.org/10.2147/OTT.S231846.
Kong Q, Guo X, Guo Z, Su T. Urinary exosome miR-424 and miR-218 as biomarkers for Type 1 diabetes in children. Clin Lab. 2019. https://doi.org/10.7754/Clin.Lab.2018.180921.
Zhang Y-L, Wang J-M, Yin H, Wang S-B, He C-L, Liu J. DACH1, a novel target of miR-218, participates in the regulation of cell viability, apoptosis, inflammatory response, and epithelial-mesenchymal transition process in renal tubule cells treated by high-glucose. Ren Fail. 2020;42:463–73. https://doi.org/10.1080/0886022X.2020.1762647.
Lin Y, Li J, Ye S, Chen J, Zhang Y, Wang L, et al. LncRNA GACAT3 acts as a competing endogenous RNA of HMGA1 and alleviates cucurbitacin B-induced apoptosis of gastric cancer cells. Gene. 2018;678:164–71. https://doi.org/10.1016/j.gene.2018.08.037.
Ling H. Non-coding RNAs: therapeutic strategies and delivery systems. Adv Exp Med Biol. 2016;937:229–37. https://doi.org/10.1007/978-3-319-42059-2_12.
Huang C-K, Kafert-Kasting S, Thum T. Preclinical and clinical development of noncoding RNA therapeutics for cardiovascular disease. Circ Res. 2020;126:663–78. https://doi.org/10.1161/CIRCRESAHA.119.315856.
Slaby O, Laga R, Sedlacek O. Therapeutic targeting of non-coding RNAs in cancer. Biochem J. 2017. https://doi.org/10.1042/bcj20170079.
Ray KK, Landmesser U, Leiter LA, Kallend D, Dufour R, Karakas M, et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N Engl J Med. 2017;376:1430–40. https://doi.org/10.1056/NEJMoa1615758.
Ray KK, Stoekenbroek RM, Kallend D, Leiter LA, Landmesser U, Wright RS, et al. Effect of an siRNA therapeutic targeting PCSK9 on atherogenic lipoproteins: prespecified secondary end points in ORION 1. Circulation. 2018;138:1304–16. https://doi.org/10.1161/CIRCULATIONAHA.118.034710.
Bellera N, Barba I, Rodriguez-Sinovas A, Ferret E, Asín MA, Gonzalez-Alujas MT, et al. Single intracoronary injection of encapsulated antagomir-92a promotes angiogenesis and prevents adverse infarct remodeling. J Am Heart Assoc. 2014;3:e000946. https://doi.org/10.1161/JAHA.114.000946.
Yang Z, Shi J, Xie J, Wang Y, Sun J, Liu T, et al. Large-scale generation of functional mRNA-encapsulating exosomes via cellular nanoporation. Nat Biomed Eng. 2020;4:69–83. https://doi.org/10.1038/s41551-019-0485-1.
Braga L, Ali H, Secco I, Giacca M. Non-coding RNA therapeutics for cardiac regeneration. Cardiovasc Res. 2020. https://doi.org/10.1093/cvr/cvaa071.
Eyileten C, Wicik Z, De Rosa S, Mirowska-Guzel D, Soplinska A, Indolfi C, et al. MicroRNAs as diagnostic and prognostic biomarkers in ischemic stroke-a comprehensive review and bioinformatic analysis. Cells. 2018. https://doi.org/10.3390/cells7120249.
Alivernini S, Gremese E, McSharry C, Tolusso B, Ferraccioli G, McInnes IB, et al. MicroRNA-155-at the critical interface of innate and adaptive immunity in arthritis. Front Immunol. 2017;8:1932. https://doi.org/10.3389/fimmu.2017.01932.
Acknowledgements
This work was written by the members of the International and Intercontinental Cardiovascular and Cardiometabolic Research Team (I-COMET; www.icomet.science). The authors thank the professional linguist Magda Fitas-Dukaczewska for her support.
Funding
This work was supported financially as part of the research Grant 2019/ABM/01/00037 from Medical Research Agency, Poland. This work was implemented with CEPT infrastructure financed by the European Union-the European Regional Development Fund within the Operational Program “Innovative economy” for 2007–2013.
Author information
Authors and Affiliations
Contributions
Writing—original draft preparation, D.J., A.S., W.L., J.J.-P., A.F., A.N., C.E.; P.C., writing-review and editing, D.J., C.E., J.S.M, S.R., M.P., visualization, C.E.; supervision, C.E., M.P., H.S. and S.R.; graphical design, C.E., Z.W. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Authors declare no conflict of interest related to this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jakubik, D., Fitas, A., Eyileten, C. et al. MicroRNAs and long non-coding RNAs in the pathophysiological processes of diabetic cardiomyopathy: emerging biomarkers and potential therapeutics. Cardiovasc Diabetol 20, 55 (2021). https://doi.org/10.1186/s12933-021-01245-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-021-01245-2